
An introduction to systems toxicology
Nick J.
Plant
School of Biosciences and Medicine, University of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: N.Plant@Surrey.ac.uk
First published on 11th August 2014
Abstract
The science of toxicology is the science of the system. Toxicologists aim to understand and predict the adverse effects of chemicals on biological systems. As biological systems are extremely complex, the challenge of predicting human toxicity early in the drug discovery process is immense. In the past decades, a huge effort has been undertaken to characterise the impact of chemicals on biological systems using in vitro, pre-clinical and clinical approaches. This has led to a vast amount of knowledge on the biology of systems, especially as a result of the data deluge from -omic level investigations. However, a lack of robust and comprehensive integration has meant that this wealth of data has still not led to accurate prediction of toxicity in a single system, or the ability to extrapolate robustly between systems. The new discipline of systems toxicology aims to take the computational approaches developed in systems biology and apply them to toxicology-related questions. This review will examine approaches ranging from relational databases that are both repositories for curated information and screening tools in their own right, to the potential of digital organisms in systems toxicology. Both the basic methodologies and how best they may be applied to safety assessment of chemicals will be covered. This integrated examination of toxicological data is predicted to herald a step-change in our ability to both understand and predict adverse effects of chemicals.
General introduction
A major driver in toxicology research is to understand, and ultimately predict, the adverse effects caused by xenobiotics. Before we can understand which chemicals may elicit a toxic response, or the mechanism by which this is achieved, we must fully define the biological phenotype that we wish to describe as an adverse event. Obviously the more detail that we can include in this description, the more robust downstream predictions are likely to be. Once we have defined the adverse event, we can begin to predict which xenobiotics will cause it. Perhaps the simplest approach for such prediction is guilt by association; chemicals with similar structures or modes of action are assumed to share similar biological effects, including toxicology. Beyond guilt by association (and this in itself is by no means simple), we can begin to include mechanistic understanding, allowing us to more fully examine the likelihood that a toxic behaviour could be caused. A final approach stratifies the population, identifies those most sensitive to the toxic effect and attempts to define the factors that predispose these individuals to the adverse effect.These approaches are often combined, with the term adverse outcome pathway1 increasingly used to describe the comprehensive risk assessment of human exposure to xenobiotics.
While there is an extensive literature identifying adverse effects, the molecular underpinnings for many of these adverse effects are poorly understood. One reason for this is the sheer complexity of the human body.
As depicted in Fig. 1, a traditional view of drug action envisages a single, specific interaction between the drug and a target protein leading to the desired pharmacology and therapeutic efficacy. Interactions that do not occur with the target protein (off-target) are often seen to be the cause of undesirable toxicity. However, in reality, any single chemical will almost certainly interact with multiple proteins, which in turn will also interact with an array of other proteins. It is this complex interaction network that underlies both the desired pharmacology and undesired toxicology of a drug, often through over-lapping mechanisms.
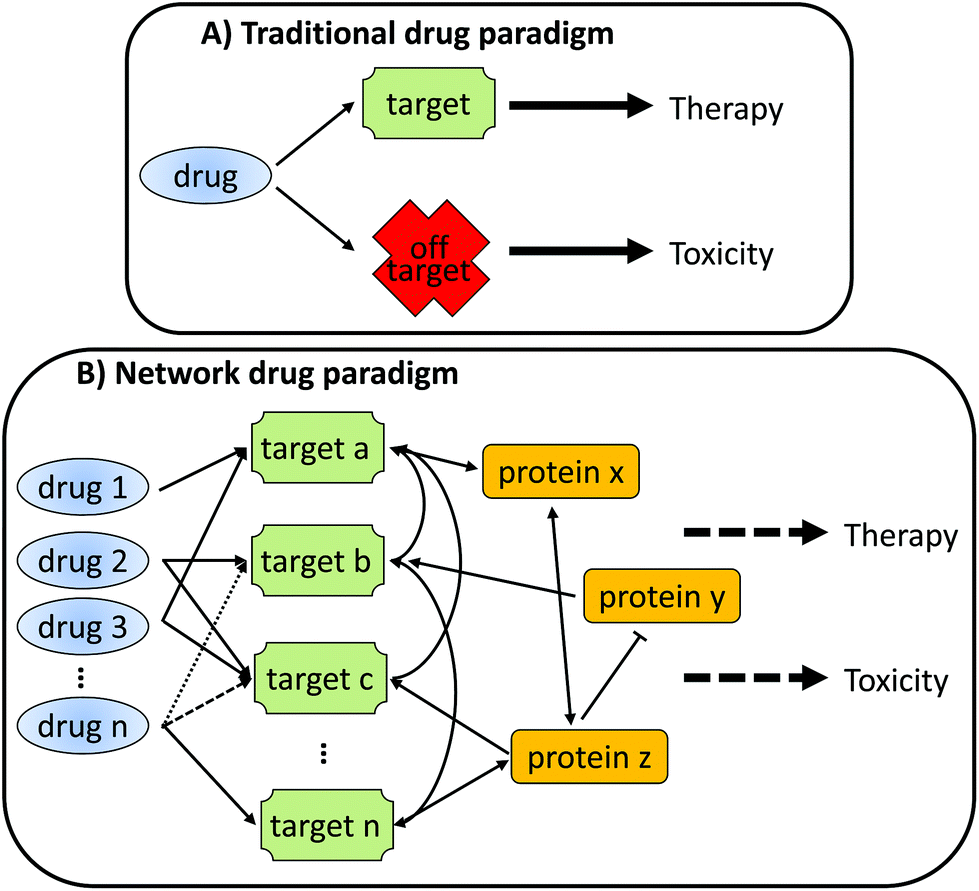 | ||
| Fig. 1 Traditional and Network paradigms for drug action. (A) In the traditional view, drugs demonstrate a specific interaction with the target protein, leading to the therapeutic effect, while an off-target interactions lead to toxicity. (B) In a network drug paradigm, drugs may interact with one or more target proteins with differing affinities. These target proteins may interact with each other, either directly or indirectly, in both positive (arrows) and inhibitory (end-bar) modes. The net effect of all these interactions may lead to both desired therapy and toxicity, with the two not being necessarily mutually exclusive. An important extension to the network drug paradigm allows for the use of combination drug cocktails to specifically tailor the net response, increasing therapeutic efficacy while minimising toxicity. | ||
To understand how this interaction network leads to either therapeutic efficacy or toxicity, or both, requires the adoption of a network view of drug action. Indeed, an extension of the network drug paradigm is that it may be more effective to use multiple drugs to drive towards a particular biological effect (e.g. therapeutic treatment of a disease). Each of the drugs in the combination may have limited effect on their own, and they will each target different aspects of the network; however, their combined effect will result in therapeutic efficacy with a reduced toxic liability.2,3
To make the most of the network drug paradigm, it is (obviously) necessary to understand the network, allowing us to predict the effect of single/multiple perturbations. As technology and biological understanding improves, we are able to design experiments of increasing complexity and resolution, further adding to the wealth of data that already exists to describe biological phenomena. However, the very nature of this data deluge means that it is becoming increasingly difficult to both identify and successfully use the relevant information, This has led to a shift in biomedical research away from producing data (the omics era) to understanding these legacy datasets (the systems era). A relatively new child of the systems era is systems toxicology, where large amounts of both de novo and legacy data are integrated to gain novel insights on the link between molecular interactions and adverse effects.4–6 This data may be derived from targeted (transgenic) animal or in vitro studies, through single omics level datasets (e.g. the transcriptomic analysis of multiple hepatotoxicants7), or multi-omic datasets (e.g. transcriptomic, proteomic and metabonomic analysis of methapyrilene hepatotoxicity8).
An important aspect of systems toxicology is the ability to study the emergent properties of biological systems by examining large scale networks rather than individual pathways or proteins. Emergent properties are those properties of a system that could not be predicated by studying the individual components in isolation; perhaps the most striking emergent property is life, as the totality of a biological organism cannot be predicted by studying a single cell in isolation.2,9 Given the poor record of scientists in predicting the adverse effects of xenobiotics from disparate datasets, one logical conclusion is that many toxicological effects are in fact emergent properties from the xenobiotic perturbation of biological systems. The rise of systems toxicology as a specific sub-discipline within toxicology can be seen by the exponential rise in publications that refer to either systems toxicology of network toxicology in the last decade (Fig. 2). Remarkably, the earliest publication using “systems toxicology” was published in 2003,10 demonstrating what a new discipline it is.
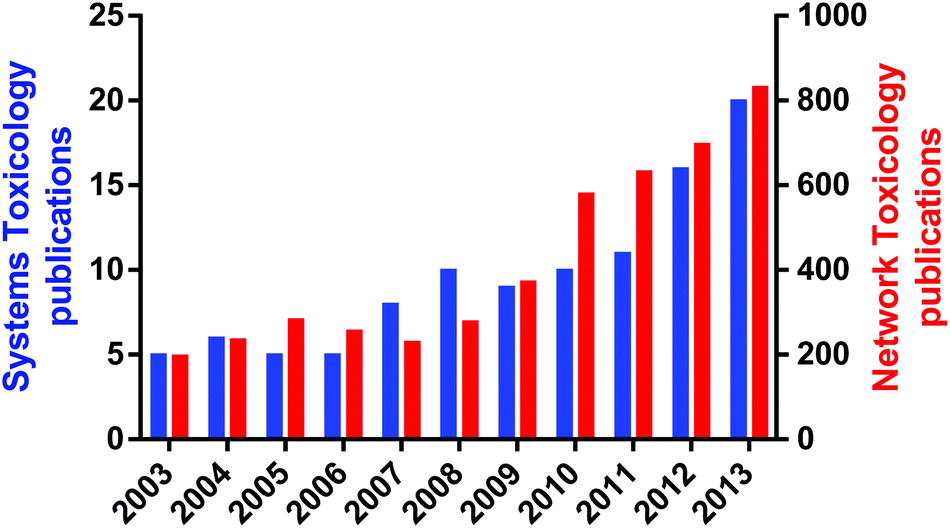 | ||
| Fig. 2 System and Network publications in Toxicology. Data are from Web of Science using “systems toxicology” (blue bars) or “network AND toxicology” (red bars) as query terms within article titles, keywords and abstract. | ||
This review will consider the computational approaches available for systems toxicology, and how they complement all aspects of the drug discovery pipeline.
Systems toxicology: multiple approaches, one aim
Perhaps the most confusing aspect of systems toxicology to the uninitiated is the sheer number of different approaches that are available to the researcher. It is thus necessary to first decide what you wish to gain from a systems toxicology approach and the data available to you. Together, these questions will help define which approach(es) are most appropriate. There are many possible decision trees, but they can be essentially encapsulated in the following questions.(1) Do I need to understand the mechanism?
(2) Is the biology to be examined well understood?
(3) Is the biology to be examined well characterised?
(4) Does the biology occur within a single cell, a single organ, or at the level of the whole organism?
If the answer to the first question is no, then a relational approach may be optimal. In a relational approach, associations between network components are predicted without necessarily understanding the mechanistic underpinning. These associations may extend from predicting chemical–protein or protein–protein interactions, through to predicting toxicity of a chemical based upon its structural fragments. Relationships may be defined by rules, statistical associations or collation of literature data.
If the answer to the second question is also no, then relational approaches may also be optimal; you cannot mechanistically model what you don't know. Providing that the answer to the second question is yes, then a modelling approach may be applicable, with the third and fourth questions setting the type of model that can be used. The answer to the third question will decide whether any model will be quantitative (i.e. based on known network connectivity and with kinetic/abundance values) or qualitative (i.e. based on known network connectivity but without kinetic/abundance values). Quantitative models can simulate biology in a highly precise manner, predicting dose- and time-courses with biologically meaningful values. In contrast, qualitative models will predict the behaviour of a system, but without ‘real’ numbers. Both approaches are of value, and the decision of which to use is often driven by the data availability. Finally, the answer to the fourth question will help to drive the degree of reductionism that is required within the model. As a model increases in complexity, essentially as it tries to reproduces more biology, then it becomes increasingly taxing both in terms of the biological data required to make the model and the computational power required to run the model. Hence, a trade-off between model size and complexity is required, with larger models often reducing complex biological sub-systems into simpler units that are more feasible to model.
The relationship of the systems toxicology approaches described within this review to the drug discovery/development pipeline is shown in Fig. 3.
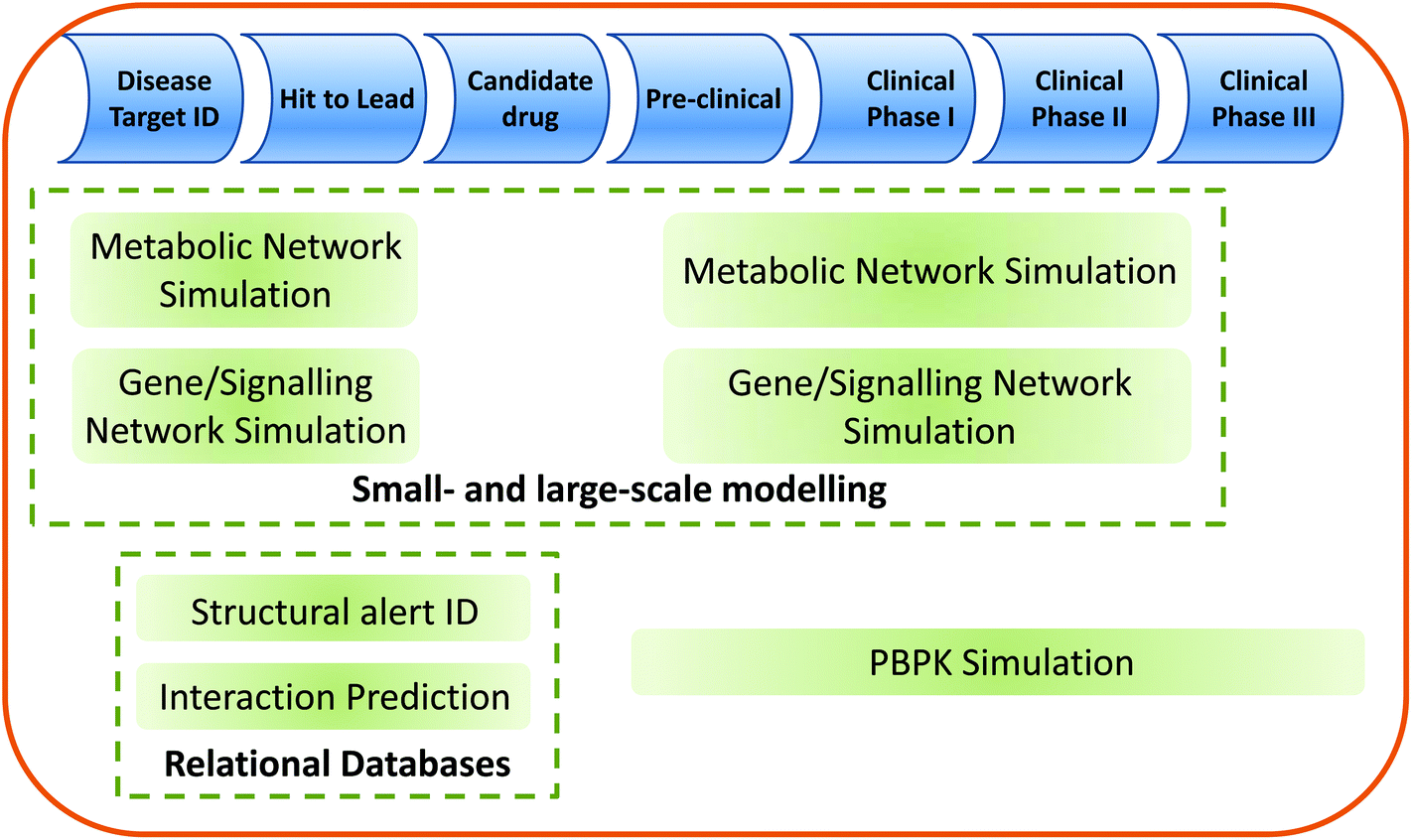 | ||
| Fig. 3 Potential roles for systems toxicology in drug discovery and development. Systems toxicology has the potential to add significant value both in the early (discovery) and later (development) phases of the drug pipeline. Potential areas of application for the approaches discussed in this review are indicated. PBPK = Physiologically-based pharmacokinetics. | ||
Relational approaches: predicting toxicity based upon prior knowledge
Relational approaches are in many ways simplistic in their methodology, but can be highly effective for identifying exposure–response relationships. The underlying paradigm for relational approaches in one of guilt by association; essentially, rule- or statistical-based approaches are used to determine the weight of evidence that two data are linked. For example, the probability that a chemical with moiety X will be a mutagen, or the probability that a chemical–protein or protein–protein interaction will occur.At one end of the spectrum, relational databases can be automatically generated using bioinformatic tools to mine data from online resources: for example, the phrase “X and Y interact to activate Z” can be identified from within the text of a journal article, creating the interaction between X and Y within the database. An automated curation process generates a very large database of potential interactions, but automation invariably leads to a reduction in quality, with interactions sometimes incorrectly assigned. To mitigate this issue and the reduced predictive power associated with it, most databases contain a degree of manual curation. This painstaking process assesses the evidence for any given interaction in a more robust manner. Obviously, a database that is manually curated will tend to provide higher quality predictions, but the biological coverage of each database may be less due to the far greater person-hours required to create such databases. Where relationships are explicitly defined in the literature, statistical approaches allow the prediction of how likely such an effect is. These models range from the purely qualitative to fully quantitative, and encompass only a few biological sub-systems to the entire metabolism of the cell. Finally, such models may be extended to examine organ- or even organism-level responses, albeit with a significant reduction in the resolution of the data. Table 1 presents some of the commonly used relational databases, covering both those specifically aimed at toxicity prediction and more general databases of interactions.
| Relational approach | Website | Reference |
|---|---|---|
| Chemical perturbation effects | ||
| LINCS: Library of integrated network-based cellular signatures | http://www.lincsproject.org/ | 18 |
| ArrayExpress: Gene expression signatures | http://www.ebi.ac.uk/arrayexpress/ | 19 |
| GEO: Gene expression omnibus | http://www.ncbi.nlm.nih.gov/geo/ | 20 |
| ToxCast | http://www.epa.gov/ncct/toxcast/ | 21 |
| Rule-based toxicity prediction | ||
| DEREK Nexus | http://www.lhasalimited.org/products/derek-nexus.htm | 11 |
| Toxtree | http://toxtree.sourceforge.net/ | 22 |
| Rule-based metabolism prediction | ||
| Meteor Nexus | http://www.lhasalimited.org/products/meteor-nexus.htm | |
| (Q)SAR-based toxicity prediction | ||
| TOPKAT: TOxicity prediction by komputer assisted technology | http://accelrys.com/solutions/scientific-need/predictive-toxicology.html | 12 |
| CEASAR: Computer-assisted evaluation of industrial chemicals according to regulations | http://www.caesar-project.eu/ | 23 |
| TEST: Toxicity estimation software tool | http://www.epa.gov/nrmrl/std/qsar/qsar.html#TEST | 24 |
| Chemical-protein interaction prediction | ||
| CARLSBAD: Drug-target interactions | http://carlsbad.health.unm.edu/wp/ | |
| ChemProt: chemical-protein predicted interactions | http://www.cbs.dtu.dk/services/ChemProt-1.0/ChemProt-2.0/ | 25 |
| SLAP: Drug-target prediction | http://cheminfov.informatics.indiana.edu:8080/slap/ | 26 |
| Protein–protein interaction and pathway prediction | ||
| Bio-entity network | http://stat.fsu.edu/~jinfeng/IBN.html | 27 |
| IMID: Integrated molecular interaction database | http://integrativebiology.org/ | 28 |
| APID: Agile protein interaction data analyzer | http://bioinfow.dep.usal.es/apid/index.htm | 29 |
| IntAct: Molecular interaction database | http://www.ebi.ac.uk/intact/main.xhtml | 30 |
| BioGRID: Biological general repository for interaction | http://thebiogrid.org/ | 31 |
| Datasets | ||
| STRING: Function protein interactions | http://string.embl.de/ | 32 |
The use of relational databases has been commonplace in the pharmaceutical industry for many decades, especially in the early stages of drug discovery/development where it is necessary to reduce a large number of potential chemical hits into a smaller number of lead compounds.
In silico screening of compound libraries can be used to identify structural alerts associated with adverse effects, allowing de-selection of chemicals likely to elicit toxicity later in the development programme. Two common approaches are rule-based systems and structure–activity relationships. Programmes such as DEREK11 use the rule-based approach, identifying chemical structural alerts based upon an extensive manually curated knowledge base of experimentally-validated effects. In contrast, programmes such as TOPKAT12 use (quantitative) structural–activity relationships systems to associate toxicological endpoints with the predicted chemical space required to elicit this endpoint. While these two approaches each have different advantages and disadvantages, their overall ability to detect common toxic endpoints, such as mutagenicity is remarkably similar.13–15 If used within the accepted bounds of the systems (structural rules16 and toxicophores,17 respectively) these approaches are very powerful and yield important information for the prioritisation of compounds drug discovery.
A recent development in SAR-based toxicity screening was presented by Lounkine et al.33 They first defined 73 biological targets associated with adverse drug outcomes ranging from sleep disorders to tachycardia. A similarity ensemble approach was then used to compare the structure of 656 marketed drugs to known ligands for each of these 73 targets, assessing if they shared greater similarity than that expected by chance. Several hundred previously annotated interactions were identified, but also 893 interactions that were previously unknown. Of these, 125 were shown to have IC50 concentrations in the micromolar range, and hence be of potential clinical significance. While such studies demonstrate the power of large-scale screening they also show some of the limitations with such an approach. In this case, while 125 new drug-adverse outcome pathway interactions were confirmed, over three quarters of the predicted interactions were disproved when tested experimentally. This is perhaps a timely reminder that as soon as tools are taken outside of a tightly constrained question-space their utility as a definitive yes/no predictor drops considerably.
Small-scale models
An alternate approach to relational databases is to build a computational model of an adverse effect of interest. The aim of such models is to better understand the biological conditions (often referred to as design principles) that underlie the adverse effect, allowing an improved prediction of when these rules will be met, and toxicity seen. Provided that at least some molecular underpinning is known for the adverse effect, then a mechanistic modelling approach is possible. The exact nature of the model(s) that can be employed will depend upon both the level of mechanistic detail known, and also the desired question. However, all these modelling approaches will utilise the same basic steps. In essence, the biological system in question is broken down into a series of reactions, each of which can be described by a mathematical formula. Perhaps the best known examples of the application of such approaches in the pharmaceutical arena are physiologically-based pharmacokinetic (PBPK) models used to predict drug disposition within the body.A key aspect of PBPK modelling is its reductionist approach, simplifying the model such that neither biological knowledge nor computational factors are limiting, but still allowing robust whole-organism predictions. A full description of PBPK is outside the scope of the review, but the interested reader is referred to reviews by Rowland34 and Bouzam.35 PBPK models are an excellent example of how meaningful predictions can be made with only a limited amount of mechanistic information.36 However, this reductionist approach, where multiple biological events are reduced to a single formula, has the limitation that the role of individual factors within a biological response may be misrepresented or even ignored. For example, drug transport across cell membranes is a complex process involving both passive and active elements in both directions. In PBPK, this process is often reduced to a single term (J), which captures the net movement across the membrane. However, as we better understand the importance of drug transporters in setting drug disposition, multiple-drug resistance and toxicity increases, it is clear that this single term may not capture the biology robustly. To address this either the differential equation that defines J must be made increasingly more complex, or multiple terms must be used to reflect each of the different processes. The former approach is effective, but rapidly leads to a complex differential equation that is unintelligible to the non-expert. The latter approach is that used by ‘bottom-up’ modelling approaches, and is described below.
The modelling of small networks, or even individual pathways, is often described as the ‘bottom-up’ approach. In such models, the level of biological knowledge required is high, including details on reaction kinetics (Km, Vmax, kcat, Hill coefficient) and robustly determined protein abundance values. This high resolution data allows the interactions of chemicals with enzymes and transporters to be described by ordinary differential equations (ODEs) describing mass action, Michaelis–Menten or Hill kinetics, for example.
Table 2 provides some useful resources for the definition of network topology, and the sourcing of kinetic and abundance data. However, it should be noted that deriving the necessary parameters for even a relatively small network will entail many hours of literature searching, and almost certainly some wet-lab experiments to fill in important data gaps.
| Database | Website | Reference |
|---|---|---|
| Network Topology | ||
| KEGG: Kyoto Encyclopaedia of Genes and Genomes | http://www.genome.jp/kegg/ | 55 |
| BioCyc: Pathway/Genome databases portal | http://biocyc.org/ | 56 |
| MetaCyc: Metabolic databases portal | http://metacyc.org/ | 57 |
| Enzyme nomenclature and annotation | ||
| ExplorEnz: The enzyme Database | http://www.enzyme-database.org/ | 58 |
| TCDB: Transporter classification database | http://www.tcdb.org/ | 59 |
| IntEnz | http://www.ebi.ac.uk/intenz/ | 60 |
| HAMAP | http://hamap.expasy.org/ | 61 |
| Protein abundance | ||
| paxdb | http://pax-db.org/#!home | 62 |
| Enzyme kinetics | ||
| BRENDA | http://www.brenda-enzymes.info/ | 63 |
| EzCatBD | http://mbs.cbrc.jp/EzCatDB/ | 64 |
| SABIO-RK | http://sabio.h-its.org/ | 65 |
These models can be easily generated using any of a large number of freely available programs. These range from programs with excellent graphical interfaces that are easily accessible to the biologist, but are limited in their analysis options (e.g. CellDesigner; http://www.celldesigner.org/37,38), to more complex equation-based inputs such as COPASI (http://www.copasi.org/39), which have more extensive network analysis tools. One common feature of these programs is that they use the systems biology mark-up language (SBML40) and systems biology graphical notation (SBGN41) formalisms. This means that generated models are transferable between over 250 different programs, allowing models to be created in one program and simulated in another. Finally, online collections of models, such as JWS Online (http://jjj.biochem.sun.ac.za/42) and BioModels (http://www.ebi.ac.uk/biomodels-main/43) provide a free searchable resource of pre-defined (curated) models, available as SBML files for simulation in a wide range of software. This allows other users to both use published models for their individual research projects, but also fosters an environment of collaborative development, where several groups may produce iterative improvements of a single biological model. The models available from both these resources simulate many aspects of biology, from regulatory gene and signalling networks such as MAPK44,45 and nuclear receptors,46,47 through drug transport48 and nuclear transport49,50 and, to more complex behaviours such as circadian rhythmicity.51,52
A good example of how small-scale modelling can be used to understand the design principles of an adverse outcome is demonstrated through the study of the glutathione anti-oxidant defence network in mammals. A quantitative model of the glutathione network was first produced by Reed et al.,53 and encompassed one-carbon metabolism, trans-sulfuration and glutathione synthesis, transport and metabolism. The model was able to reproduce some of the known biology associated with the glutathione network, such as the sensitivity of glutathione pools to oxidative stress. In addition, Reed et al., used the model to examine the emergent properties of the network, such as the impact of trisomy Ch21 (Down's syndrome) on cellular oxidative stress. As several genes involved in the network are present on Ch21, Down's syndrome leads to over-expression of the encoded proteins.
When these increased levels were simulated within the model, biochemical alterations known to occur in people with Down's syndrome were predicted, such as a functional folate deficiency. This model was recently expanded by Geenen et al., adding in the γ-glutamyl cycle, ophthalmic acid synthesis and the detoxification of paracetamol.54 This provides an ideal example of the iterative nature of computational modelling; the original model of Reed et al., was unable to reproduce the experimental observations of Geenen et al., when THLE-2E1 cells were exposed to paracetamol. Addition of a regulatory signal in the model allowing increased γ-glutamyl cysteine synthetase in response to oxidative stress did allow the model to reproduce the experimental data robustly, and this up-regulation was confirmed experimentally. Hence, not only did an iterative cycle of model development → experimental testing → model refinement allow the model to better capture the known biology, but it also revealed an important adaptive response to oxidative stress.
In the above example, a computational model was used to understand a mechanism of toxicity. Essentially, the model identifies all the molecular components that are required to reproduce the toxicity observed in vitro/in vivo. A related question is to understand the relative importance of different parts of a network in any given biological scenario. To answer such a question requires the use of a technique such as Metabolic Control Analysis (MCA). The original view that any given pathway is controlled through the activity of a single, rate-limiting enzyme is now known to be an over-simplification.66 In reality, each reaction within a pathway will exert some control on the metabolite flow through the entire pathway, with the level of control varying between reactions. This concept becomes even more important when considering networks where several interlinked pathways may impact upon each other. MCA is an important tool for studying such effects, identifying emergent properties based on the individual reaction properties and their interactions. In the example of glutathione detoxification, MCA was used to predict the importance of methionine influx on the capacity of the anti-oxidant defence system.54 Methionine is a major input into the glutathione network, being a precursor for glutathione synthesis. The model correctly predicted the existence of a threshold for intracellular methionine levels, below which the network was unable to safely detoxify even relatively low exposures of paracetamol (5 mM).
MCA also has an important application in the identification of metabolic chokepoints. These are the proteins within the network that if disrupted are most likely to lead to a dramatic shift in network behaviour. For toxicologists, this would be the shift in biological phenotype that results in an adverse event. However, it is easy to see how such an approach can be reversed to identify therapeutic targets. A good example of such an approach can be seen through the modelling of central carbon metabolism, including cholesterol. In the model of Maier et al., MCA predicts HMG-CoA reductase to have an exceptionally high control coefficient (0.5) in the production of cholesterol.67 This is consistent with the enormous clinical success of the statin class of drugs, which act as HMG-CoA reductase inhibitors.68
The model of the glutathione defence network described above simulates reactions occurring in a specific cell type, in this case hepatocytes. However, before the drug reaches the target cell, it must enter the body and be subject to the processes of absorption, distribution, metabolism and excretion. As such, an obvious extension for such cellular models is to combine them with PBPK models; the PBPK model predicts the concentration of paracetamol within the liver, and the glutathione defence network model predicts the impact of this exposure. These two modelling approaches use different scales of information, both in terms of the detail of the parameters (individual reactions versus composite reactions) and the scale of the reactions (small volumes, fast time versus large volumes, mid- to slow times). Due to this, such combination models are usually referred to as multi-scale. With regard to paracetamol toxicity, multi-scale models have been proposed by Ben-Shachar et al.,69 and Geenen et al.70 Both models combine classical PBPK models of paracetamol disposition with deterministic model of liver metabolism. In doing so, they are able to both replicate experimental data and provide insight into the impact of enzyme polymorphisms and glutathione metabolic capacity on the outcome of paracetamol overdose, respectively. These models provide important proof-of-concept studies demonstrating that it is possible to create multi-scale computational models that can capture the complex biology observed during toxicity, and provide novel insights into risk and/or mechanism. However, it is now important that these approaches are fully developed so that they can not only reproduce classical toxicity paradigms, but also add value to the safety assessment of novel chemicals.
Not all small-scale models need to be fully quantitative; indeed, it is quite possible that for even a small network the required abundance and kinetic parameters may not be completely known. In such cases, it is necessary to use a qualitative model, which simulates the behaviour of a network rather than trying to reproduce faithfully the time and concentration curves for all species within the network. Indeed, for the larger-scale models required to reproduce whole-cell, -organ, or -organism behaviour, such an approach may be the only viable option. Qualitative models are of particular value if one wishes to either understand the emergent properties of the network (i.e. the global biological response to a stimulus), or to examine if a particular scenario is possible (i.e. following a particular stimulus can this behaviour happen?). The latter case has obvious applications in toxicology, where we must first identify if something is possible (i.e. hazard), and then assess the likelihood that it will occur (i.e. risk). A common approach for such qualitative models is the use of Petri nets. Petri nets are a directed bipartite graph consisting of places and transitions, which are connected by arcs that define their relationship. As such they are ideal for representing decision trees, and this approach has been employed to undertake environmental risk assessments.71,72 For example, Ozbek and Pinder developed a fuzzy-Petri net to represent an expert knowledgebase on benzene contaminated groundwater. This knowledge base was formed of textual statements from public-health professionals, and utilised to allow refinement of responses to potential benzene contamination.71 In addition to their use to describe decision trees, Petri nets can be used to describe biological networks, with places representing chemicals, transitions representing reactions and arcs defining the relationships between the two.73 Using this approach, Petri net formalism has been used to model small networks such as JAK/STAT signalling74 and apoptosis,75 as well as examine more complex phenotypes such as metabolic disorders.76 This type of biology-orientated Petri net has yet to be employed to understand or predict toxicity on its own, but it is being integrated into larger-, multi-scale models to create dynamic biological models, as will be described below.
Large-scale and multiscale modelling
The study of small networks has obvious advantages with regards to the high quality experimental data that can be used to generate and then validate each model. However, there are also a number of disadvantages, with perhaps the most challenging being that there is always a chance that something important has been excluded from the model. Within biological networks there exists considerable redundancy, such that there is often several ways to achieve a particular biological phenotype.77 Due to this, it is possible that a model will faithfully reproduce an in vivo behaviour, but will be unable to predict the results of perturbations to this network. For example, a simple kinetic model could reproduce the metabolism of paracetamol in the liver, describing its conversion to non-toxic and toxic metabolites. However, unless the model includes gene expression processes, it could not predict the interaction between alcohol and paracetamol as this relies on protein stabilisation of CYP2E1 by alcohol. Careful and rigorous validation of models against experimental data can mitigate this risk to some degree, but can never completely remove it. The obvious solution to this problem is to make the network larger; so large that it encompasses all the known interactions within the system under study.The commonest approach for large-scale modelling is the reconstruction of genome-scale metabolic networks (GSMNs), which essentially capture all enzymatic and transport reactions possible within a cell. GSMNs were originally reconstructed for prokaryotes such as E. coli, which have smaller genomes and considerably simpler metabolic networks. However, in the past few years advances both in computational power and biological understanding has allowed reconstructions of genome-scale metabolic networks for mammals to be generated. Such reconstructions may be general, capturing all the possible metabolic reactions that could occur in any cell type (e.g. ReconX78,79), or represent a specific target cell (e.g. the hepatocyte-specific Hepatonet180). As with the small-scale models previously discussed, the majority of these reconstructions are publicly available through websites such as BioModels, as well as sites specifically aimed at GSMNs (e.g. MetaNetX81). Table 3 lists some web resources with valuable information for genome-scale network reconstruction.
| Relational Approach | Website | Reference |
|---|---|---|
| Metabolic network reconstruction | ||
| MetaNetX | http://metanetx.org/ | 81 |
| BiGG: Biochemical, genetic and genomic database | http://bigg.ucsd.edu/ | 93 |
| Model SEED | http://seed-viewer.theseed.org/seedviewer.cgi?page=ModelView | 94 |
| General network reconstruction | ||
| Pathway tools software | http://bioinformatics.ai.sri.com/ptools/ | 95 |
| KEGG mapper | http://www.genome.jp/kegg/tool/map_pathway.html | 55 |
| ERGO: Genome analysis and discovery | http://www.igenbio.com/ergo_bioinformatics_and_analysis | 96 |
| Cytoscape | http://www.cytoscape.org/ | 97 |
| IPA: Ingenuity pathway analysis | http://www.ingenuity.com/products/ipa | 98 |
One current gap in the reconstructed GSMNs is that they are predominantly human. While this is ideal for examining human physiology (normal and diseased) and the impact of toxicant exposure, it does not allow the examination of pre-clinical species responses. The development of GSMNs for the major pre-clinical species is an important future direction to ensure that this powerful technology can be used to improve species extrapolation during the pre-clinical to clinical translation, or to aid human risk-assessment where the knowledge base relies considerably upon animal data.
Reconstructions of whole cell metabolism are ideally placed to examine the metabolic landscape of the cell. This landscape reflects not only what reactions are possible, but which are likely to occur. As such it is akin to a national power grid, where the flow of electricity through different parts of the network alters to meet demand. Furthermore, this landscape will change in response to stimulus (e.g. high demand in one part of the power grid) or disruption (e.g. loss of certain power lines). The metabolic landscape may, therefore, provide important novel insights into disease progression and toxicity, showing how the body's metabolism changes in response to the disease (either globally or locally). In addition, once these changes are understood they present obvious target for drug discovery. The metabolic landscape of a cell can be predicted by exploring all the possible combinations of reactions that could occur in the cell. Obviously, such a list would be very large, so the solution space is made smaller by adding constraints to the possible solution, such as reaction stoichiometry and thermodynamic considerations; hence, GSMNs are often referred to as constraint-based models (CBM). A standard method to identity the metabolic landscape of a cell is flux balance analysis (FBA), which is used to explore the solution space of reaction fluxes within a CBM (reviewed by Orth, Thiele and Palsson82). In addition to the use of stoichiometric and thermodynamic constraints, FBA uses an objective function to determine the desired phenotype of the cell (e.g. the production of a particular set of metabolites). Reaction fluxes are set to optimise the production of this objective given the constraints of the system. Such analysis can predict which metabolic subsystems are most likely to be active to meet a given objective function (biological behaviour). FBA has been used extensively in the field of biotechnology to optimise production processes. For example, production of an antibiotic can be optimised by growing bacteria in the correct nutrient mixture to both optimise antibiotic production per bacterium (antibiotic production as an objective function)83 and total bacterial growth (biomass as an objective function). In the former case, the objective function is simply the antibiotic in question. In the latter case, the objective function is more complex, being a combination of all factors required for bacteria to divide (amino acids, nucleotides, ATP etc.). Recently, the same approach was used to define the optimal medium for hepatocyte growth.84 Yang et al., derived an objective function containing key components of the hepatocyte phenotype (e.g. albumin and urea production), and then used FBA to determine the optimal amino acid composition of growth medium to support this objective function. Such an approach provides a novel solution to the long-standing issue that hepatocyte-specific features rapidly disappear in vitro, as the cells quickly de-differentiate and lose their phenotype.85
In keeping with the central tenet of systems biology that models should be constantly refined through the integration of new information, general GSMNs can be refined through the use of systems level data such as transcriptomics or proteomics. This data can be used to further constrain the FBA, producing a flux distribution reflecting those genes that are expressed and those that are not. Essentially, flux distributions are explored that optimise production of the objective function while meeting the stoichiometric and thermodynamic constraints of the system, as normal. In addition, these flux distributions are further constrained to maximise flux through reactions catalysed by proteins whose transcripts are highly expressed, and minimize flux through reactions catalysed by low/non-expressed proteins.86 This approach has been particularly useful for exploring genotype–phenotype relationships that diverge during disease development. For example, Folger et al. used transcriptomic data to generate a ‘cancer GSMN’ based upon the NCI-60 cell line resource.87 Critically, the increasing number of clinically-derived – omic level datasets raises the tantalizing prospect of personalised GSMNs to understand both general mechanisms and inter-individual variation in disease progression and toxicity. For example, Agren et al. generated personalised GSMNs for 27 hepatocellular carcinoma patients based upon proteomic data. Examination of these personalised GSMNs allowed the prediction of therapies that would work for the entire cohort, but also those therapies were patient stratification would produce optimal response.88 The application of CBMs to understand and optimise treatment of human disease is a rapidly developing field, with the real prospect of significant clinical benefits being delivered in a relatively short time frame.
By contrast, the use of such approaches in systems toxicology is still in its infancy, with examples currently limited to classical toxicants such as paracetamol.89 However, given the novel insights already provided by CBMs for the understanding of disease, it is clear that systems toxicology will likewise benefit in the next few years.
A major difference between small-scale and large-scale modelling approaches is their treatment of the cell as a dynamic environment. Small-scale models often encompass gene regulation (transcription and translation) and signalling pathways (e.g. MAPK etc.). As such, they are ideally placed to simulate the dynamic response of cells to chemical stimuli. By contrast, large-scale models rely upon the assumption of a steady-state. This assumption states that the levels of chemicals do not change within the system over the time of analysis. To achieve this, the sum of all fluxes producing a chemical must be equal to the sum of all fluxes removing that chemical. While this is an important assumption to permit complex analyses such as FBA, it does deviate from the true, dynamic nature of biological systems, where the levels of enzymes (and the bounds of their associated reaction fluxes) constantly alter in response to an ever-changing chemical environment. We rationalise the use of a steady-state model by considering the time separation between chemical reactions (fast; microsecond time-scale) and gene regulation and signalling networks (slow; minutes to hours). Hence, in large-scale models we explore the metabolic landscape of a cell in discrete time slices, such that the slower processes of transcription and translation do not have time to occur.
The assumption of a steady-state presents a significant problem to the systems toxicologist. By its very nature toxicity is a dynamic event, and thus should be modelled as such. Future developments must allow CBMs to be dynamic, incorporating both fast (enzyme reaction) and slow (transcription/translation and signalling) components. This will produce a truly dynamic, ‘living’ cell that is able to respond to alterations in the chemical environment. Under such a paradigm, levels of an external chemical (e.g. input to or output from the GSMN) would activate regulatory gene or signalling pathways, which would in turn alter the levels of metabolic/transport proteins. These changes would alter specific reaction bounds within the GSMN, altering the metabolic landscape. Such a living cell is critical in predicting the outcome of the chemical challenge; no effect, hormesis, adaptation or toxicity.
Various modification of FBA have been proposed to address this issue, including rFBA,90 iFBA91 and dFBA92 (regulatory, integrated and dynamic FBA, respectively). While all of these approaches have some merit, they also each have their own limitations. For example, rFBA does not differentiate between transcription and translation, reducing the biological resolution of the model, while iFBA and dFBA require deterministic models of regulation, meaning that they are limited to biological areas that are data dense. However, such approaches have been used successfully to examine toxicological mechanisms. In their model of the hepatocyte metabolic response to paracetamol, Krauss et al. used the dFBA approach to integrate an hepatocyte GSMN (Hepatonet1) with a PBPK model for paracetamol disposition.89 Essentially, they used a bow-tie design to provide dynamic simulation, whereby input and output PBPK models are ‘tied’ together through a GSMN; first, the PBPK model is solved for a specific time unit; second, the CLint from the PBPK model informs bounds within Hepatonet1; third, FBA explores flux distributions within Hepatonet1, and determines the flux towards paracetamol degradation; fourth, this flux informs CLint for the PBPK model.
The Krauss et al. model is a good demonstration of the linkage of drug ADME at the cellular- and organism-level allowing the reproduction of the response to a classical toxicant. However, as previously noted, dFBA is limited by the fact that it does not directly reproduce the transcriptional and translational processes that underlie the metabolic adaptation. This reduction in resolution within the simulation may lead to the misinterpretation, or complete loss, of important phenotypic behaviours.
A novel approach to undertake dynamic simulation of metabolism at the genome-scale was recently presented by Fisher et al. They proposed a novel framework, QSSPN (quasi steady-state Petri net) that uses a multi-scale approach to integrate the dynamic gene and signal regulatory components captured by small-scale networks and the genome-scale metabolic analysis of large-scale models.99 In this approach, a Petri net is used to represent the regulatory gene and signalling network, including transcriptional, translational and post-translational regulatory processes. Changes in the external chemical environment are sensed through the Petri net, which ultimately leads to alterations in the activity levels of target proteins; these alterations result in changes to the flux bounds of specified reactions within the GSMN. The metabolic landscape of the GSMN is then predicted through FBA, with the outputs from the GSMN providing feedback into the Petri net to complete the regulatory loop. Using this system, Fisher et al., were able to reproduce the dynamic behaviour of hepatocytes in response to cholesterol loading. Upon addition of excess cholesterol, nuclear receptors within the Petri net are activated, leading to activation of enzymes that metabolise cholesterol. However, this rapidly leads to an increased production of bile acids, undesirable as this can lead to the adverse outcome cholestasis. The QSSPN is able to sense this potentially toxic accumulation and initiate a feedback loop to reduce flux through cholesterol metabolism and increase transporter-mediated cholesterol efflux instead. This allows the effective clearance of cholesterol without excess production of toxic by-products. Finally, once cholesterol levels return to the pre-challenge level, the metabolic system resets to its original state. This demonstration of how a multi-scale simulation can capture the dynamic response of cells to chemical stimuli opens the way for more complex models that can understand and predict more complex pathologies. A particularly exciting development in the QSSPN framework is the ability to combine both qualitative and quantitative models. This opens the possibility of multi-scale models that contain different levels of resolution depending upon the available information. This will be particularly important to capture some of the more complex biological behaviours such as circadian rhythmicity, which can really only be described with a deterministic model. Their incorporation into qualitative large-scale models is crucial if we wish to fully understand the impact of, for example, circadian rhythms on drug action.100
Network drug targeting: a mechanism to reduce toxicity?
The established paradigm for drug discovery requires the identification of a therapeutic target, followed by the identification and subsequent optimization of compounds that are able to potently and selectively interact with this target. However, such an approach is often limited by several factors: first, it is often difficult to design compounds that will interact with a specific target while minimizing interactions with highly related molecules (off-target effects). This is further compounded by the fact that as compound selection is driven towards those highly potent molecules, even a small degree of off-target interaction can result in significant biological consequences. Second, even if a highly specific, potent compound can be identified, its properties will invariably have to be altered to improve pharmacokinetic parameters, leading to a necessary compromise between pharmacodynamic and pharmacokinetic optimization.101 Third, biological systems often possess redundancy, such that pharmacological modification of a single target can be compensated for by the use of an alternate system.102,103Even if such a series of hurdles can be safely navigated, it is necessary to use several test systems (in vitro and pre-clinical models) during drug development. This requires the ability to robustly extrapolate data from one system to another, which in turn requires an understanding of the differences between each network, and how this will impact on drug behaviour. Given the highly complex (and not fully understood) nature of the human biological network, it is thus not surprising that we do not always predict the effects of even a single, highly selective/potent drug. The result of poor prediction may be a lack of therapeutic efficacy in later stages of drug development, unexpected adverse drug events, or the emergence of drug resistance.104 The discipline of systems toxicology should increase our ability to better predict the impact of drugs on biological systems, and the extrapolation of these effects from one system to another. This should permit the identification of toxic liabilities of drug molecules much earlier in the drug discovery/development pipeline.
Systems approaches may present a further solution to reduce the risk of adverse events. The use of drug cocktails in certain therapeutic areas is well established, and relies on the use of multiple marketed drugs together to target several biological pathways at once, with the aim of increasing efficacy while reducing the risk of adverse effects and/or the development of resistance.105 Given that the design of optimal drug cocktails requires an innate understanding of network behaviour, the use of computational approaches is a natural partner to aid selection of optimal combinations.2,106 Proof-of-principle for such an approach was presented by Folger et al., who utilised a whole-genome scale metabolic network model of cancer metabolism to predict ‘synthetic lethality’ for drug pairings.87 The central paradigm of synthetic lethality relies on the adaptive nature of biological networks, and their robustness to chemical perturbation. For example, the efficacy of a drug against therapeutic target A may be mitigated by the expression of protein B, which represents a network survival adaptation. Combinations targeting both A and B would demonstrate increased efficacy, as both the therapeutic target and rescue system are targeted. Folger et al. not only demonstrated that such combinations could be identified computationally, but also that these predictions could predict responses of the NCI-60 cancer cell line collection to combination therapy.
An exciting extension of the network drug targeting paradigm is that if combination therapy is the aim from the start, then the biological effect of each drug alone might be minimal, and indeed may not even be seen as therapy under traditional definitions. However, the effect of these chemicals in combination on the network will be such that a significant therapeutic effect will be observed.2 On this basis, it may be necessary to alter the drug discovery paradigm, such that effective combinations are designed at the start of the discovery process, rather than through the combination of existing drugs. Such novel approaches are already being developed, using novel computational software to search for de novo combination therapies.107,108
The computationally-led design of drug cocktails appears to hold great promise for the generation of optimised therapies, including at a personalised level. However, an important unanswered question is whether this approach may generate not only the desired synergistic pharmacology, but also undesired synergistic toxicology. Indeed, for network drug therapy to be truly successful, it could be argued that there should be a reduction in adverse events compared to standard combination therapy. The answer to this question has been partly addressed by Lehar et al., who undertook large-scale metabolic simulations of over 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 multi-dose experiments relevant to a range of disease phenotypes. As expected, they demonstrated that combination therapies were generally more specific than single agent therapies. Critically, they also demonstrated that this additional specificity was because the combinations were able to exploit unique properties of the disease phenotype of the cell, a key claim of the network drug paradigm. With regards to toxicity, their simulations again supported the network drug paradigm, demonstrating a reduction in toxic liability for the majority of combinations, rather than increase.109
000 multi-dose experiments relevant to a range of disease phenotypes. As expected, they demonstrated that combination therapies were generally more specific than single agent therapies. Critically, they also demonstrated that this additional specificity was because the combinations were able to exploit unique properties of the disease phenotype of the cell, a key claim of the network drug paradigm. With regards to toxicity, their simulations again supported the network drug paradigm, demonstrating a reduction in toxic liability for the majority of combinations, rather than increase.109
Conclusions
Predicting the impact of chemicals on the human body is an extremely complex process due the sheer number of interactions possible. One solution to this problem has been to drive drug development towards more selective drugs, with the aim that this will remove large parts of the network from consideration. However, this approach has not been fully successful due to a number of complicating factors: First, even highly selective compounds will possess some off-target interactions, albeit at a low level. Second, therapeutic efficacy is often desired in only a single tissue, but the drug is distributed throughout the body, meaning on-interactions in non-target tissues may lead to undesirable effects. Third, inter-individual variation in the factors controlling drug action, both pharmacokinetic and pharmacodynamic, may result in adverse effects in specific groups within the patient population.To address these limitations it is necessary to view the biological system as a whole, and this has led to the development of systems biology in general, and systems toxicology specifically. It is important to stress that system approaches are not dramatic departures from the drug discovery/development and safety assessment paradigms currently in use. Rather, they build upon the vast experimental knowledgebase already present, integrating the data into a more easily accessible format. As depicted in Fig. 4, system toxicology uses existing experimental data to generate and validate a number of complimentary approaches. Relational databases provide an integrated home for these data, with the additional advantage that they include important information on how the data fits together. As such, relational databases are an important tool for systems toxicology in their own right. However, they also act as important sources for the parameters needed for systems toxicology modelling approaches, whether these are the more traditional PBPK modelling or newer genotype–phenotype modelling. Perhaps the most important aspect of systems toxicology, and indeed all systems approaches is the iterative nature of the process. Experimental data on ‘real’ biology is used to populate relational databases and construct models. These databases and models can then be validated through their ability to predict known biological phenomena. Identified gaps or boundaries in the models are then addressed through incorporation of new experimental data. Using this cycle of data generation → data integration → model/database validation → gap identification, allows the generated models/databases to continually develop and expand their biological coverage.
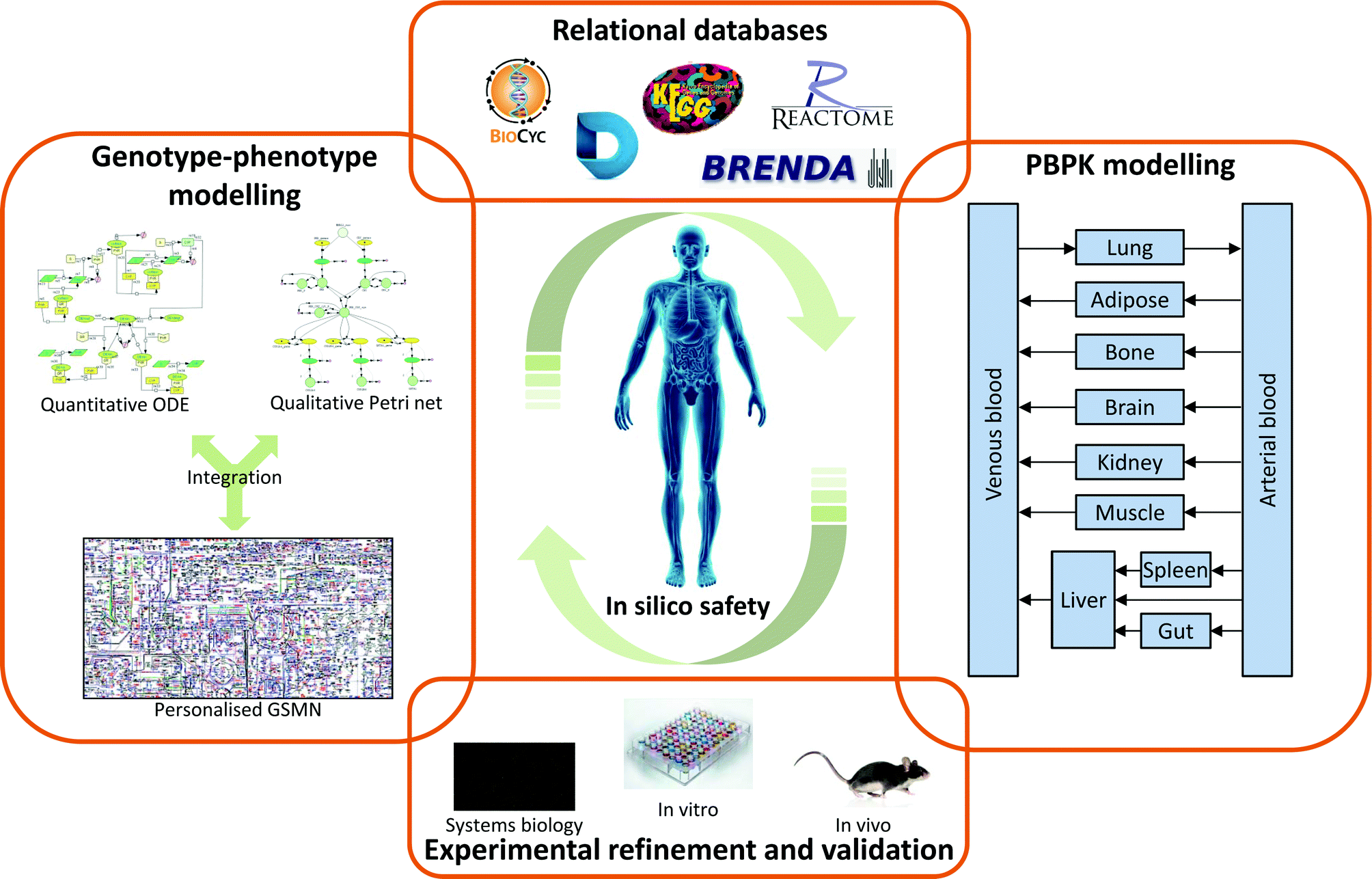 | ||
| Fig. 4 The iterative cycle of systems toxicology leading to improved safety assessment. Systems toxicology approaches (relational databases, genotype–phenotype modelling, PBPK modelling) are underpinned by experimental data. Iterative cycles of model refinement and validation lead to increasingly predictive models that will provide high value for safety assessment throughout the drug discovery/development pipeline. | ||
Systems toxicology represents one of the new biological disciplines that aim to build comprehensive digital organisms. These models will integrate experimental data from both standard biological and toxicological experiments, as well as the vast legacy datasets generated through -omic level analysis. The integration of these data into digital cells, organs and ultimately organisms will result in a step-change in our ability to both understand disease progression and to develop safe, efficacious network-based drugs to treat these diseases.
Notes and references
- M. Vinken, Toxicology, 2013, 312, 158–165 CrossRef CAS PubMed.
- A. Kolodkin, F. C. Boogerd, N. Plant, F. J. Bruggeman, V. Goncharuk, J. Lunshof, R. Moreno-Sanchez, N. Yilmaz, B. M. Bakker, J. L. Snoep, R. Balling and H. V. Westerhoff, Eur. J. Pharm. Sci., 2012, 46, 190–197 CrossRef CAS PubMed.
- H. Kitano, Nature, 2002, 420, 206–210 CrossRef PubMed.
- M. D. Waters and J. M. Fostel, Nat. Rev. Genet., 2004, 5, 936–948 CrossRef CAS PubMed.
- W. H. M. Heijne, A. S. Kienhuis, B. van Ommen, R. H. Stierum and J. P. Groten, Expert Rev. Proteomics, 2005, 2, 767–780 CrossRef CAS PubMed.
- S. Geenen, P. N. Taylor, J. L. Snoep, I. D. Wilson, J. G. Kenna and H. V. Westerhoff, Arch. Toxicol., 2012, 86, 1251–1271 CrossRef CAS PubMed.
- J. F. Waring, R. A. Jolly, R. Ciurlionis, P. Y. Lum, J. T. Praestgaard, D. C. Morfitt, B. Buratto, C. Roberts, E. Schadt and R. G. Ulrich, Toxicol. Appl. Pharmacol., 2001, 175, 28–42 CrossRef CAS PubMed.
- A. Craig, J. Sidaway, E. Holmes, T. Orton, D. Jackson, R. Rowlinson, J. Nickson, R. Tonge, I. Wilson and J. Nicholson, J. Proteome Res., 2006, 5, 1586–1601 CrossRef CAS PubMed.
- S. Huang, Briefings Funct. Genomics Proteomics, 2004, 2, 279–297 CrossRef CAS.
- M. Waters, G. Boorman, P. Bushel, M. Cunningham, R. Irwin, A. Merrick, K. Olden, R. Paules, J. Selkirk, S. Stasiewicz, B. Weis, B. Van Houten, N. Walker and R. Tennant, Environ. Health Perspect., 2003, 111, 15–28 CAS.
- D. M. Sanderson and C. G. Earnshaw, Hum. Exp. Toxicol., 1991, 10, 261–273 CAS.
- K. Enslein, H. H. Borgstedt, B. W. Blake and J. B. Hart, In Vitro Toxicol., 1987, 1, 129–147 CAS.
- N. F. Cariello, J. D. Wilson, B. H. Britt, D. J. Wedd, B. Burlinson and V. Gombar, Mutagenesis, 2002, 17, 321–329 CrossRef CAS.
- A. Hillebrecht, W. Muster, A. Brigo, M. Kansy, T. Weiser and T. Singer, Chem. Res. Toxicol., 2011, 24, 843–854 CrossRef CAS PubMed.
- N. G. Bakhtyari, G. Raitano, E. Benfenati, T. Martin and D. Young, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Revi., 2013, 31, 45–66 CrossRef CAS PubMed.
- P. N. Judson, S. A. Stalford and J. Vessey, Toxicol. Res., 2013, 2, 70–79 RSC.
- J. Kazius, R. McGuire and R. Bursi, J. Med. Chem., 2005, 48, 312–320 CrossRef CAS PubMed.
- B. L. Millard, M. Niepel, M. P. Menden, J. L. Muhlich and P. K. Sorger, Nat. Methods, 2011, 8, 487–U2255 CrossRef CAS PubMed.
- A. Brazma, H. Parkinson, U. Sarkans, M. Shojatalab, J. Vilo, N. Abeygunawardena, E. Holloway, M. Kapushesky, P. Kemmeren, G. G. Lara, A. Oezcimen, P. Rocca-Serra and S. A. Sansone, Nucleic Acids Res., 2003, 31, 68–71 CrossRef CAS PubMed.
- T. Barrett, S. E. Wilhite, P. Ledoux, C. Evangelista, I. F. Kim, M. Tomashevsky, K. A. Marshall, K. H. Phillippy, P. M. Sherman, M. Holko, A. Yefanov, H. Lee, N. Zhang, C. L. Robertson, N. Serova, S. Davis and A. Soboleva, Nucleic Acids Res., 2013, 41, D991–D995 CrossRef CAS PubMed.
- D. J. Dix, K. A. Houck, M. T. Martin, A. M. Richard, R. W. Setzer and R. J. Kavlock, Toxicol. Sci., 2007, 95, 5–12 CrossRef CAS PubMed.
- G. Patlewicz, N. Jeliazkova, R. J. Safford, A. P. Worth and B. Aleksiev, SAR QSAR Environ. Res., 2008, 19, 495–524 CrossRef CAS PubMed.
- E. Benfenati, Chem. Cent. J., 2010, 4 Search PubMed.
- T. M. Martin, P. Harten, R. Venkatapathy, S. Das and D. M. Young, Toxicol. Mech. Methods, 2008, 18, 251–266 CrossRef CAS PubMed.
- S. K. Kjaerulff, L. Wich, J. Kringelum, U. P. Jacobsen, I. Kouskoumvekaki, K. Audouze, O. Lund, S. Brunak, T. I. Oprea and O. Taboureau, Nucleic Acids Res., 2013, 41, D464–D469 CrossRef PubMed.
- B. Chen, X. Dong, D. Jiao, H. Wang, Q. Zhu, Y. Ding and D. J. Wild, BMC Bioinf., 2010, 11 CAS.
- L. Bell, R. Chowdhary, J. S. Liu, X. Niu and J. Zhang, PLoS One, 2011, 6 Search PubMed.
- S. Balaji, C. McClendon, R. Chowdhary, J. S. Liu and J. Zhang, Bioinformatics, 2012, 28, 747–749 CrossRef CAS PubMed.
- C. Prieto and J. D. L. Rivas, Nucleic Acids Res., 2006, 34, W298–W302 CrossRef CAS PubMed.
- S. Kerrien, B. Aranda, L. Breuza, A. Bridge, F. Broackes-Carter, C. Chen, M. Duesbury, M. Dumousseau, M. Feuermann, U. Hinz, C. Jandrasits, R. C. Jimenez, J. Khadake, U. Mahadevan, P. Masson, I. Pedruzzi, E. Pfeiffenberger, P. Porras, A. Raghunath, B. Roechert, S. Orchard and H. Hermjakob, Nucleic Acids Res., 2012, 40, D841–D846 CrossRef CAS PubMed.
- A. Chatr-aryamontri, B.-J. Breitkreutz, S. Heinicke, L. Boucher, A. Winter, C. Stark, J. Nixon, L. Ramage, N. Kolas, L. O'Donnell, T. Reguly, A. Breitkreutz, A. Sellam, D. Chen, C. Chang, J. Rust, M. Livstone, R. Oughtred, K. Dolinski and M. Tyers, Nucleic Acids Res., 2013, 41, D816–D823 CrossRef CAS PubMed.
- A. Franceschini, D. Szklarczyk, S. Frankild, M. Kuhn, M. Simonovic, A. Roth, J. Lin, P. Minguez, P. Bork, C. von Mering and L. J. Jensen, Nucleic Acids Res., 2013, 41, D808–D815 CrossRef CAS PubMed.
- E. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J. Hamon, J. L. Jenkins, P. Lavan, E. Weber, A. K. Doak, S. Cote, B. K. Shoichet and L. Urban, Nature, 2012, 486, 361–367 CAS.
- H. Jones and K. Rowland-Yeo, CPT: Pharmacometrics Syst. Pharmacol., 2013, 2, e63–e63 CrossRef PubMed.
- F. Bouzom, K. Ball, N. Perdaems and B. Walther, Biopharm. Drug Dispos., 2012, 33, 55–71 CrossRef CAS PubMed.
- C. C. Hunault, R. Z. Boerleider, B. G. H. Hof, J. J. G. Kliest, M. Meijer, N. J. Nijhuis, I. de Vries and J. Meulenbelt, Clin. Toxicol., 2014, 52, 121–128 CrossRef CAS PubMed.
- A. Funahashi, Y. Matsuoka, A. Jouraku, M. Morohashi, N. Kikuchi and H. Kitano, Proc. IEEE, 2008, 96, 1254–1265 CrossRef.
- A. Funahashi, N. Tanimura, M. Morohashi and H. Kitano, BIOSILICO, 2003, 1, 159–162 CrossRef.
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- M. Hucka, A. Finney, H. M. Sauro, H. Bolouri, J. C. Doyle, H. Kitano, A. P. Arkin, B. J. Bornstein, D. Bray, A. Cornish-Bowden, A. A. Cuellar, S. Dronov, E. D. Gilles, M. Ginkel, V. Gor, I. I. Goryanin, W. J. Hedley, T. C. Hodgman, J. H. Hofmeyr, P. J. Hunter, N. S. Juty, J. L. Kasberger, A. Kremling, U. Kummer, N. Le Novere, L. M. Loew, D. Lucio, P. Mendes, E. Minch, E. D. Mjolsness, Y. Nakayama, M. R. Nelson, P. F. Nielsen, T. Sakurada, J. C. Schaff, B. E. Shapiro, T. S. Shimizu, H. D. Spence, J. Stelling, K. Takahashi, M. Tomita, J. Wagner and J. Wang, Bioinformatics, 2003, 19, 524–531 CrossRef CAS PubMed.
- N. Le Novere, M. Hucka, H. Mi, S. Moodie, F. Schreiber, A. Sorokin, E. Demir, K. Wegner, M. I. Aladjem, S. M. Wimalaratne, F. T. Bergman, R. Gauges, P. Ghazal, H. Kawaji, L. Li, Y. Matsuoka, A. Villeger, S. E. Boyd, L. Calzone, M. Courtot, U. Dogrusoz, T. C. Freeman, A. Funahashi, S. Ghosh, A. Jouraku, S. Kim, F. Kolpakov, A. Luna, S. Sahle, E. Schmidt, S. Watterson, G. Wu, I. Goryanin, D. B. Kell, C. Sander, H. Sauro, J. L. Snoep, K. Kohn and H. Kitano, Nat. Biotechnol., 2009, 27, 735–741 CrossRef CAS PubMed.
- J. L. Snoep and B. G. Olivier, Mol. Biol. Rep., 2002, 29, 259–263 CrossRef CAS.
- C. Li, M. Donizelli, N. Rodriguez, H. Dharuri, L. Endler, V. Chelliah, L. Li, E. He, A. Henry, M. I. Stefan, J. L. Snoep, M. Hucka, N. Le Novere and C. Laibe, BMC Syst. Biol., 2010, 4 Search PubMed.
- B. N. Kholodenko, Eur. J. Biochem., 2000, 267, 1583–1588 CrossRef CAS.
- N. I. Markevich, J. B. Hoek and B. N. Kholodenko, J. Cell Biol., 2004, 164, 353–359 CrossRef CAS PubMed.
- I. Bailey, G. G. Gibson, K. Plant, M. Graham and N. Plant, PLoS One, 2011, 6, e16703 CAS.
- A. Kolodkin, N. Sahin, A. Phillips, S. R. Hood, F. J. Bruggeman, H. V. Westerhoff and N. Plant, Nat. Commun., 2013, 4, 1972 CrossRef PubMed.
- K. Howe, G. G. Gibson, T. Coleman and N. Plant, Drug Metab. Dispos., 2009, 37, 391–399 CrossRef CAS PubMed.
- S. Kim and M. Elbaum, Biophys. J., 2013, 105, 565–569 CrossRef CAS PubMed.
- A. N. Kolodkin, F. J. Bruggeman, N. Plant, M. J. Mone, B. M. Bakker, M. J. Campbell, J. P. T. M. van Leeuwen, C. Carlberg, J. L. Snoep and H. V. Westerhoff, Mol. Syst. Biol., 2010, 6, 446 CrossRef CAS PubMed.
- J. C. Leloup and A. Goldbeter, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7051–7056 CrossRef CAS PubMed.
- P. Achermann and A. A. Borbely, Front. Biosci., Landmark Ed., 2003, 8, S683–S693 CrossRef.
- M. C. Reed, R. L. Thomas, J. Pavisic, S. J. James, C. M. Ulrich and H. F. Nijhout, Theor. Biol. Med. Modell., 2008, 5 Search PubMed.
- S. Geenen, F. B. du Preez, J. L. Snoep, A. J. Foster, S. Sarda, J. G. Kenna, I. D. Wilson and H. V. Westerhoff, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 4943–4959 CrossRef CAS PubMed.
- M. Kanehisa and S. Goto, Nucleic Acids Res., 2000, 28, 27–30 CrossRef CAS.
- P. D. Karp, C. A. Ouzounis, C. Moore-Kochlacs, L. Goldovsky, P. Kaipa, D. Ahren, S. Tsoka, N. Darzentas, V. Kunin and N. Lopez-Bigas, Nucleic Acids Res., 2005, 33, 6083–6089 CrossRef CAS PubMed.
- R. Caspi, T. Altman, J. M. Dale, K. Dreher, C. A. Fulcher, F. Gilham, P. Kaipa, A. S. Karthikeyan, A. Kothari, M. Krummenacker, M. Latendresse, L. A. Mueller, S. Paley, L. Popescu, A. Pujar, A. G. Shearer, P. Zhang and P. D. Karp, Nucleic Acids Res., 2010, 38, D473–D479 CrossRef CAS PubMed.
- A. G. McDonald and K. F. Tipton, FEBS J., 2014, 281, 583–592 CrossRef CAS PubMed.
- M. H. Saier Jr., V. S. Reddy, D. G. Tamang and A. Vaestermark, Nucleic Acids Res., 2014, 42, D251–D258 CrossRef PubMed.
- A. Fleischmann, M. Darsow, K. Degtyarenko, W. Fleischmann, S. Boyce, K. B. Axelsen, A. Bairoch, D. Schomburg, K. F. Tipton and R. Apweiler, Nucleic Acids Res., 2004, 32, D434–D437 CrossRef CAS PubMed.
- I. Pedruzzi, C. Rivoire, A. H. Auchincloss, E. Coudert, G. Keller, E. de Castro, D. Baratin, B. A. Cuche, L. Bougueleret, S. Poux, N. Redaschi, I. Xenarios, A. Bridge and C. UniProt, Nucleic Acids Res., 2013, 41, D584–D589 CrossRef CAS PubMed.
- M. Wang, M. Weiss, M. Simonovic, G. Haertinger, S. P. Schrimpf, M. O. Hengartner and C. von Mering, Mol. Cell. Proteomics, 2012, 11, 492–500 CAS.
- I. Schomburg, A. Chang, S. Placzek, C. Soehngen, M. Rother, M. Lang, C. Munaretto, S. Ulas, M. Stelzer, A. Grote, M. Scheer and D. Schomburg, Nucleic Acids Res., 2013, 41, D764–D772 CrossRef CAS PubMed.
- N. Nagano, T. Noguchi and Y. Akiyama, Proteins: Struct., Funct., Bioinf., 2007, 66, 147–159 CrossRef CAS PubMed.
- U. Wittig, R. Kania, M. Golebiewski, M. Rey, L. Shi, L. Jong, E. Algaa, A. Weidemann, H. Sauer-Danzwith, S. Mir, O. Krebs, M. Bittkowski, E. Wetsch, I. Rojas and W. Mueller, Nucleic Acids Res., 2012, 40, D790–D796 CrossRef CAS PubMed.
- H. V. Westerhoff, A. K. Groen and R. J. A. Wanders, Biosci. Rep., 1984, 4, 1–22 CrossRef CAS.
- K. Maier, U. Hofmann, A. Bauer, A. Niebel, G. Vacun, M. Reuss and K. Mauch, Metab. Eng., 2009, 11, 292–309 CrossRef CAS PubMed.
- J. C. LaRosa, J. He and S. Vupputuri, JAMA, J. Am. Med. Assoc., 1999, 282, 2340–2346 CrossRef CAS.
- R. Ben-Shachar, Y. Chen, S. Luo, C. Hartman, M. Reed and H. F. Nijhout, Theor. Biol. Med. Modell., 2012, 9 Search PubMed.
- S. Geenen, J. W. T. Yates, J. G. Kenna, F. Y. Bois, I. D. Wilson and H. V. Westerhoff, Integr. Biol., 2013, 5, 877–888 RSC.
- M. M. Ozbek and G. F. Pinder, in Computational Methods in Water Resources, Vols 1 and 2, Proceedings, ed. S. M. Hassanizadeh, R. J. Schotting, W. G. Gray and G. F. Pinder, 2002, vol. 47, pp. 779–786 Search PubMed.
- Y. Li, H. Wang and Z. Qiang, Modeling of WEEE Recycling Process in China with Petri Net, 2008 Search PubMed.
- P. J. E. Goss and J. Peccoud, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6750–6755 CrossRef CAS.
- M. A. Blatke, A. Dittrich, C. Rohr, M. Heiner, F. Schaper and W. Marwan, Mol. BioSyst., 2013, 9, 1290–1307 RSC.
- M. Heiner, I. Koch and R. Will, BioSystems, 2004, 75, 15–28 CrossRef PubMed.
- M. Chen and R. Hofestadt, J. Biomed. In., 2006, 39, 147–159 CAS.
- H. Kitano, Nat. Rev. Genet., 2004, 5, 826–837 CrossRef CAS PubMed.
- N. C. Duarte, S. A. Becker, N. Jamshidi, I. Thiele, M. L. Mo, T. D. Vo, R. Srivas and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1777–1782 CrossRef CAS PubMed.
- I. Thiele, N. Swainston, R. M. T. Fleming, A. Hoppe, S. Sahoo, M. K. Aurich, H. Haraldsdottir, M. L. Mo, O. Rolfsson, M. D. Stobbe, S. G. Thorleifsson, R. Agren, C. Bölling, S. Bordel, A. K. Chavali, P. Dobson, W. B. Dunn, L. Endler, D. Hala, M. Hucka, D. Hull, D. Jameson, N. Jamshidi, J. J. Jonsson, N. Juty, S. Keating, I. Nookaew, N. Le Novère, N. Malys, A. Mazein, J. A. Papin, N. D. Price, E. Selkov Sr., M. I. Sigurdsson, E. Simeonidis, N. Sonnenschein, K. Smallbone, A. Sorokin, J. H. G. M. van Beek, D. Weichart, I. Goryanin, J. Nielsen, H. V. Westerhoff, D. B. Kell, P. Mendes and B. O. Palsson, Nat. Biotechnol., 2013, 31, 419 CrossRef CAS PubMed.
- C. Gille, C. Boelling, A. Hoppe, S. Bulik, S. Hoffmann, K. Huebner, A. Karlstaedt, R. Ganeshan, M. Koenig, K. Rother, M. Weidlich, J. Behre and H.-G. Holzhuetter, Mol. Syst. Biol., 2010, 6, 411 CrossRef PubMed.
- M. Ganter, T. Bernard, S. Moretti, J. Stelling and M. Pagni, Bioinformatics, 2013, 29, 815–816 CrossRef CAS PubMed.
- J. D. Orth, I. Thiele and B. O. Palsson, Nat. Biotechnol., 2010, 28, 245–248 CrossRef CAS PubMed.
- C. Khannapho, H. Zhao, B. K. Bonde, A. M. Kierzek, C. A. Avignone-Rossa and M. E. Bushell, Metab. Eng., 2008, 10, 227–233 CrossRef CAS PubMed.
- H. Yang, C. M. Roth and M. G. Ierapetritou, Biotechnol. Bioeng., 2009, 103, 1176–1191 CrossRef CAS PubMed.
- N. Plant, Drug Discovery Today, 2004, 9, 328–336 CrossRef CAS.
- T. Shlomi, M. N. Cabili, M. J. Herrgard, B. O. Palsson and E. Ruppin, Nat. Biotechnol., 2008, 26, 1003–1010 CrossRef CAS PubMed.
- O. Folger, L. Jerby, C. Frezza, E. Gottlieb, E. Ruppin and T. Shlomi, Mol. Syst. Biol., 2011, 7 Search PubMed.
- R. Agren, A. Mardinoglu, A. Asplund, C. Kampf, M. Uhlen and J. Nielsen, Mol. Syst. Biol., 2014, 10 Search PubMed.
- M. Krauss, S. Schaller, S. Borchers, R. Findeisen, J. Lippert and L. Kuepfer, PLoS Comput. Biol., 2012, 8 Search PubMed.
- M. W. Covert, C. H. Schilling and B. Palsson, J. Theor. Biol., 2001, 213, 73–88 CrossRef CAS PubMed.
- M. W. Covert, N. Xiao, T. J. Chen and J. R. Karr, Bioinformatics, 2008, 24, 2044–2050 CrossRef CAS PubMed.
- A. Varma and B. O. Palsson, Appl. Environ. Microbiol., 1994, 60, 3724–3731 CAS.
- J. Schellenberger, J. O. Park, T. M. Conrad and B. O. Palsson, BMC Bioinf., 2010, 11 Search PubMed.
- C. S. Henry, M. DeJongh, A. A. Best, P. M. Frybarger, B. Linsay and R. L. Stevens, Nat. Biotechnol., 2010, 28, 977–U922 CrossRef CAS PubMed.
- P. D. Karp, S. Paley and P. Romero, Bioinformatics, 2002, 18(Suppl 1), S225–S232 CrossRef PubMed.
- R. Overbeek, N. Larsen, T. Walunas, M. D'Souza, G. Pusch, E. Selkov, K. Liolios, V. Joukov, D. Kaznadzey, I. Anderson, A. Bhattacharyya, H. Burd, W. Gardner, P. Hanke, V. Kapatral, N. Mikhailova, O. Vasieva, A. Osterman, V. Vonstein, M. Fonstein, N. Ivanova and N. Kyrpides, Nucleic Acids Res., 2003, 31, 164–171 CrossRef CAS PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- A. Kraemer, J. Green, J. Pollard Jr. and S. Tugendreich, Bioinformatics, 2014, 30, 523–530 CrossRef CAS PubMed.
- C. P. Fisher, N. J. Plant, J. B. Moore and A. M. Kierzek, Bioinformatics, 2013, 29, 3181–3190 CrossRef CAS PubMed.
- R. Dallmann, S. A. Brown and F. Gachon, in Annual Review of Pharmacology and Toxicology, Vol 54, ed. P. A. Insel, Annual Reviews, Palo Alto, 2014, vol. 54, pp. 339–361 Search PubMed.
- J. P. Hughes, S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CrossRef CAS PubMed.
- G. M. Edelman and J. A. Gally, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13763–13768 CrossRef CAS PubMed.
- R. Albert, H. Jeong and A. L. Barabasi, Nature, 2000, 406, 378–382 CrossRef CAS PubMed.
- H. M. Coley, Cancer Treat. Rev., 2008, 34, 378–390 CrossRef CAS PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- A. L. Hopkins, Nat. Chem. Biol., 2008, 4, 682–690 CrossRef CAS PubMed.
- M. L. Radhakrishnan and B. Tidor, J. Chem. Inf. Model., 2008, 48, 1055–1073 CrossRef CAS PubMed.
- A. A. Borisy, P. J. Elliott, N. W. Hurst, M. S. Lee, J. Lehar, E. R. Price, G. Serbedzija, G. R. Zimmermann, M. A. Foley, B. R. Stockwell and C. T. Keith, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7977–7982 CrossRef CAS PubMed.
- J. Lehar, A. S. Krueger, W. Avery, A. M. Heilbut, L. M. Johansen, E. R. Price, R. J. Rickles, G. F. Short, J. E. Staunton, X. W. Jin, M. S. Lee, G. R. Zimmermann and A. A. Borisy, Nat. Biotechnol., 2009, 27, 659–U116 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |