 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient deep blue fluorescent polymer light-emitting diodes (PLEDs)†
Javan H.
Cook
a,
José
Santos
b,
Haiying
Li‡
b,
Hameed A.
Al-Attar
a,
Martin R.
Bryce
*b and
Andrew P.
Monkman
*a
aDepartment of Physics, Durham University, Durham, DH1 3LE, UK. E-mail: a.p.monkman@durham.ac.uk
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 12th June 2014
Abstract
A new series of deep blue/blue emitting co-polymers are reported. Poly(9,9-dihexylfluorene-3,6-diyl and 2,7-diyl-co-2,8-dihexyldibenzothiophene-S,S-dioxide-3,7-diyl) derivatives p(F-S) of varying composition have been synthesised. The effects of two different S derivatives with dialkoxy sidechains, the F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S monomer feed ratio, and meta versus para conjugation with respect to the F units have all been investigated in terms of photophysics and polymer light-emitting diode (PLED) device performance in the architecture ITO/PEDOT:PSS/polymer/TPBi/LiF/Al. The meta polymers poly(9,9-dihexylfluorene-3,6-diyl-co-2,8-di(O-methylenecyclohexyl)dibenzothiophene-S,S-dioxide-3,7-diyl) p(Fm-SOCy) in three different co-monomer ratios, P1–3, give deep blue electroluminescence peaking at 415 nm, with the ratio of 70:30 p(Fm:SOCy) producing a maximum external quantum efficiency (EQE) ηext, max 2.7%, whilst the ratio of 85:15 gave the highest maximum brightness Lmax of 81 cd m−2, with CIE coordinates (0.17, 0.12) The analogous para series poly(9,9-dihexylfluorene-2,7-diyl-co-2,8-di(O-methylenecyclohexyl)dibenzothiophene-S,S-dioxide-3,7-diyl) p(Fp-SOCy) and poly(9,9-dihexylfluorene-2,7-diyl-co-2,8-dihexyloxydibenzothiophene-S,S-dioxide-3,7-diyl) p(Fp-SO6) in two different ratios, P4–7, produced blue emission peaking at ca. 450 nm. The ratio of 70:30 F:S units consistently gave better devices than the corresponding 50:50 co-polymers. It was also observed that co-polymers incorporating the bulkier SOCy derivatives gave more efficient and brighter devices, with polymer P5 attaining a remarkable ηext, max 3.2%, 4.4 cd A−1, 3.4 lm W−1 and maximum brightness 2500 cd m−2 with CIE (0.16, 0.18).
:S monomer feed ratio, and meta versus para conjugation with respect to the F units have all been investigated in terms of photophysics and polymer light-emitting diode (PLED) device performance in the architecture ITO/PEDOT:PSS/polymer/TPBi/LiF/Al. The meta polymers poly(9,9-dihexylfluorene-3,6-diyl-co-2,8-di(O-methylenecyclohexyl)dibenzothiophene-S,S-dioxide-3,7-diyl) p(Fm-SOCy) in three different co-monomer ratios, P1–3, give deep blue electroluminescence peaking at 415 nm, with the ratio of 70:30 p(Fm:SOCy) producing a maximum external quantum efficiency (EQE) ηext, max 2.7%, whilst the ratio of 85:15 gave the highest maximum brightness Lmax of 81 cd m−2, with CIE coordinates (0.17, 0.12) The analogous para series poly(9,9-dihexylfluorene-2,7-diyl-co-2,8-di(O-methylenecyclohexyl)dibenzothiophene-S,S-dioxide-3,7-diyl) p(Fp-SOCy) and poly(9,9-dihexylfluorene-2,7-diyl-co-2,8-dihexyloxydibenzothiophene-S,S-dioxide-3,7-diyl) p(Fp-SO6) in two different ratios, P4–7, produced blue emission peaking at ca. 450 nm. The ratio of 70:30 F:S units consistently gave better devices than the corresponding 50:50 co-polymers. It was also observed that co-polymers incorporating the bulkier SOCy derivatives gave more efficient and brighter devices, with polymer P5 attaining a remarkable ηext, max 3.2%, 4.4 cd A−1, 3.4 lm W−1 and maximum brightness 2500 cd m−2 with CIE (0.16, 0.18).
Introduction
Organic light-emitting devices (OLEDs) are a rapidly advancing technology.1 Current applications, such as mobile displays and lighting, offer numerous benefits2 over their inorganic competitors, including lower power consumption, faster response times, increased viewing angles, and greater ranges of colour and contrast, along with compatibility with large area and flexible substrates. The vast majority of OLED research has concentrated on the visible spectrum, primarily for display and lighting applications.3 More recently OLEDs which emit from the deep blue through to the violet range of the spectrum have received increased investigation.4 Deep blue light is essential to achieve good colour rendering and high colour temperature in white OLEDs for lighting applications, and for greater colour contrast and a wider colour gamut in displays.5 Maximum external quantum efficiencies (EQEs) of ηext, maxca. 3–6% for emission in the range 400–480 nm have been reported for both small molecule and polymer based devices.6 Very recently an alternating copolymer of 9,9-(dioctylfluorene) and tetrafluorophenylene with λELmax 405 nm and CIE coordinates (0.17, 0.06) was reported to achieve ηext, max 5.03%.7 Whilst this is a higher maximum efficiency than that reported here, our polymers produce efficient deep blue/blue devices, combined with low turn-on voltages and high brightnesses, providing a significant advance in the field.Derivatives of poly(9,9-dialkylfluorene-2,7-diyl)s (pFs) are well established as emitters in electroluminescent devices. They possess many desirable properties, namely: blue emission, high charge-carrier mobilities, good thermal and electrochemical stability, high photoluminescence quantum yields (PLQY), and facile chemical modification and co-polymerisation.8 Our group9 and others10 have incorporated dibenzothiophene-S,S-dioxide-3,7-diyl (S) units into a poly(9,9-dialkylfluorene) main chain and shown that these co-polymers possess enhanced blue spectral stability [compared to homo-poly(9,9-dialkylfluorene)] and colour tunability, including green and white emission by chemical modifications. The electron-deficient S units are topologically similar to fluorene; they lower the lowest unoccupied molecular orbital (LUMO) which improves electron injection and makes charge injection and transport more balanced. We previously reported that F-S copolymers with hexyl substituents attached to the S units display improved solubility and spectrally stable blue-shifted emission arising from a twisted backbone structure which disrupts the extended π-conjugation.11
The aim of the present work is to build on these results and study a series of F-S copolymers incorporating new substituents on the S framework, namely O-methylenecyclohexyl (SOCy) or O-hexyl (SO6), designed to produce efficient, solution-processable, deep blue polymer LEDs (PLEDs) with emission at wavelengths shorter than λmax 450 nm. Seven new copolymers have been synthesised; ratios of the monomers have been varied, as has the position of linkage to the fluorene units, i.e. 2,7 (para, conjugated) or 3,6 (meta, broken conjugation). The photophysics of the copolymers is reported. They function as both carrier transporters and fluorescent emitters in PLEDs, whose performance is improved (increased efficiency and brightness, and reduced turn-on voltage) by an additional electron injecting layer of 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi).
Results and discussion
Synthesis
The synthetic routes and structures of the co-polymers P1–P7 are shown in Scheme 1. Monomer synthesis is reported in the ESI.† The co-polymers were synthesised by Suzuki polycondensation reactions,12 using different monomer feed ratios, followed by end-capping with phenyl groups. The polymers were isolated as white or very pale yellow solids; their molecular weights are stated in Table 1. | ||
| Scheme 1 Synthesis and structures of co-polymers P1–P7. | ||
| m | n | M w /Da | M n /Da | |
|---|---|---|---|---|
|
a Based on monomer feed ratios of F:SOCy or SO6 units.
b Estimated by GPC analysis using polydispersity polystyrene standards obtained from Polymer Laboratories.
|
||||
| P1 | 50 | 50 | 25700 |
13600 |
| P2 | 70 | 30 | 15200 |
7800 |
| P3 | 85 | 15 | 17500 |
7000 |
| P4 | 50 | 50 | 20900 |
7500 |
| P5 | 70 | 30 | 88900 |
27200 |
| P6 | 50 | 50 | 26800 |
10400 |
| P7 | 70 | 30 | 62000 |
21400 |
Optical properties
The spectroscopic data for the polymers P1–7 is summarised in Table 2 and thin film photoluminescence spectra are shown in Fig. 1a and b. The polymers are bright blue emitters. They exhibit very little solvatochromism and the differences observed in the λPLmax can be attributed to a change in refractive index between the solvents. This agrees well with the previous results for analogous materials.11 For polymers P1–3, the SO6 units and the end-capping phenyl groups are bonded through the 3 and 6 positions of the F units (meta conjugated), which results in the polymer conjugation being broken. This yields a blue-shifted emission (by ca. 30 nm in thin films) compared to the para analogues, P4–7, and to the related (para) polymers with 2,8-dihexyldibenzothiophene-S,S-dioxide (Shexyl) units.11| Polymer | Solvent/film | λ absmax/nm | λ PLmax/nm | PLQY, ΦPLa | E onsetT/eV |
|---|---|---|---|---|---|
| a Error ±10%. | |||||
| P1 | Ethyl acetate | 345 | 414 | ||
| Cyclohexane | 351 | 427 | |||
| Film | 350 | 424 | 0.59 | 2.44 | |
| P2 | Ethyl acetate | 346 | 412 | ||
| Cyclohexane | 347 | 413 | |||
| Film | 350 | 420 | 0.77 | 2.45 | |
| P3 | Ethyl acetate | 346 | 412 | ||
| Cyclohexane | 346 | 422 | |||
| Film | 350 | 420 | 0.75 | 2.44 | |
| P4 | Ethyl acetate | 379 | 426 | ||
| Cyclohexane | 377 | 424 | |||
| Film | 390 | 448 | 0.58 | 2.48 | |
| P5 | Ethyl acetate | 381 | 429 | ||
| Cyclohexane | 404 | 442 | |||
| Film | 390 | 450 | 0.62 | 2.42 | |
| P6 | Ethyl acetate | 382 | 425 | ||
| Cyclohexane | 394 | 432 | |||
| Film | 395 | 450 | 0.58 | 2.42 | |
| P7 | Ethyl acetate | 384 | 426 | ||
| Cyclohexane | 379 | 429 | |||
| Film | 390 | 452 | 0.55 | 2.42 | |
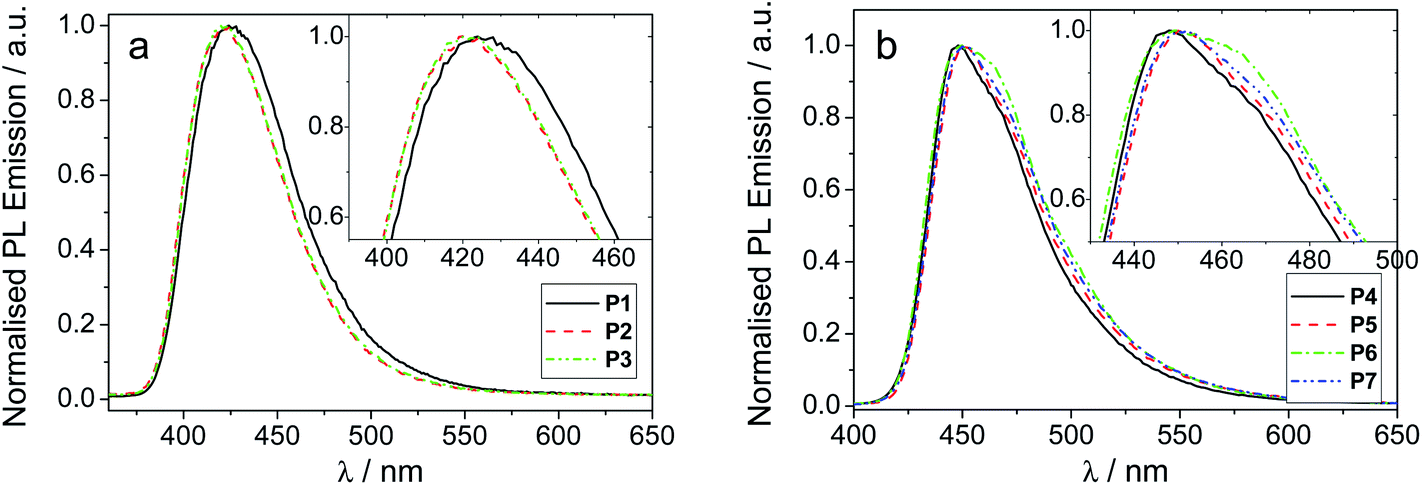 | ||
| Fig. 1 Normalised PL emission spectra for (a) polymers P1–3 and (b) polymers 4–7 in thin film. Insets show an expansion of the λmax region. | ||
We previously showed that in related co-polymers with Shexyl instead of SOCy or SO6 side-chains an increasing ratio of the Shexyl component with respect to the F component (F:Shexyl ratios 85:15; 70:30; 50:50) results in a significant blue shift in both the absorption and emission maxima (in solution and thin film), ascribed to an increase in the dihedral angle breaking the polymer's extended π conjugation.11 However, within the present two series of meta (P1–3) and para (P4–P7) polymers with SOCy or SO6 side-chains there is only a small change in absorption and emission maxima for solution and thin films as the ratios of SOCy or SO6 units are varied. From these data it can be deduced that the SOCy and SO6 side-chains lead to less sterically-induced twisting of the backbone compared to the previous Shexyl analogues,11 presumably due to the presence of the oxygen atom, rather than a CH2 unit, attached to the S unit.
The emission of these polymers is, however, substantially red shifted due to the change from meta (P1–3) to para (P4–P7) linkages (Fig. 1). A more subtle trend is that with increased ratio of the SOCy and SO6 moiety (50:50 ratio, P4 and P6) there is a very small (2 nm) blue shift in λmax of thin film emission compared to the 70:30 F:S analogues (P5 and P7). Also, by increasing the size of the pendant group from SO6 (P6 and P7) to SOCy (P4 and P5) the λmax value blue shifts by 2 nm for both ratios. More notably, the larger SOCy groups reduce the shoulder at the red edge (seen especially in P6; Fig. 1b), leading to a narrower emission peak for P4 and P5, compared to P6 and P7. No significant change is observed in the film PLQY values for polymers P1–7 which are all within the range 0.55–0.77.
The observed triplet levels of P1–7 (EonsetT 2.42–2.48 eV) correspond well to the reported values for similar polymers which possess restricted backbone π-conjugation.11
Deep blue and blue PLED devices
Devices were fabricated for polymers P1–7 using the structure: glass|ITO (150 nm)|PEDOT:PSS HIL 1.5 (70 nm)|LEP (60 nm)|TPBi (20 nm)|LiF (1 nm)|Al (100 nm). For all of the devices, the light emitting polymer (LEP) layer consisted of one of the polymers P1–7 without any other dopants or transporters added. The results for each device are represented in Table 3, Fig. 2 and 3. For each of the meta conjugated polymers P1–3 electroluminescence is observed in the deep blue (Fig. 4a), with λmax at ca. 415 nm. The exact peak wavelength is difficult to identify due to the noise present in the spectra due to the low relative brightness; nonetheless for P1, P2 and P3, respectively, the CIE coordinates at the turn-on voltage (10 cd m−2) are (0.17, 0.11), (0.17, 0.10) and (0.16, 0.09) and at maximum brightness are (0.18, 0.13), (0.17, 0.11) and (0.17, 0.12). CIE diagrams are shown in the ESI.†| V on /V | Brt/cd m−2 | EQE/% | Dev Eff/cd A−1 | Brtmax/cd m−2 | EQEmax% | Dev Effmax/cd A−1 | Lummax/lm W−1 | CIE (x, y)e | CIE (x, y)f | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Device architecture: ITO/PEDOT:PSS/polymer/TPBi/LiF/Al. b V on is the turn-on voltage, defined here as the voltage at which the device reached a brightness of 10 cd m−2. c A comparison current density of 5 mA cm−2 was selected. d A comparison current density of 50 mA cm−2 was selected. e CIE coordinates at the turn-on voltage (10 cd m−2). f CIE coordinates at the maximum brightness. CIE diagrams are shown in the ESI. | ||||||||||
| P1 | 7.2 | 23c | 0.60c | 0.47c | 28 | 1.7 | 0.91 | 0.47 | 0.17, 0.11 | 0.18, 0.13 |
| P2 | 6.8 | 30c | 0.83c | 0.58c | 37 | 2.7 | 1.2 | 0.70 | 0.17, 0.10 | 0.17, 0.11 |
| P3 | 6.6 | 33c | 1.2c | 0.66c | 81 | 1.3 | 0.72 | 0.34 | 0.16, 0.09 | 0.17, 0.12 |
| P4 | 4.4 | 640d | 0.8d | 1.3d | 740 | 2.1 | 2.7 | 1.7 | 0.16, 0.15 | 0.17, 0.17 |
| P5 | 3.4 | 1500d | 2.2d | 3.1d | 2500 | 3.2 | 4.4 | 3.4 | 0.16, 0.15 | 0.16, 0.18 |
| P6 | 4.3 | 320d | 0.4d | 0.6d | 320 | 2.5 | 2.1 | 1.5 | 0.16, 0.18 | 0.17, 0.20 |
| P7 | 3.7 | 1000d | 1.4d | 2.1d | 1400 | 2.8 | 3.7 | 2.7 | 0.16, 0.17 | 0.16, 0.18 |
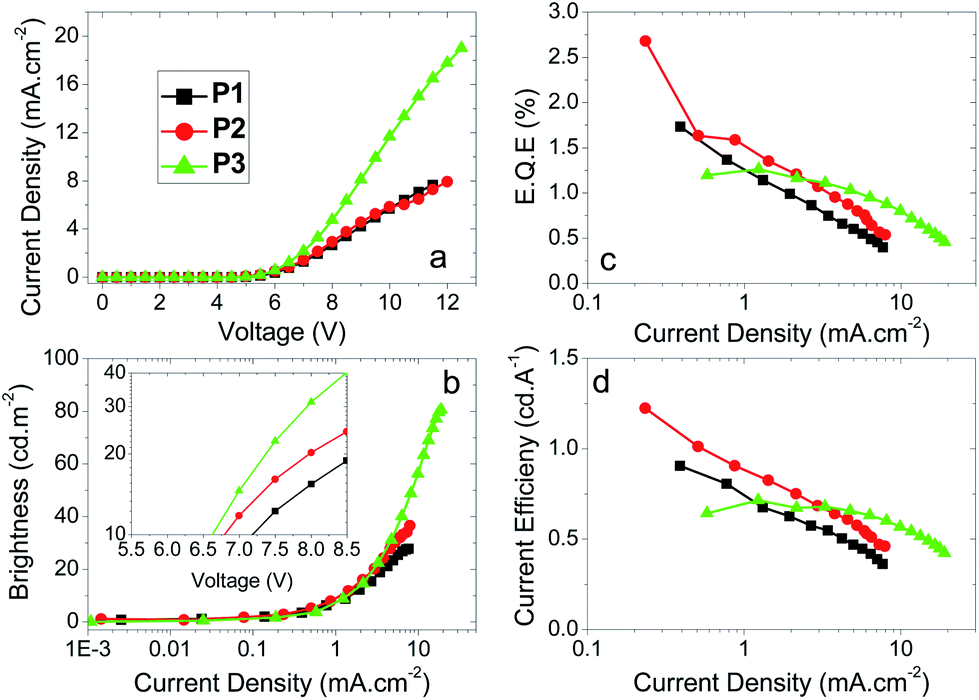 | ||
| Fig. 2 Plots of (a) J–V curves, (b) luminance vs. J, (c) EQE vs. J and (d) device efficiency vs. J for the polymers P1–3. Inset to (b) shows the turn-on voltages for the devices in a plot of luminance vs. V. | ||
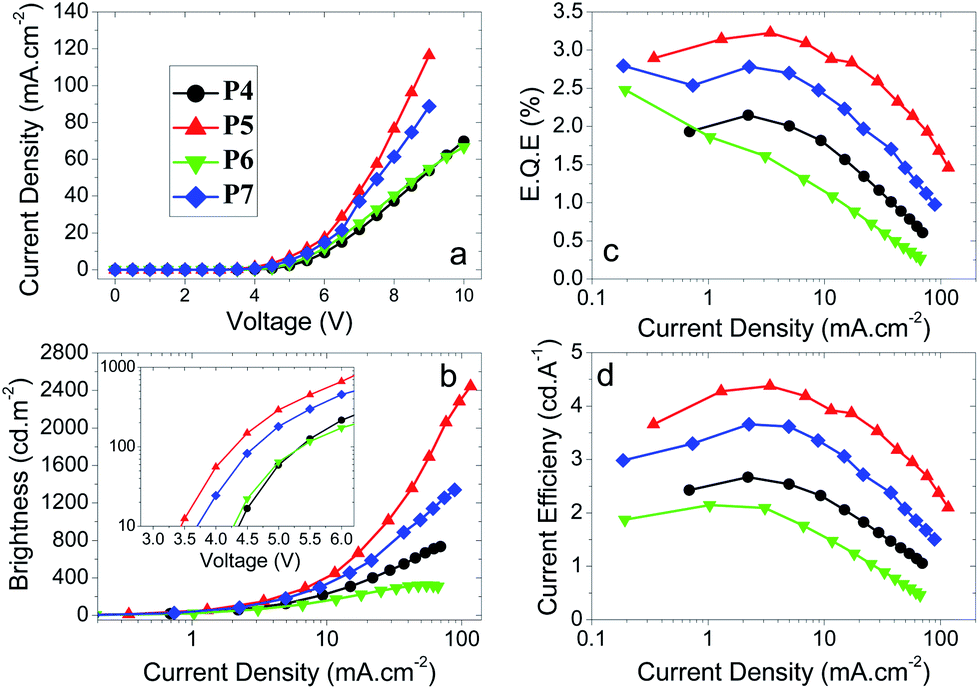 | ||
| Fig. 3 Plots of (a) J–V curves, (b) luminance vs. J, (c) EQE vs. J and (d) device efficiency vs. J for the polymers P4–7. Inset to (b) shows the turn-on voltages for the devices in a plot of luminance vs. V. | ||
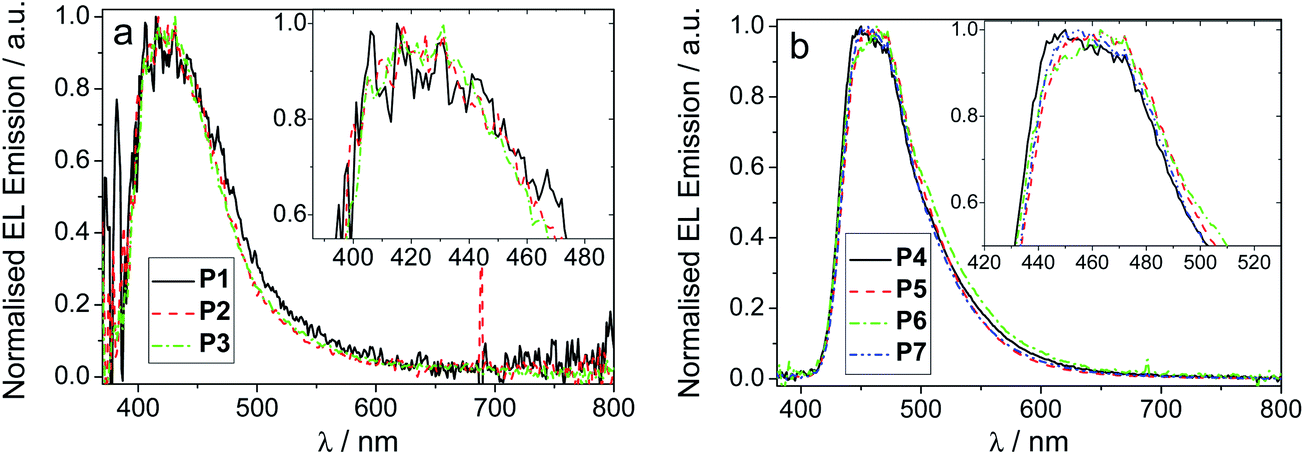 | ||
| Fig. 4 Normalised EL spectra for (a) polymers P1–3 and (b) polymers P4–7 in thin film. Insets show an expansion of the λmax region. | ||
From Table 3 and Fig. 2 it can be seen that sequentially decreasing the ratio of SOCy to F, from 50:50 to 30:70, to 15:85 (i.e.P1, P2 and P3, respectively) has the effect of increasing the maximum brightness from 28 cd m−2 to 37 cd m−2 to 81 cd m−2, respectively. The maximum external quantum efficiencies initially increase when the SOCy ratio decreases from 50:50 (P1, EQE 1.7%) to 30:70 (P2, 2.7%) and then decreases again when the ratio is decreased to 15:85 (P3, 1.3%). It can also be observed that increasing the amount of SOCy present in the polymer decreases the current density passing through the device. This is consistent with the increased proportion of SOCy decreasing the extent of π conjugation in the polymer and thus reducing its carrier transporting properties up to a certain threshold. Once this threshold is reached the current density remains more or less constant, hence the similarity between P1 and P2 in Fig. 2a. It is also observed that increasing the ratio of SOCy reduces the amount of emissive species (oligo-F domains) formed, accounting for the drop in brightness at comparable current densities and thus the drop in efficiency (P3 > P2 > P1). Our devices are not fully optimized for efficient out-coupling in the deep blue, which explains the lower device efficiencies for P1–3 compared to P4–7.
Regarding the para polymers P4–7, similar trends to the meta polymers P1–3 are observed (Fig. 3). The major differences between the two sets of polymers are the maximum current densities and the emission spectra. Whilst polymers P1–3 have EL peak emission at λmaxca. 415 nm, polymers P4–7 have λmaxca. 450 nm, as seen in Fig. 4b. Both this and the change in maximum current density can be explained by the broken conjugation of the polymers P1–3 due to the meta-linkage. Similar to polymers P1–3, reducing the concentration of SOCy (P4, P5) or SO6 (P6, P7) component from 50% to 30% increases the maximum brightness and efficiency of the devices as well as substantially decreasing the turn-on voltage. A decrease of SOCy or SO6 ratio in the polymer has the effect of increasing the current density through the device, which can be explained by substantially increasing the quantity of emissive species (longer domains of oligo-F units) with P5 showing the highest efficiency. This indicates that subtle electronic and/or structural factors influence PLED performance in this series.
Conclusions
New polyfluorene based co-polymers have been synthesised with varying ratios of two different functionalised dibenzothiophene-S,S-dioxide derivatives. Also investigated was the effect of meta and para conjugation within the polymers with respect to the position of backbone linkage in the F units. For the meta polymers P1–3 deep blue emission peaking at ca. 415 nm is observed. The ratio of 70:30 F to SOCy in polymer P2 leads to the highest maximum efficiency reaching 2.7% EQE, whilst the ratio of 85:15 in polymer P3 gives the highest maximum brightness of 81 cd m−2. For the para polymers P4–7 blue emission peaking at ca. 450 nm is observed. The ratio of 70:30 F to SOCy or SO6 (P5 and P7) produces better devices than the analogous 50:50 polymers (P4 and P6). It was also observed that the bulkier SOCy derivative gives more efficient and brighter devices, with polymer P5 attaining a remarkable ηext, max 3.2%, 4.4 cd A−1, 3.4 lm W−1 and Lmax 2500 cd m−2 with CIE (0.16, 0.18) in a simple PLED architecture. As noted above, it has recently been reported that polymer-based devices have exceeded 5% EQE for deep blue emission.7 Whilst this is a higher maximum EQE than we observe for P1–7, our polymers produce efficient deep blue/blue devices with good colour stability, and combined with low turn-on voltages and high brightnesses, this is a significant advance in the field.
Experimental
General Suzuki co-polymerisation procedure
All monomers were dissolved in dry toluene (200 mg of the corresponding boronic ester in 7 mL) and degassed by bubbling argon into the solution for 30 min, then PdCl2[P(o-tol)3]2 (1% mol) was added and reaction degassed for additional 15 min. A degassed aqueous Et4NOH solution (4 mL, 20% w/w) was added and the reaction mixture was stirred at 115 °C for 24 h. All co-polymers were end-caped with phenyl units by addition of bromobenzene (1 mL), followed by benzeneboronic acid (100 mg) 1 h later. After 1 h, upon cooling, the mixture was poured into methanol (300 mL) and the resulting precipitate was filtered and sequentially washed with methanol, water and methanol. After drying, the solid was redissolved in chloroform (20 mL) and a solution of palladium scavenger (1.0 g of sodium diethyldithiocarbamate trihydrate) in water (20 mL) was added, and the mixture was stirred overnight at 60 °C. The organic layer was separated and sequentially washed with dilute aqueous HCl (5%), concentrated sodium acetate and water. The resulting organic extracts were filtered through a plug of celite and concentrated under vacuum. The co-polymers were isolated as white or very pale yellow fibrous solids by precipitation when the concentrate was added dropwise to methanol.Optical characterisation
Absorption spectra for solution and solid state samples were obtained using a Shimadzu UV-vis-NIR spectrophotometer whilst emission spectra were acquired using a Jobin-Yvon fluoromax spectrofluorimeter. The excitation wavelengths were determined from the maximum absorbance of the polymers as obtained from the absorption spectra. The triplet energies of solid state samples at 16 K were calculated using a gated luminescent measurement of the phosphorescence technique.13 Solutions were produced in ethyl acetate or cyclohexane, and the OD kept below 1.0. Solid state samples were drop-cast from a 1:1 mixture of 175 mg mL−1 zeonex and 0.5 mg mL−1 of the polymer, both in chlorobenzene, and had a maximum absorbance of 2.0 OD.
Device fabrication and characterisation
The devices featured an ITO anode (150 nm, 16 Ω) commercially pre-coated on a glass substrate (24 mm × 24 mm). The substrates were cleaned with acetone, isopropanol and acetone sequentially in a sonic bath for a period of 9 min each. They were then exposed to low pressure plasma for a period of 3 min and treated with UV-ozone for a further 4 min. A hole-injection layer (HIL) of PEDOT:PSS of thickness 70 nm was deposited by spin coating and then baked on a hotplate at 200 °C for 3 min to remove any leftover moisture. The PEDOT:PSS used was the commercially available HIL 1.5 from Heraeus Precious Metals, Germany. Active layers of polymers P1–7 were prepared in a solution of chlorobenzene, concentration varied to produce layers of 60 nm, and then spun on top of the PEDOT:PSS. The device was then annealed at 120 °C on a hotplate for 10 min to remove residual chlorobenzene. An electron injection layer (EIL) consisting of a 20 nm layer of 1,3,5-tris(N-phenyl benzimidazol-2-yl)benzene (TPBi) was thermally evaporated directly on top of the polymer layer. This was followed by a 1 nm thick lithium fluoride (LiF) cathode, which was thermally evaporated using a shadow mask to produce parallel strips perpendicular to the ITO anodes, forming four individually addressable pixels per substrate each of area 5 mm × 4 mm. The LiF was capped with a 100 nm thick layer of aluminium to protect it from oxidation. An evaporation pressure of ca. 10−6 mbar and a rate of ca. 0.1 nm s−1 was used for all of evaporated layers. The devices were then encapsulated with DELO UV curable epoxy (Katiobond) and a 12 × 12 mm glass cover slide.The devices were characterised in a calibrated Labsphere LMS-100 integrating sphere, connected to a USB 4000 CCD spectrometer supplied by a 30 μm UV/Vis fibre optic cable, under steady state conditions. Layer thicknesses were measured using a J. A. Woolam VASE Ellipsometer after having been spin coated onto SiSiO2 substrates. The non-uniformity of the organic layer thicknesses across the samples leads to a 5–10% error in device efficiencies and all measurements were averages over at least four devices. A summary of the materials used and the device configuration can be found in Table 3.
Acknowledgements
The authors would like to acknowledge the financial support of the Durham Energy Institute and EPSRC.References
- (a) Organic Light Emitting Devices: Synthesis, Properties and Applications, ed. K. Mullen and U. Scherf, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) Z. Li and H. Meng, Organic Light-Emitting Materials and Devices, CRC, Boca Raton, FL, 2006 Search PubMed.
- (a) S. C. Stinson, Chem. Eng. News, 2000, 78, 22–23 CrossRef PubMed; (b) W. E. Howard, Sci. Am., 2004, 290, 76–81 CrossRef CAS PubMed; (c) R. Friend, J. Burroughes and T. Shimoda, Phys. World, 1999, 12, 35–40 CAS.
- (a) K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef PubMed; (b) P.-L. T. Boudrealt, S. Beaupre and M. Leclerc, Polym. Chem., 2010, 1, 127–136 RSC; (c) C. Rothe, Laser Photonics Rev., 2007, 1, 303–306 CrossRef CAS; (d) N. T. Kalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696–2723 CrossRef PubMed; (e) H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621–630 CrossRef CAS; (f) P. L. Burn, S. C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675–1688 CrossRef CAS; (g) K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater, 2010, 22, 572–582 CrossRef CAS PubMed; (h) L. Ying, C.-L. Ho, H. Wu, Y. Cao and W.-Y. Wong, Adv. Mater, 2014, 26, 2459–2473 CrossRef CAS PubMed.
- (a) X. J. Feng, S. F. Chen, Y. Ni, M. S. Wong, M. M. K. Lam, K. W. Cheah and G. Q. Lai, Org. Electron., 2014, 15, 57–64 CrossRef CAS PubMed; (b) F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed; (c) S. H. Jeong and J. Y. Lee, J. Mater. Chem., 2011, 21, 14604–14609 RSC; (d) S. Nau, N. Schulte, S. Winkler, J. Frisch, A. Vollmer, N. Koch, S. Sax and E. J. W. List, Adv. Mater., 2013, 25, 4420–4424 CrossRef CAS PubMed; (e) A. Sergent, G. Zucchi, R. B. Pansu, M. Chaigneau, B. Geffroy, D. Tondelier and M. Ephritikhine, J. Mater. Chem. C, 2013, 1, 3207–3216 RSC; (f) S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, J. Mater. Chem. C, 2014, 2, 421–424 RSC; (g) Y. X. Yang, P. Cohn, A. L. Dyer, S. H. Eom, J. R. Reynolds, R. K. Castellano and J. G. Xue, Chem. Mater., 2010, 22, 3580–3582 CrossRef CAS; (h) J. Ye, Z. Chen, M.-K. Fung, C. Zheng, X. Ou, X. Zhang, Y. Yuan and C.-S. Lee, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS.
- J. R. Koo, S. J. Lee, G. W. Hyung, B. Y. Kim, D. H. Lee, W. Y. Kim, K. H. Lee, S. S. Yoon and Y. K. Kim, Thin Solid Films, 2013, 544, 234–237 CrossRef CAS PubMed.
- Y. Yang, P. Cohn, S.-H. Eom, K. A. Abboud, R. K. Castellano and J. Xue, J. Mater. Chem. C, 2013, 1, 2867–2874 RSC.
- U. Giovanella, C. Botta, F. Galeotti, B. Vercelli, S. Battiato and M. Pasinbi, J. Mater. Chem. C, 2013, 1, 5322–5329 RSC , in this paper brightness data are presented in terms of irradiance (maximum value 450 μW cm−2 at 405 nm) rather than in the standard units (cd m−2) used in the present work and related papers in ref. 4. Therefore, direct comparison of device brightness cannot be made.
- (a) S.-A. Chen, H.-H. Lu and C.-W. Huang, Adv. Polym. Sci., ed. U. Scherf and D. Neher, Springer-Verlag, Berlin, Heidelberg, 2008, vol. 212, pp. 49–84 Search PubMed; (b) U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- (a) F. B. Dias, S. King, A. P. Monkman, I. I. Perepichka, M. A. Kryuchkov, I. F. Perepichka and M. R. Bryce, J. Phys. Chem. B, 2008, 112, 6557–6566 CrossRef CAS PubMed; (b) S. M. King, I. I. Perepichka, I. F. Perepichka, F. B. Dias, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 586–591 CrossRef CAS; (c) F. B. Dias, K. T. Kamtekar, T. Cazati, G. Williams, M. R. Bryce and A. P. Monkman, ChemPhysChem, 2009, 10, 2096–2104 CrossRef CAS PubMed.
- (a) J. Liu, J. Zou, W. Yang, H. Wu, C. Li, B. Zhang, J. Peng and Y. Cao, Chem. Mater., 2008, 20, 4499–4506 CrossRef CAS; (b) Y. Li, H. Wu, J. Zou, L. Ying, W. Yang and Y. Cao, Org. Electron., 2009, 10, 901–909 CrossRef CAS PubMed; (c) H. Xiao, L. Yu, Y. Li, W. Yang, B. Zhang, W. Yang, H. Wu and Y. Cao, Polymer, 2012, 53, 2873–2883 CrossRef CAS PubMed; (d) L. Yu, J. Liu, S. Hu, R. He, W. Yang, H. Wu, J. Peng, R. Xia and D. D. C. Bradley, Adv. Funct. Mater., 2013, 23, 4366–4376 CrossRef CAS.
- K. T. Kamtekar, H. L. Vaughan, B. P. Lyons, A. P. Monkman, S. U. Pandya and M. R. Bryce, Macromolecules, 2010, 43, 4481–4488 CrossRef CAS.
- (a) J. Murage, J. W. Eddy, J. R. Zimbalist, T. B. McIntyre, Z. R. Wagner and F. E. Goodson, Macromolecules, 2008, 41, 7330–7338 CrossRef CAS; (b) J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schluter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS PubMed.
- (a) V. Jankus, C. Winscom and A. P. Monkman, J. Chem. Phys., 2009, 130, 074501 CrossRef PubMed; (b) C. Rothe and A. P. Monkman, Phys. Rev. B: Condens. Matter, 2003, 68, 075208 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of the monomer units; CIE diagrams. See DOI: 10.1039/c4tc00896k |
| ‡ Present address: College of Chemistry and Materials Science, Liaoning Shihua University, Fushun City, 113001, P. R. China. |
| This journal is © The Royal Society of Chemistry 2014 |