 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interacting networks of purely organic spin–1/2 dimers†
Yulia B.
Borozdina‡
a,
Evgeny
Mostovich
ab,
Volker
Enkelmann
a,
Bernd
Wolf
c,
Pham T.
Cong
c,
Ulrich
Tutsch
c,
Michael
Lang
c and
Martin
Baumgarten
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
bNovosibirsk Institute of Organic Chemistry Siberian Branch of the Russian Academy of Science, Lavrentiev avenue 9, 630090 Novosibirsk, Russia
cPhysikalisches Institut, Johann Wolfgang Goethe- University Frankfurt, 60438 Frankfurt am Main, Germany
First published on 26th June 2014
Abstract
In the present study we report the synthesis of some novel nitronyl nitroxide biradical systems 1–4c with various π-bridges between the radical centres. UV-Vis, IR, EPR and X-ray diffraction studies, along with MS and NMR data where appropriate, are described. Magnetic measurements revealed that the biradicals 1c, 3c and 4c exhibit a moderately strong antiferromagnetic intra-molecular exchange, whereas nitroxide 2c shows a significantly higher exchange coupling, which can only be explained by the presence of strong inter-molecular interactions. From DFT calculations performed on the basis of the X-ray crystal structure of compound 4c, a theoretical value of the intra-dimer coupling constant Jintra = −8.6 K is obtained. Direct proof also for inter-molecular arrangement with Jinter ∼ −2 K was provided by the low temperature AC studies of biradical 4c. According to the magnetic characterization, the nitronyl biradical 4c is a promising candidate for a purely organic-based low-dimensional quantum magnet.
Introduction
Recently, coupled spin–dimer systems composed of closely-spaced pairs of spin-1/2 carrying entities, have been used as model systems for exploring critical phenomena under well-controlled conditions.1 As a prominent example of special features observed here we mention the field-induced delocalization of bosonic excitations in reduced dimensions. Likewise, for three-dimensional spin–dimer networks, these systems can be used to study the phenomenon of Bose-Einstein condensation (BEC) of magnetic excitations.1The most commonly used spin carriers for constructing magnetic materials are metal ions and nitroxide radicals.2 Hence, various approaches toward molecular magnets can be classified into inorganic and organic ones, where the spins are provided by metal ions, the so-called “metal-radical” or hybrid approach, which combines magnetically active metal ions and persistent organic radicals as ligands,3,4 and the organic approach,5 with purely organic radical entities. Inorganic materials have some drawbacks, as the size of the cluster, which can be compressed only till a certain limit, and that the structure of the complex is difficult to predict depending on serendipity. On the other hand, organic compounds are usually transparent in many regions and could be obtained in optical active chiral forms.4 Thus, they might be used as magneto-optical switches and for the manipulation of polarized light in optical devices. Latterly they were extensively studied due to their potential application as radical-based batteries,6–8 electric conductors6,9,10 and spintronic devices.6,9,11
Importantly, chemistry techniques permits to perform structural modifications in organic materials in order to control the intra- and inter-molecular exchange interactions between the spin carriers.5 Thus, the former can be designed and adjusted through synthesis of different conjugated bridges between the spin-containing sites, although the latter requires supramolecular approaches employing hydrogen bonding, π-stacking, and metal complexation. Such tunability is unprecedented in most of the traditional inorganic materials and is the key to obtain materials with unique magnetic properties.
As the spin carrier we have chosen nitronyl nitroxide (NN) radicals,1 due to their thermal and environmental stability and chelating properties. As in other biradical species, such as imino nitroxide, verdazyl or tert-butyl nitroxide radicals, the spin density of the unpaired electron is delocalized over two sites of coordination, leaving open the possibility to arrange molecular units into a supramolecular network with unprecedented magnetic structure.12,13
In our study we considered organic spin–dimer systems with singlet ground state and moderate intra-dimer interactions in the range of −2 to −8 K (the numbers refer to the exchange coupling defined in accordance with the Hamiltonian used in the DFT calculations). In addition, the compounds should form interacting networks due to small inter-dimer interactions, giving rise to field-induced magnetic states, depending on the dimensionality of the interactions, at fields below the saturation field.2 Since it is difficult to predict the precise magnitude of the intra- and inter-molecular exchange interactions, which can further affect the strength of the bulk magnetic coupling, we started from approximate computational estimations based on the coordination geometry and the bond distances in the molecules, and have targeted four biradicals for in-depth characterization.
Herein we report the synthesis of biradicals 1c–4c, designed for the fine tuning of spin exchange interactions, their characterization, including X-ray structural analysis, and, finally, the magnetic studies, which are discussed with respect to the quantum chemical calculations based on a broken symmetry DFT approach.
Results and discussion
Synthesis of biradicals
The synthesis of the nitronyl nitroxide radicals (NN) relies, in general, on the condensation between 2,3-bis(hydroxyamino)-2,3-dimethylbutane (BHA) with an aldehyde functional group, followed by the oxidation reaction of the condensation products (Fig. 1). In order to achieve fine tuning of the magnetic exchange interactions in biradical systems, we used conjugation control of the bridging unit with topologically aligned radicals in the para-position. Theoretical studies emphasized the importance of the mutual orientation and relative distances in the crystal packing for the unimpeded magnetic coupling propagation.13 We engineered four novel π-conjugated formyl derivatives (1a–4a), the key precursors towards NN synthesis, via diverse multi-steps approaches, as outlined in Schemes 1–4. These π-conjugated systems vary from each other, and can be divided into three different classes. The first group (i) contains electron-donor substituents, which are able to increase the electron density along the spin-coupling backbone (radical 1a–c, dimethoxyphenyl-derivatives). Class (ii) is made by rigid and planar molecules (compounds 3 and 4, in other words, acetylene derivatives). Finally, group (iii), which includes five-membered hetero-rings as functional part of the coupling unit, which provides non-alternate spin-polarization paths (compound 2a–c, pyrazole derivatives).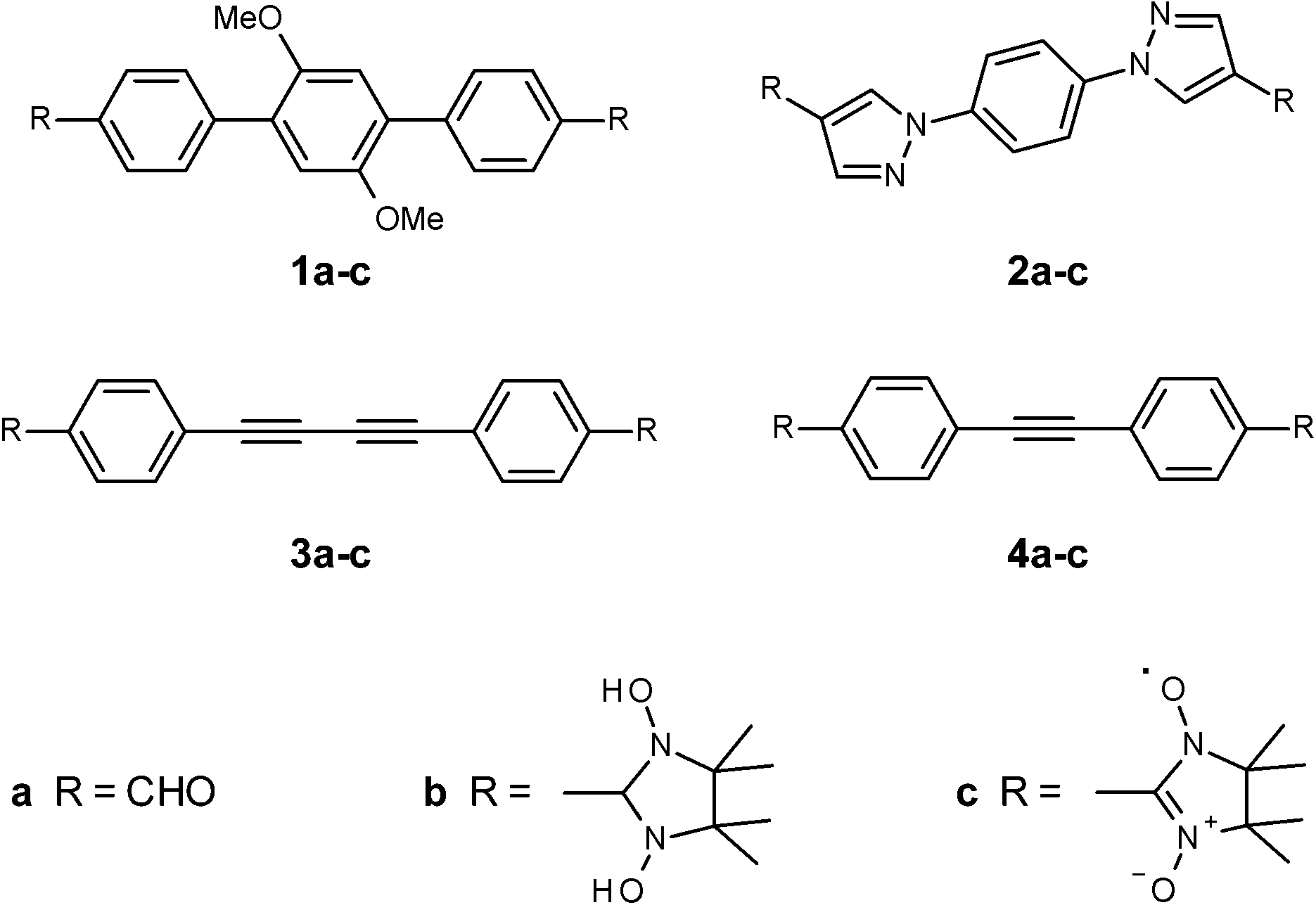 | ||
| Fig. 1 Model compounds 1–4 with emphasized bridging units. | ||
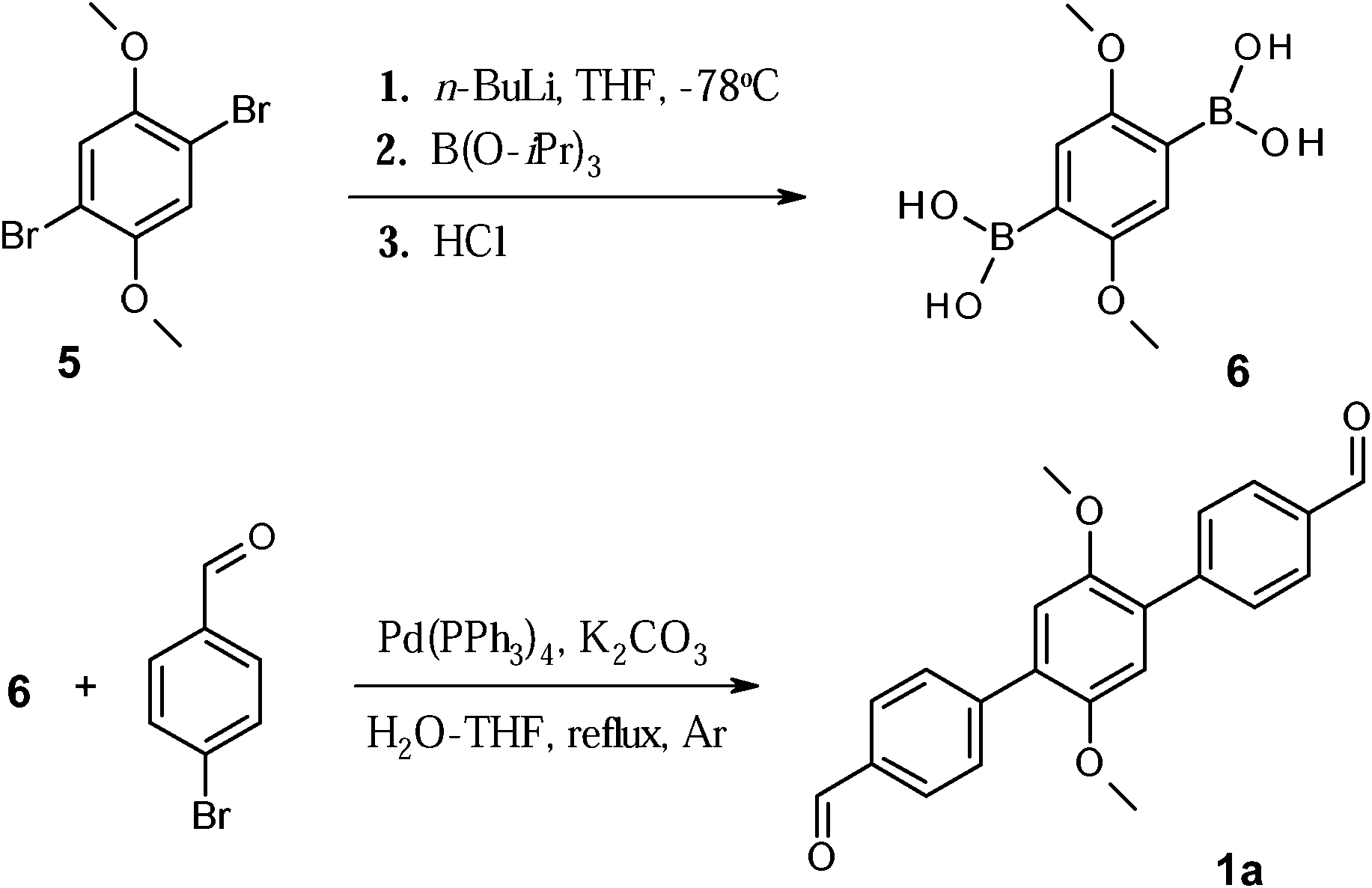 | ||
| Scheme 1 Synthetic route towards dialdehyde 1a. | ||
Synthesis of the first target dialdehyde 1a (class (i)) is represented in Scheme 1. Regardless of the different coupling conditions tested, the 4-[4-(4-formylmethyl)-2,5-dimethoxyphenyl]benzaldehyde (compound 1a) could not be synthesised via direct coupling of the dibromo derivative 5 with 4-formylbenzeneboronic acid. In fact, only the mono-substituted product was isolated at best. Thus, an alternative route was explored successfully, as shown in Scheme 1. Initially the dimethoxyether 5 was treated with n-BuLi in dry THF at low temperature (T = −78 °C), and reaction mixture was left stirring under argon atmosphere at the same temperature for 5 hours. At the end of this time period excess of boron triisopropoxide (tri(propan-2-yloxy)borane) was added dropwise. The mixture was treated with 5% HCl, which afforded diboronic acid derivative (compound 6) as white solid. Here, the resonance 1H-NMR signal of the hydroxy groups (OH) present in the isolated product falls at 7.82 ppm and confirmed, therefore, formation of the desired derivative. The compound was furthermore recrystallized from H2O–CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture, affording highly pure derivative 6 in a fairly good yield (45%).
:1) mixture, affording highly pure derivative 6 in a fairly good yield (45%).
Diboronic acid 6 was then used in the Suzuki coupling reaction with 4-bromobenzaldehyde. The dialdehyde 1a was obtained only after prolonged reaction under reflux (5 days) in 62% yield. The 1H-NMR spectrum of 1a shows the appearance of new signals in the aromatic region, at 7.80 and 7.95 ppm, together with 1H-resonance signal at 10 ppm, which is the clear fingerprint for the presence of undamaged aldehyde moieties.
Scheme 2 outlines the synthetic route towards 4-formyl-1-[4-(4-formyl-1H-pyrazol-1-yl)phenyl]-1H-pyrazole (compound 2a, class (iii)). The reaction of 1,4-dibrombenzene 7 with excess of pyrazole in the presence of CuI and potassium carbonate (nitrobenzene, T = 210 °C) afforded 1,4-bis(1-pyrazol-1H-yl)benzene 8 in a high yield (76%). Subsequent iodination reaction was carried out using a mixture of 8, iodic acid, and iodine. The reactants were heated at 80 °C in acetic acid and let to react for three days. The process progress was monitored by TLC and FD-mass analysis of the reaction mixture aliquots. Crude product precipitated upon cooling as colourless plates. Although the reaction yield appeared initially very good, the crude contained not only the target di-iodo derivative 9, but also substantial amount of mono-iodinated species (∼25%). Extension of the reaction time did not significantly improve the ratio between mono- and di-iodo derivatives. However, the mono-iodinated compound could be easily separated from the mother solution by filtration, leading to the di-iodinated compound 9 in fairly good yield (∼60%). Notably, the mono-iodinated compound could be used itself as a starting material, in order to force the completion of the iodinating reaction, in other words, to functionalize the position 4 of the left unsubstituted pyrazole ring. Here, the disappearance of the pyrazole proton signal at 4th position (6.50 ppm), is accompanied by a significant down-shift of the H signal at the 5th position, from 7.75 ppm to 8.76 ppm in 1H NMR spectra, and those resonances can be used as fingerprints to probe the progresses of conversion into compound 9.
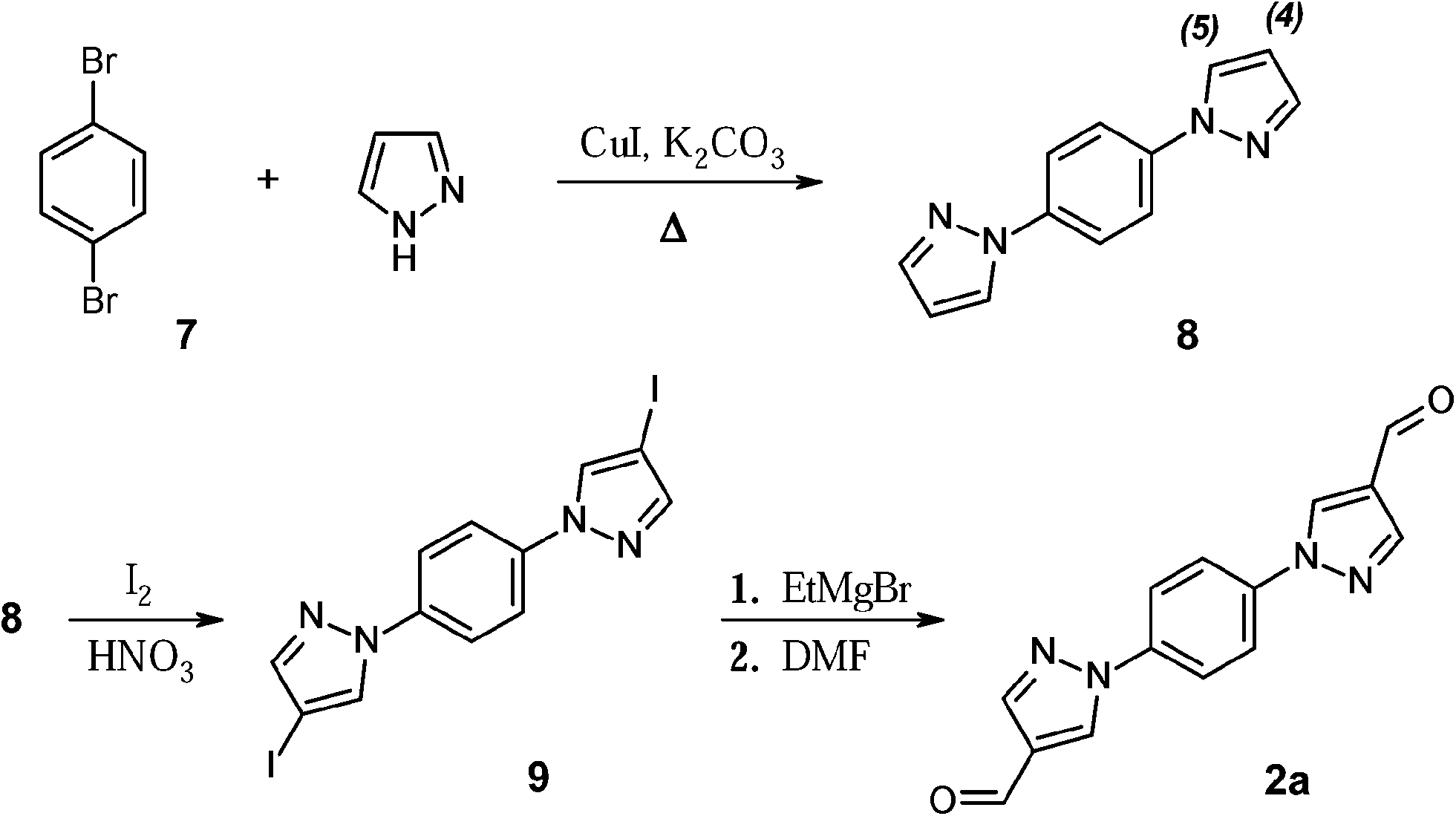 | ||
| Scheme 2 Synthetic route towards dialdehyde 2a. | ||
The final step towards dialdehyde 2a, containing pyrazole-phenyl bridging unit, was accomplished via formylation reaction of the di-iodo derivative 9 through initial formation of the corresponding Grignard reagent. Compound 9 reacted smoothly with ethylmagnesium bromide. The magnesium intermediate was quenched with the stoichiometric amounts of DMF and afforded the target dialdehyde 2a in high yield (89%).
The diacetylene fragment was introduced as the spacer into the π-system, giving rise to the biradical 3c (class (ii)). It is expected that 3c should feature little or no torsion within the molecule, which is essential for the weak intra-molecular coupling. One of the most efficient methods for the assembly of the molecules containing acetylene fragments relies on Glaser homo-coupling.14 Regardless of the different coupling conditions tested, we could achieve only traces amounts of the dialdehyde 3a employing direct Glaser coupling methodology to 4-ethynylbenzaldehyde 10 (Scheme 3). Thus, another route was undertaken, where the aldehyde function was at first protected with ethylene glycol affording the acetal precursor 11.15 The 1H NMR spectrum of 11 showed characteristic signals of dioxolane ring at 4.0 ppm (–CH3, methylene groups) and at 6.7 ppm (–CH). The signal associated to the aldehyde group, on the contrary, was absent, hence ensuring the complete conversion into the aldehyde-protected intermediate. The dioxolane derivative 11 was subjected to Glaser coupling. The reaction required excess of copper (I) and (II) chlorides (7 eq. and 5 eq., respectively) in pyridine media, followed by acidic cleavage of the dioxolane ring in THF–water mixture (5:3). Here, appearance in the 1H, 13C NMR spectra of strong signals typical for the aldehyde fragments at 10.0 (1H) and 191 (13C) ppm, confirmed successful completion of the coupling reaction and formation of the desired 4,4′-(buta-1,3-diyne-1,4-diyl)dibenzaldehyde 3a.
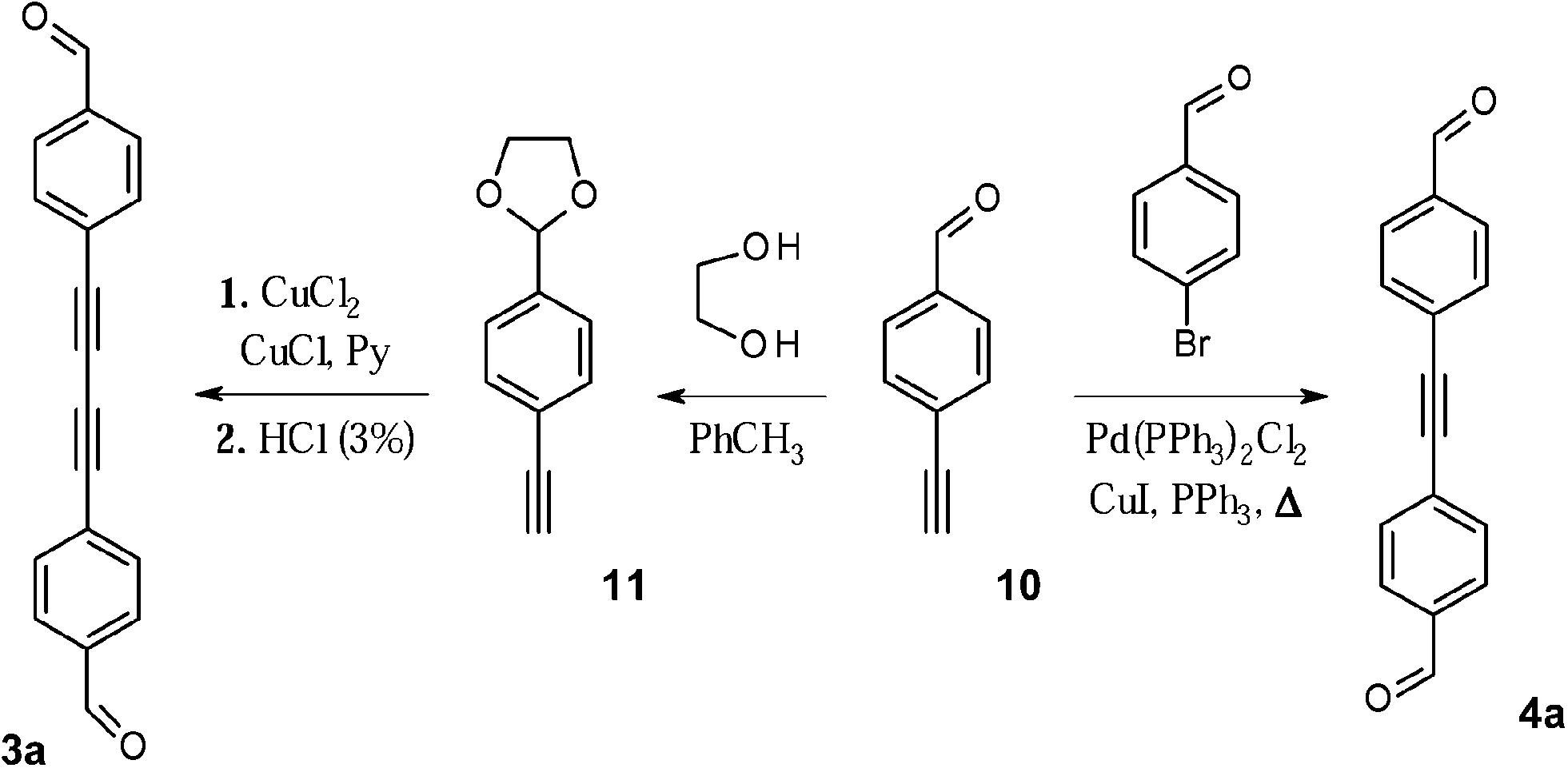 | ||
| Scheme 3 Synthetic route towards dialdehydes 3a and 4a. | ||
To explore the potential of another closely associated derivative, the tolane dialdehyde 4a was synthesized via Sonogashira–Hagihara cross-coupling reaction.16 This system contains a shorter π-conjugated linker between the radical centres, hence the resulting through-bond exchange interaction is expected to be stronger in comparison to 3a. The cross-coupling reaction between 4-bromobenzaldehyde and 4-ethynylbenzaldehyde 10 was carried out employing catalytic mixture of Pd(PPh3)2Cl2, CuI, PPh3 in the presence of base (Et3N) and led to the formation of the dialdehyde 4a in high yield (70%).
The target nitronyl nitroxide molecules could be routinely prepared as illustrated in Scheme 4. The condensation of the 2,3-diamino-N,N′-dihydroxy-2,3-dimethylbutane with carbal-dehydes is an air-sensitive process and required, therefore, strict anaerobic conditions and degassed solvents.
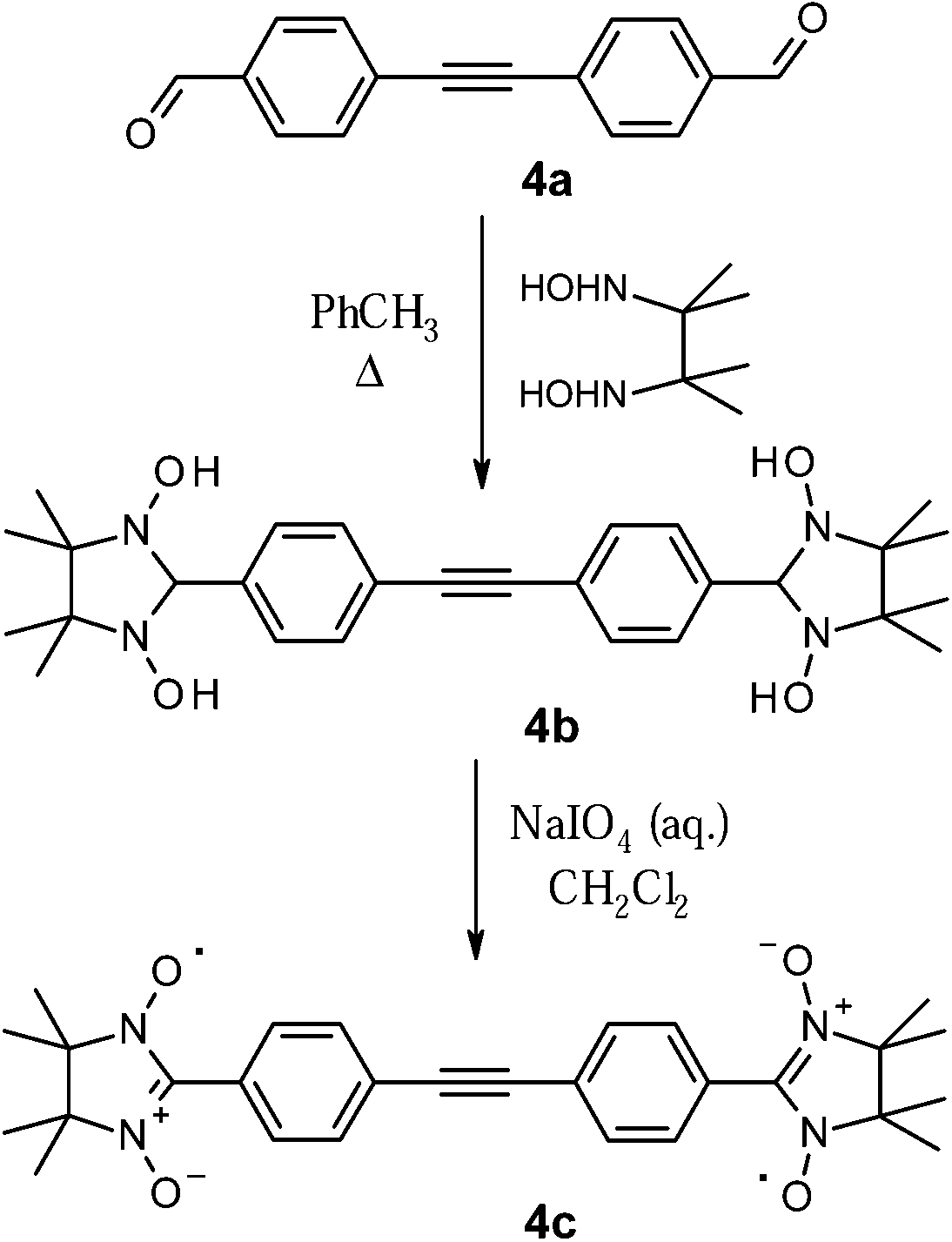 | ||
| Scheme 4 Synthesis of the imidazolidine 4b and biradical 4c. | ||
As a general rule, reaction with dialdehydes 1–4a was performed in deaerated toluene under argon at 110 °C, and afforded the corresponding N,N′-dihydroxyimidazolidines 1b–4b in quantitative yields. Once formed, the radical precursors (e.g.4b in Scheme 4) precipitated during the condensation step, and they were isolated by filtration as analytically pure (from 1H-NMR spectra) white or yellowish powders. The 1H-NMR spectra of these intermediates exhibited characteristic peaks of the hydroxy groups (∼7.8–8.0 ppm, marked with the letter a in Fig. 2.) and the nodal proton located at the second position of the imidazolidine ring (∼4.7 ppm, –CH group, labelled with the letter d). To illustrate this, the 1H NMR spectrum of the condensation product 4b is represented in Fig. 2. Here, the remaining in the sample toluene is specified with an asterisk.
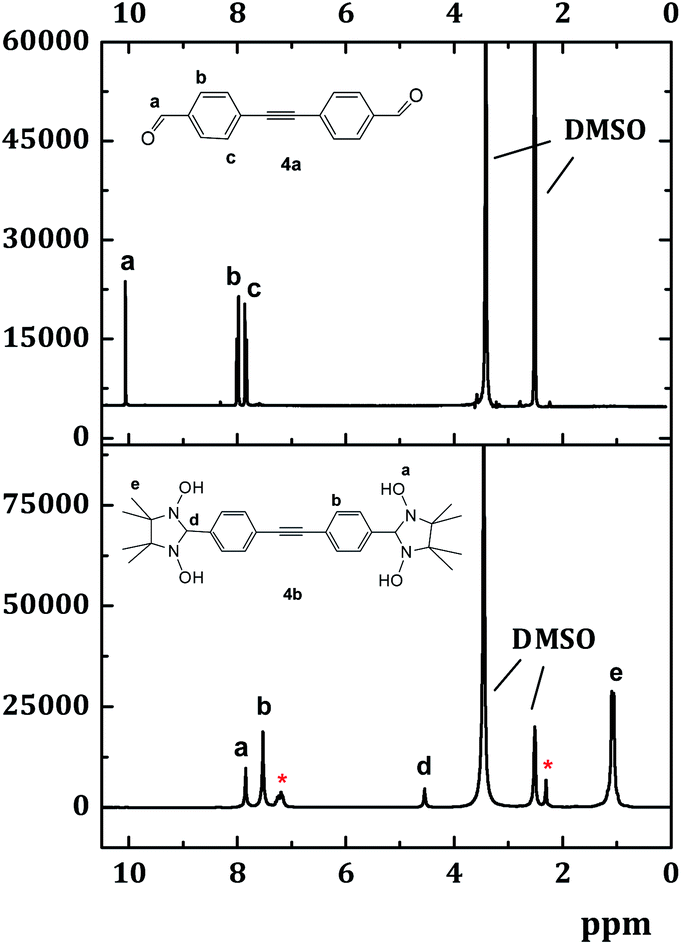 | ||
| Fig. 2 NMR spectrum of 4a, b in DMSO at 250 MHz (128 and 105 scans, respectively). The intensities of the solvent signals differ so much due to the significantly lower solubility of the dialdehyde 4a in DMSO. The asterisk indicates toluene. | ||
Oxidation of the N,N′-dihydroxyimidazolidines 1b–4b with sodium periodate in a biphasic media of dichloromethane and water afforded nitronyl biradicals 1c–4c (Scheme 4). In order to avoid both further oxidation of the radicals to imino nitroxides, and to diminish the loss of the radical units, the reactions were carried out at T = 0–5 °C using an ice bath. This method was preferred over the one, where solid metal oxides were applied as oxidizing agents,17,18 because it allowed a better control of the oxidation reaction by using stoichiometric amounts of periodate in solution. The progress of the reaction was monitored by TLC of the reaction mixture aliquots. All the radicals were purified using flash column chromatography on silica (SiO2).
The synthesized nitroxide systems 1c–4c were found fairly well soluble in most of the organic solvents tested (e.g. MeOH, CHCl3, THF). However, in aprotic media (e.g. toluene), longer stability of the radical molecules over the time was observed. In protic solvents (e.g. CH2Cl2 or CHCl3), partial decomposition of the radicals into diamagnetic products occurred within a day, due to sequential release of water molecules (dehydration), as witnessed from the decrease of the radical signal intensity in both UV-Vis absorption and in the EPR spectra (UV-Vis spectroscopy with absorption signals around 600 nm, and EPR resonance signals around giso = 2.0063).
Absorption spectra (UV-Vis and IR spectroscopic studies)
The relevant UV-Vis and IR data recorded for 1c–4c are given together in Table 1 for a better comparison.| Biradical | IRaνNO/cm−1 | UV-Visbλmax nm−1 (εb M−1 cm−1) | Massc g mol−1 | EPR aN/mT |
|---|---|---|---|---|
| a Measured in solid state at room temperature (T = 293 K). b Measured in toluene solution except for compound 2c that was measured in freshly prepared CHCl3 solution. c Measured in dichloromethane by FAB. The m/z+ (100%) peak corresponds to [M]+. | ||||
| 1c | 1358 | 627 (557) | 599 | 0.752 |
| 2c | 1354 | 599 (3808), 654 (4011) | 519 | 0.768 |
| 3c | 1365 | 622 (578) | 511 | 0.744 |
| 4c | 1356,1364 | 611 (868) | 487 | 0.762 |
The UV-Vis absorption spectra of the biradicals systems 1c–4c were recorded in toluene solutions and exhibited the characteristic deep blue colour, which is typical for this class of radicals. Two distinctive set of absorption bands dominate the optical spectra (see ESI Fig. S1†). The first set of bands is characterized by differently enhanced vibronic components, and takes place at a longer wavelength, around 600 nm. Those signals correspond to the n–π* transitions of the aminoxyl oxides moiety. The second set of absorption values includes the π–π* transitions of the aromatic systems, and falls at much higher energy in the 300–380 nm range. Complementary to UV-Vis, the appearance of a novel signal after oxidation of the imidazolidine derivatives at around υ ∼1350 cm−1 (Table 1) was assumed to arise from the N–O stretching vibration of the imidazoline moiety, which was in harmony with comparable signals observed for several other NN systems described in the literature.19,20 Other strong signals appear at ∼1550 cm−1, but those are less characteristic since they belong to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations. In addition, the IR spectra ensured absence of the traces of the dialdehyde precursors or imidazolidine condensation products in the purified biradical powders (see for comparison Fig. S2 and S3†).
N stretching vibrations. In addition, the IR spectra ensured absence of the traces of the dialdehyde precursors or imidazolidine condensation products in the purified biradical powders (see for comparison Fig. S2 and S3†).
EPR spectroscopy
The solution EPR spectra of the biradical systems 1c–4c were recorded in oxygen-free toluene solutions (see ESI Fig. S4–S7†). at room temperature (T = 298 K). The nine lines splitting pattern witnessed in the studied systems highlights efficient through-bond coupling of the two nitronyl nitroxide residues via spin-polarization effects of the π-spacer. Regardless of the different distances between the radical centres, as well as in the molecular identity of the coupling units employed, this property hold for 1c, 2c and 4c. Thus, within the limit of X-band EPR spectroscopy, the attainment of strong exchange interactions (J) |J/aN| ≫ 1 between the spin carriers were experimentally verified in all studied biradical systems. The spin-concentrations for 1c–4c at T = 298 K were always consistent with two spin centres, where singlet and triplet states are thermally populated and statistically distributed according to the Boltzmann law. The calculated values of the hyperfine coupling constants (aN) for 1c–4c are given in Table 1 and fall between 0.7 and 0.8 mT (giso ≈ 2.0063). These values are in harmony with those typically observed in the nitronyl nitroxide radical systems found in the literature.Crystal structures analysis
Single-crystals suitable for X-ray analysis were obtained by slow diffusion of dichloromethane–hexane mixture (1:4) for biradicals 1c, 3c and 4c at room temperature. Deep-blue needle crystals of biradicals 1c, 3c and 4c were analysed by X-ray diffraction and their ORTEP drawing as given in Fig. 3–5. Pertinent crystallographic parameters and refinement data are listed in Table 2, while selected torsional angles for the studied biradicals are reported in Table 3.
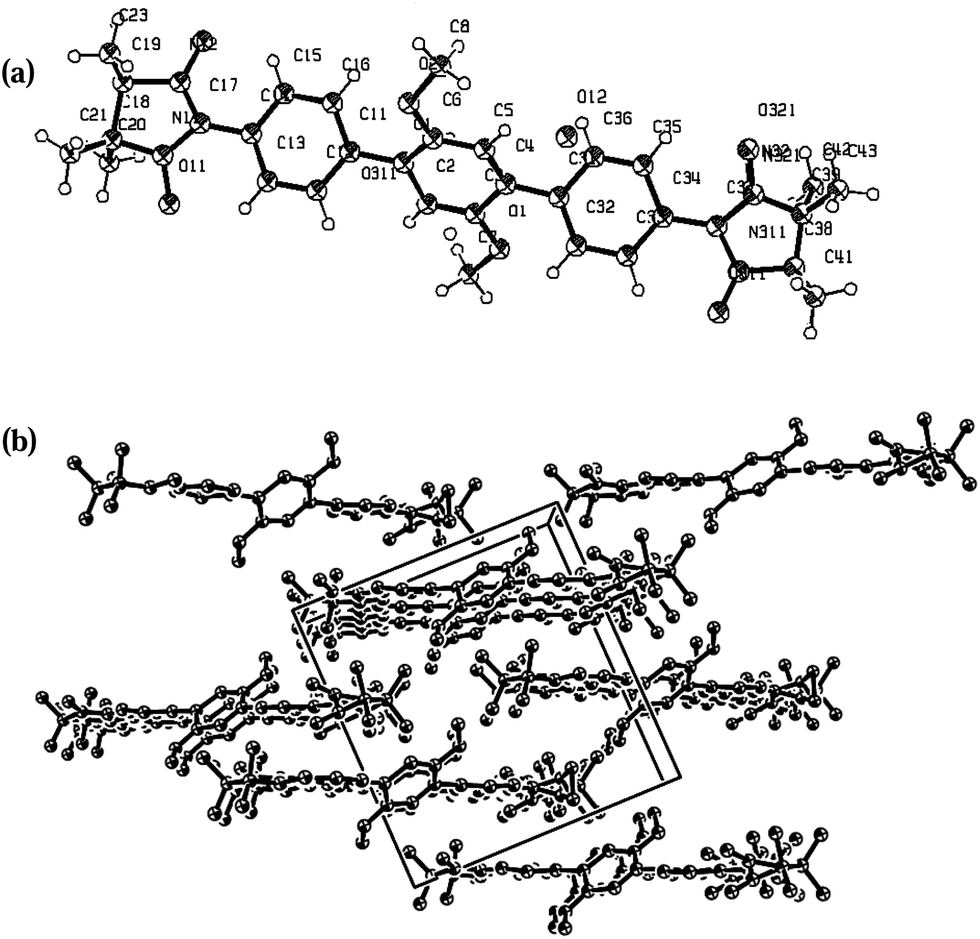 | ||
| Fig. 3 X-ray structure and the crystal packing of 1c with ORTEP drawn at the 50% of the probability level: (a) molecule; (b) crystal packing. | ||
| 1c | 3c | 4c | |
|---|---|---|---|
| Formula | C34H40N4O6 | C30H32N4O4 | C28H32N4O4 |
| M | 600 | 512 | 488 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | Pn (no. 7) | P21/c (no. 14) | P21/n (no. 14) |
| a/Å | 13.045(5) | 8.1768(2) | 6.0834(4) |
| b/Å | 8.447(4) | 19.0191(8) | 11.2581(5) |
| c/Å | 13.703(5) | 8.6983(3) | 19.1236(9) |
| β/deg | 91.593(12) | 103.4(2) | 90.832(3) |
| V/Å3 | 1509.3(11) | 1315.89(8) | 1309.59(12) |
| Z | 4 | 4 | 4 |
| R factor(%) | 9.41 | 7.32 | 4.53 |
| ρ c/g × cm−3 | 1.322 | 1.294 | 1.239 |
| Mo kα/mm−1 | 0.711 | 0.711 | 0.711 |
| N ref | 4156 | 3687 | 3784 |
| N par | 193 | 173 | 163 |
| S | 5.633 | 0.985 | 1.081 |
| CCDC | 822066 | 816633 | 816635 |
As shown in the ORTEP diagram (Fig. 3), radical 1c is symmetric to the central phenyl ring and has C2 symmetry axis through C1–C4 atoms of the benzene ring. The dihedral angles between the phenyl ring and radical plane are 11.2° (N11⋯C17⋯C14⋯C13) and 5.75° (N12⋯C17⋯C14⋯C15). The O–N–C–N–O moiety is planar, with nearly equivalent O–N bond lengths of 1.31(O11⋯N11), 1.33 (O12⋯N12), 1.26 (O31⋯N31), 1.24 (O32⋯N32) Å. Closer analysis of the crystal packing shows that the N–O entities of neighboring molecules are attached to the benzene ring (O11⋯H161* 2.48, O12⋯H51* 2.6, O12⋯C36* 3.18 Å) and methoxy group of phenyl moiety to a radical fragment of the neighboring unit (O32⋯O1* 2.94, O32⋯C7* 3.02 Å); the shortest intersheet contact is found between the nitronyl oxygen and methyl group (O312⋯H203* 2.36 Å). Therefore, the intermolecular distances are short enough to favour magnetic interactions.
The crystallographic data were collected on Nonius Kappa CCD (Mo kα, μ = 0.71073 Å) diffractometer equipped with a graphite monochromator.
Compound 3c is crystallized in a transoid arrangement with a center of symmetry located on the central C1–C1* bond (Fig. 4). The derivative is fairly planar, and there is a dihedral angle between the mean plane of the C6 ring and the imidazolidine moiety of 25°. In the present structure the distances between the N–O and C–N atoms of the radical subunits are similar: N(16)–O(18) 1.276; N(17)–O(19) 1.281; N(16)–C(9) 1.36; N(17)–C(9) 1.345 Å. This symmetry is due to the complete delocalization of the odd electron in the non-bonding orbitals of the ‘ONCNO’ core. The N–O bond and the C–N bond distances are between the values of a single and a double bond. Closer examination of the crystal packing shows that the radicals are organized in a zig-zag fashion (O18⋯H133* 2.53 Å, Fig. 4(b)) with additional hydrogen bonds between the sheets formed by C5 atom of the benzene ring with hydrogen atoms (C5⋯H71* 2.8, C5⋯H152* 2.86 Å, Fig. 4).
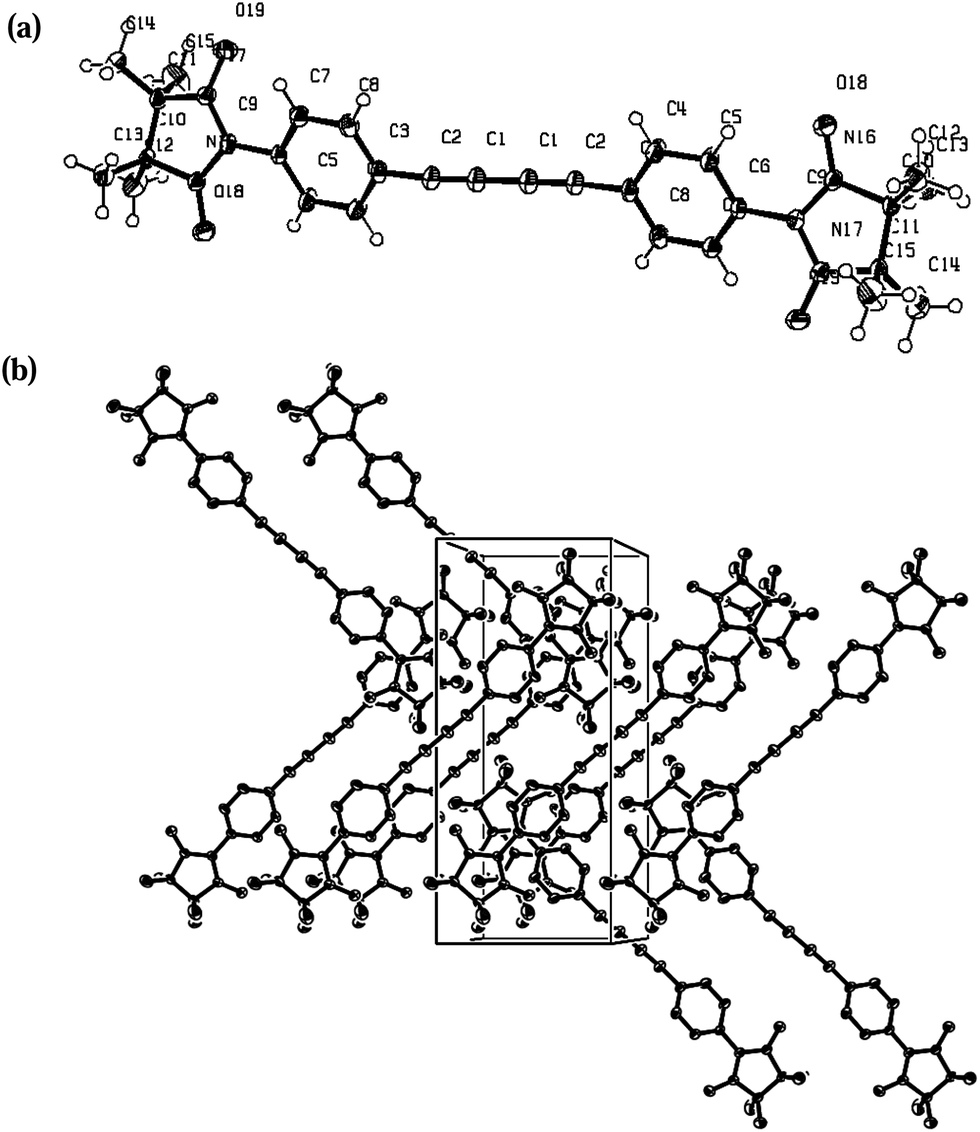 | ||
| Fig. 4 X-ray structure and the crystal packing of 3c with ORTEP drawn at the 50% of the probability level: (a) molecule; (b) crystal packing. View along the c axe of the crystal packing of compound 3c. | ||
The molecular structure of compound 4c (Fig. 5) is similar to the one previously reported21 which has the same space group (P21/n). Probably, such similar cell organization can be explained by crystallization conditions. The torsion angle between the plane of the benzene ring and the radical moiety is 25°. Adjacent radicals are organized in a head-to-tail fashion forming 1D network: the first N–O fragment of each radical unit is attached to C7, H71 atoms of the benzene ring, the second NO binds to the hydrogen atoms of the methyl group of an imidazolidine fragment. Geometrical parameters are: O2⋯H71* 2.52, O1⋯H141* 2.59, O2⋯C7* 3.16, C7⋯C13 3.4 Å; the distance between H-bonded sheets totals 2.36 Å (O1⋯H101*).
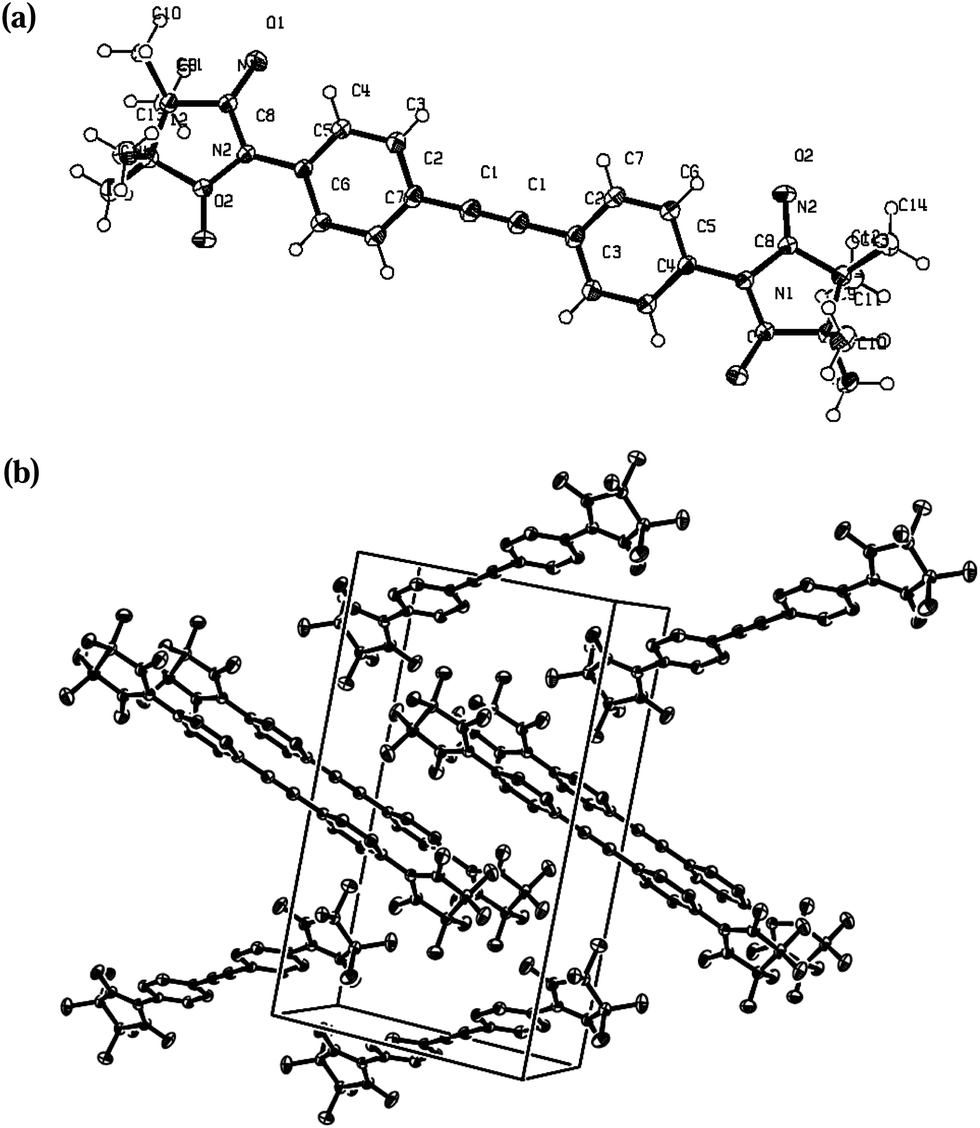 | ||
| Fig. 5 X-ray structure of 4c with ORTEP drawn at the 50% of the probability level: (a) molecule; (b) crystal packing. | ||
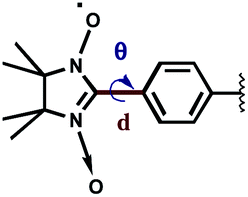
Reasonably to assume, that the intermolecular interaction in two radicals oriented in a head-to-tail fashion is at maximum when the two NO groups are arranged in such a way that the O–N⋯O* angle α is equal to 90° and the angle β between the π* orbitals of the nitronyl nitroxide radicals with a vector normal to the N2O2 plane is also 90°.22,23 Thus, not only the intermolecular distances but the whole molecular structure of the biradical 4c favours intermolecular magnetic interactions.
In order to rationalize the magnetic properties of molecular compounds, analysis of the molecular packing (so, as to find out the possible magneto-structural correlations) was carried out. The studied biradical NN systems shared nearly planar arrangements of the coupling-core. Structural analyses revealed rather small torsions between the imidazolyl and the aromatic core (<30°), which facilitates efficient communication between the radical moieties through the coupler. Additionally, the intersheet contacts are found to be short enough (∼2.5 Å) to favour magnetic interactions.
DFT calculations and magnetic measurements
In order to understand the influence of the bridging unit on the electronic properties and especially on the exchange coupling, DFT calculations were performed. We thus carried out a geometry optimization with the B3LYP hybrid function and 6-31G* basis set and we then applied the broken symmetry approach (BS) for the singlet and triplet states with BLYP (to avoid Hartree–Fock contamination) and 6-31G* basis set according to an approximate projection technique as suggested by Yamaguchi.24 For the simplest case of two spins S = 1/2 this form of the spin Hamiltonian (H = −2J12S1S2, here the exchange parameter J will be negative in the case of antiferromagnetic interaction) gives the exchange interaction as J = (E(BS) − E(T))/(S2(T) − S2(BS)), where E(BS) is the energy of the broken symmetry approach, E(T) is the energy of the triplet state and S2 are the eigenvalues of the spin operator for these states. In case of weak interaction without spin contamination S2(T) = 2 and S2(BS) = 1 the direct exchange yields J = E(BS) − E(T). The obtained theoretical exchange coupling constants J are listed in Table 4 together with the experimental values for a better comparison.| Biradical | T max/K | J calc/Ka | J calc/Kb | J exp/Kc |
|---|---|---|---|---|
| a Optimized geometries were obtained from DFT calculations (B3LYP, 6-31G*), then the broken symmetry approach was applied (BLYP, 6-31G*) for the evaluation of J. b The exchange interactions were evaluated for X-ray crystal structures applying the broken symmetry approach with BLYP functionals and 6-31G* basis set. c Expected from the isolated dimer model, where 2J/kBTmax = 1.599. | ||||
| 1c | 0.7 | −2.7 | −2.0 | −0.9 |
| 2c | 19.5 | −2.5 | — | −16.0! |
| 3c | 3.2 | −14.4 | −5.8 | −2.4 |
| 4c | 6.3 | −16.8 | −8.6 | −4.8 |
The magnetic properties of the samples 1c–4c were determined in the temperature range 2 K ≤ T ≤ 270 K and in magnetic fields B ≤ 5 T by using a Quantum Design SQUID magnetometer. All data have been corrected for the temperature-independent diamagnetic core contribution of the constituents25 and the magnetic contribution of the sample holder. For temperatures below 2 K the ac-susceptibility data shown in Fig. 7 were recorded using an ultra-high resolution ac-susceptometer adapted to a 3He–4He top-loading dilution refrigerator. The compensated-coil susceptometer is optimized for measuring small single crystals in the mg range. The system is equipped with a 12 T superconducting magnet.
Generally, all the biradicals under investigation (1c–4c) featured a similar magnetic behaviour in the studied temperature range. The high-quality single crystalline materials 1c, 3c, and 4c exhibit magnetic contributions of Curie type, assigned to uncoupled spins, the concentration of which is much less than 1%. The Curie contribution for the biradical 2c was slightly larger. Furthermore the small Curie contribution for 2c is indicated by the broken line in the inset of Fig. S9.†
As an example for the magnetic behaviour of the biradicals 1c–4c, we show the magnetic susceptibility, plotted as χmolT, for the system 4c in Fig. 6. The data reveal an almost temperature-independent behaviour in the range from about 100 K to 270 K, with a value χmolT = (0.734 ± 0.005) cm3 Kmol−1. This value is very close to the theoretically expected value of 0.75 cm3 Kmol−1 for two independent spin S = 1/2 entities (broken line in Fig. 6). Very similar values of χmolT = (0.731 ± 0.017) cm3 Kmol−1 were observed for the biradicals 1c–3c in the same temperature range. On cooling χmolT becomes progressively reduced below about 140 K, a clear signature of dominant antiferromagnetic through-bond interactions between radical centers (intra-dimer coupling). An estimate of the mean value for the magnetic exchange coupling in the high-temperature regime can be obtained by fitting the inverse magnetic susceptibility 1/χmol by a Curie–Weiss model in the temperature range 70K ≤ T ≤ 270 K, cf. straight line in the inset of Fig. 6. The fit yields an antiferromagnetic Weiss temperature θW = −(8.2 ± 0.5) K. For a more precise determination of the dominant magnetic interactions, the temperature dependence of the molar magnetic susceptibility over the whole temperature range 2 K ≤ T ≤ 270 K was fitted using the Bleaney–Bowers equation26 for isolated dimers, cf. inset to Fig. 6. The intra-dimer magnetic exchange coupling constant Jintra between two S = 1/2 spins used in this expression refers to a Hamiltonian of the form H = −2J12S1S2, which was taken also for the DFT calculations. The inset of Fig. 6 demonstrates that the isolated-dimer model provides a reasonable description of the overall behaviour of χmol(T) for the biradical 4c. The biradicals 1c and 3c exhibited a similar behaviour, in accordance with the theoretical prediction, and their χmol(T) data were analyzed by an analogous procedure. Their intra-dimer exchange constants are listed in Table 4.
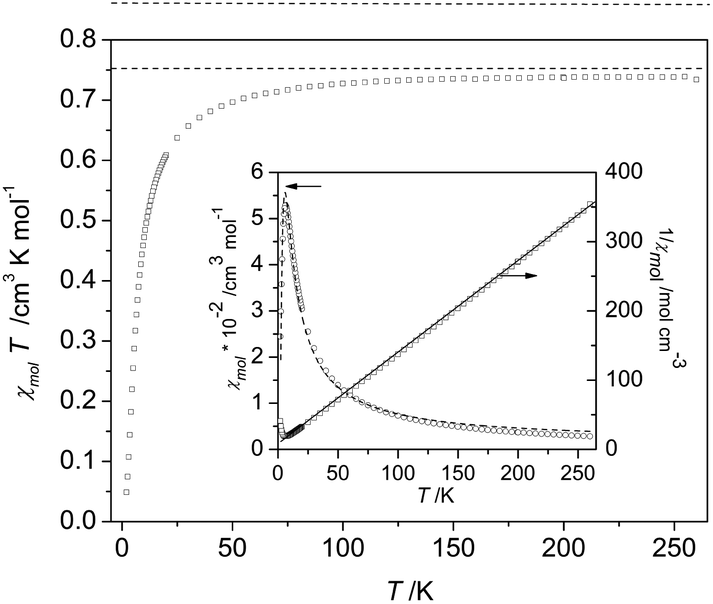 | ||
| Fig. 6 Magnetic susceptibility in a representation χmolT of biradical 4c. The broken line indicates the theoretical value of 0.75 cm3 K mol−1 for a dimer system formed by two spin S = 1/2 entities. Inset: molar susceptibility χmol (left scale) and its inverse 1/χmol (right scale) as a function of temperature together with fits performed by using a Curie–Weiss expression (solid line) and a Bleaney–Bowers equation26 (broken line). For fitting parameter and details see text. | ||
However, a closer inspection of the low-temperature (T < 4 K) susceptibility data as a function of temperature, and at T = const. as a function of magnetic field, for the various biradicals (see Fig. 7 below for 4c) revealed significant deviations from the isolated-dimer model. These deviations were attributed to the small magnetic intra- and inter-molecular interactions between neighboring biradicals mediated via hydrogen bonds (1c, 3c, 4c). The strength of these interactions depends on the distance and relative orientation of the biradicals.27–29
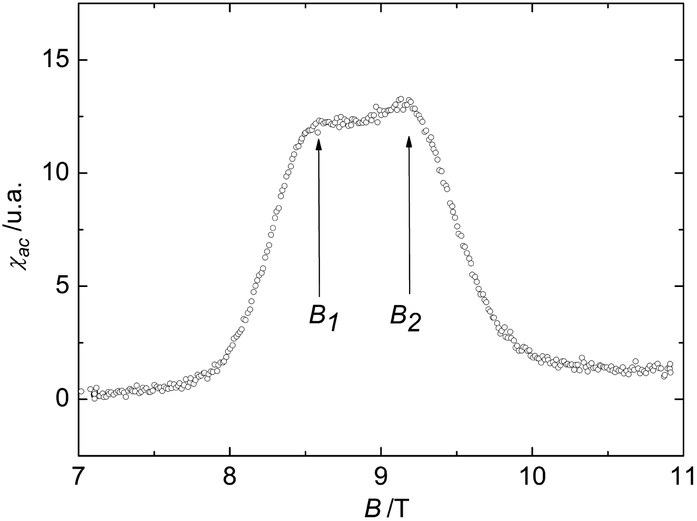 | ||
| Fig. 7 AC-susceptibility of the biradical 4c at T = 191 mK. The data feature rounded peaks at B1 ≈ 8.6 T and B2 ≈ 9.2 T. | ||
The results of AC-susceptibility measurements as a function of magnetic field at a constant temperature of 191 mK are shown in Fig. 7 on example of biradical 4c. The rounded double-peak structure in the data, with the peak positions B1 ≈ 8.6 T and B2 ≈ 9.2 T, indicates a field-induced magnetic state for B1 ≤ B ≤ B2. Note that B2 roughly corresponds to the saturation field Bs. Similar anomalies in the susceptibility are found in other spin ladder compounds, like (C5H12N)2CuBr4,30 which represent a class of low-dimensional materials with structural and physical properties lying between those of 1D and 2D systems. While a rounded double-peak structure is expected when the inter-dimer couplings are one- or quasi two- dimensional, sharp peaks reflect three-dimensional interactions. In fact, a quasi two-dimensional magnetic behaviour for the biradical 4c is also suggested by its crystal structure.
In leading order, the fields B1 and B2 (≈ Bs) are related to the dominant Jintra and an averaged Jinter by gμBB1/2 = Jintra − (Jinter)aver and gμBB2/2 = Jintra + 2(Jinter)aver (see, e.g. ref. 30 and references cited therein), resulting in (Jinter)aver of the order of −1 to −2 K for 4c. These are typical values for magnetic interactions mediated by hydrogen bridges.27–29 Thus, according to our magnetic characterization, the nitronyl biradical 4c is a promising candidate for a purely organic-based quasi two-dimensional quantum magnet.
Remarkably, the biradical 2c shows significant deviations from the theoretical expectation, cf.Table 4. For this system the maximum of χmol was located at a temperature Tmax = 19.5 K, distinctly higher than expected from DFT calculations for an isolated single biradical entity. This is different for all other biradicals shown in Table 4 where the agreement between experimental value and theoretical prediction is reasonably good. This might be the result of a peculiar magneto-structural correlation influence and/or strong inter-molecular exchange interactions existing in the material 2c. For instance, attraction of a neighboring radical to the lone pair of the nitrogen atom of the pyrazolyl moiety in 2c or stronger orbital overlap between the radical centres might cause such a drastic change in the magnetic properties of this biradical. In fact, the magnetic interactions based on structural peculiarities of this type are difficult to predict. The strength of these interactions strongly depends on the relative geometry between the interacting magnetic orbitals which is also determined by the crystal packing. Better understanding of the origin of such an unexpected behaviour in 2c therefore requires a detailed analysis of the crystal structure. Work on obtaining a suitable crystal for X-ray measurement is in progress.
Conclusions
In this study a series of novel nitronyl nitroxide biradicals 1c–4c coupled via modified tolane linkers was synthesized and their magnetic behaviour was studied. The tolane bridge seemed to be perfectly suited for the requirement of intra-dimer exchange coupling J ∼ 10 K. The structural analyses showed that the small torsions between the radical moieties and the modified diacetylene bridging subunit assist unimpeded through-bond exchange, as well as the small intersheet distances (π-stacking) favour efficient magnetic interactions. Magnetic measurements and their interpretation revealed that the biradical 2c exhibits surprisingly strong antiferromagnetic interactions, much larger than predicted for intra-molecular exchange interaction. In all other cases weak through-space antiferromagnetic interactions are predominant.Summarizing, we found promising candidates for the quest to synthesize a purely organic molecular magnet. Quantum Monte Carlo simulations for analysing the low-temperature susceptibility data are underway in order to derive the underlying effective exchange coupling topology of this compound. Efforts towards the preparation of extended networks through the formation of complexes with different metal ions coordinated to the radical unit and/or π-bridge are presently on-going. It would be of interest to extend these studies to other series of tolane-bridged biradicals, in particular the idea to introduce additional chelating sites within the core is considered.
Experimental
General Remarks
All chemicals and reagents were used as received from commercial sources (Acros Organics, Aldrich, Fluka, Lancaster, Merck and Strem) without additional purification. Solvents for synthesis were used as received, unless otherwise mentioned. ESR spectra were recorded in dilute, oxygen-free solutions in toluene, concentration ∼10−4 mol L, using a Bruker X-band spectrometer ESP300 E, equipped with an NMR gaussmeter (Bruker ER035), a frequency counter (Bruker ER041XK) and a variable temperature control continuous flow N2 cryostat (Bruker B-VT 2000). The g-factor corrections were obtained using the DPPH (g = 2.0037) as standard. UV-Vis spectra were recorded in toluene solutions with a Perkin-Elmer spectrometer (UV/Vis/NIR Lambda 900) using a 1 cm optical path quartz cell at room temperature, unless otherwise specified.1H and 13C NMR spectra were recorded on a Bruker DPX 250, Bruker DMX 300 spectrometer. Solid powders were pressed and IR spectra of the samples were recorded as they were (Nicolet 730 FT-IR spectrometer). Mass spectra were obtained on FDMS, VG Instruments ZAB-2 mass spectrometer. Elemental analyses were performed at the University of Mainz, Faculty of Chemistry and Pharmacy on a Foss Heraeus Varieo EL. The melting points were measured on a Büchi B-545 apparatus (uncorrected) by using open-ended capillaries.ESI.†A. General procedure of the condensation reaction between 2,3-dimethyl-2,3-bis(hydroxylamino)butane with aldehydes. A suspension of BHA (1.1 eq. for each aldehyde group) and an aldehyde (1.0 eq.) in toluene (5 mL mmol−1) was carefully deaerated using argon bubbling for ∼20 minutes. The flask was provided with a condenser containing an argon balloon, and moved into an oil bath. The mixture was heated under argon near reflux for 2–18 h. The progress of the reaction was monitored by TLC (SiO2, hexane/ethyl acetate or dichloromethane with 5% of methanol). After the process was completed and the flask was cooled down till room temperature formed precipitate (white or yellowish) was filtered off, washed with toluene (2 × 10 mL), heptane (1 × 10 mL) and dried on air. The product was used for the next step without additional purification.
B. General procedure for oxidation of 4,4,5,5-tetramethylimidazolidine-1,3-diol derivatives to nitronyl nitroxides. A suspension of 4,4,5,5-tetramethylimidazolidine-1,3-diol (1 mmol) in H2O–CH2Cl2 (1:2) mixture (∼50 mL) was cooled down to 0–5°C (to avoid overoxidation and formation of imino nitroxide species) using an ice bath. To that mixture NaIO4 5% aqueous solution (0.8 eq.) was added dropwise. After reaction was complete (0.5–3 h), dark-blue organic layer was separated. The aqua phase was extracted with dichloromethane (3 × 25 mL). The combined organic extracts were additionally washed with water (2 × 30 mL), NaCl saturated aqueous solution (1 × 50 mL), dried over MgSO4 (in some cases – Na2SO4). The residue after filtration was evaporated and purified using a chromatographic column (SiO2, hexane/ethyl acetate or CH2Cl2/MeOH).
:1, 100 mL) mixture, affording white needles of the product 6 in 45% yield (1.02 g). 1H-NMR ((CD3)2SO, 250 MHz, 298 K, 16 scan), δ ppm: 3.78 (s, 6H, –OCH3), 7.16 (s, 2H, Ar-H), 7.82 (s, 4H, –B(OH)2). 13C-NMR ((CD3)2SO, 63 MHz, 298K, 256 scan), δ ppm: 55.6, 116.8, 124.6, 157.4. FAB+m/z 225.9 [M]+.
:1) mixture and dried (0.71 g, 62%). 1H-NMR ((CD3)2SO, 250 MHz, 298 K, 64 scan), δ ppm: 3.80 (s, 6H, –OCH3), 7.12 (s, Ar-H), 7.80, 7.95 (both dd, 4H, 3J = 8.15 Hz, Ar-H), 10.04 (s, 2H, –CHO). 13C-NMR ((CD3)2SO, 176 MHz, 323 K, 51200 scan), δ ppm: 56.4, 114.8, 129.2, 129.4, 130.0, 134.9, 143.8, 150.5, 192.7. FAB+m/z 346.52 [M]+.
:1) eluent afforded dark-blue prisms of biradical 1c in 54% yield (0.15 g). UV-Vis (toluene): λ/nm (ε/M−1 cm−1) 627 (557), 356 (20425). (Calc. for C34H40N4O6: C, 67.98; H, 6.71; N, 9.33C. Found: C, 67.90; H, 6.66; N, 9.43%).
:1) was added through the rubber septum. Solution was degassed using argon bubbles for 20 min, and then catalytic mixture of Pd(PPh3)2Cl2 (54 mg, 0.077 mmol), PPh3 (40 mg, 0.154 mmol), CuI (15 mg, 0.077 mmol) was added at once. The flask was sealed, moved into an oil bath and heated to 70 °C under argon for 12 h. The solvents were concentrated in vacuo. Purification using column chromatography (SiO2, CH2Cl2) afforded the titled compound 4a in 92% yield (0.33 g). 1H-NMR (CDCl3, 250 MHz, 298 K, 64 scan), δ ppm: 7.71, 7.90 (both dd 2H, 3J = 8.2 Hz, Ar-H), 10.04 (s, 2H, –CHO). 13C-NMR (THF-d8, 75 MHz, 298 K, 1725 scan), δ ppm: 92.4, 129.0, 130.1, 132.9, 137.5, 191.3. FAB+m/z 234.063 [M]+.
Acknowledgements
The authors are pleased to acknowledge continued support from the Max Planck Society. This work was also supported by SFB TR49. We warmly thank Dr Giorgio Zopellaro for lively and helpful discussions.Notes and references
- Ch. Rüegg, K. Kiefer, B. Thielemann, D. F. McMorrow, V. Zapf, B. Normand, M. B. Zvonarev, P. Bouillot, C. Kollath, T. Giamarchi, S. Capponi, D. Poilblanc, D. Biner and K. W. Krämer, Phys. Rev. Lett., 2008, 101, 247202 CrossRef CAS; V. Gurarie and J. T. Chalker, Phys. Rev. Lett., 2002, 89, 136801 CrossRef; T. Giamarchi, Ch. Rüegg and O. Tchernyshyov, Nat. Phys., 2008, 4, 198 CrossRef.
- J. H. Osiecki and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 1078 CrossRef CAS.
- A. Caneschi, D. Gatteschi, P. Rey and R. Sessoli, Inorg. Chem., 1988, 27, 1756 CrossRef CAS; A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Acc. Chem. Res., 1989, 22, 392 CrossRef; C. Trein, L. Norel and M. Baumgarten, Coord. Chem. Rev., 2009, 253, 2342 CrossRef PubMed.
- K. Inoue, T. Hayamizu, H. Iwamura, D. Hashizume and Y. Ohashi, J. Am. Chem. Soc., 1996, 118, 1803 CrossRef CAS; S. Hiraoka, T. Okamoto, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, J. Am. Chem. Soc., 2004, 126, 58 CrossRef PubMed; C. Hirel, J. Pécaut, S. Choua, P. Turek, D. B. Amabilino, J. Veciana and P. Rey, Eur. J. Org. Chem., 2005, 348 CrossRef.
- T. Makarova and F. Palacio, Carbon Based Magnetism, Elsevier B. V., 2006 CrossRef CAS; M. Tamura, Y. Nakazawa, D. Shiomi and M. Kinoshita, Chem. Phys. Lett., 1991, 186, 401 CrossRef CAS.
- Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, Wiley, Chichester, 2010 Search PubMed.
- S. Nishida, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Angew. Chem., 2005, 117, 7443 ( Angew. Chem., Int. Ed. , 2005 , 44 , 7277 ) CrossRef CAS; K. Sato, S. Nakazawa, R. Rahimi, T. Ise, S. Nishida, T. Yoshino, N. Mori and T. Takui, J. Mater. Chem., 2009, 19, 3739 RSC; Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197 CrossRef PubMed.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737 CrossRef CAS PubMed.
- T. Sugawara, H. Komatsu and K. Suzuki, Chem. Soc. Rev., 2011, 40, 3105 RSC.
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281 CrossRef CAS PubMed; A. Iwasaki, L. Hu, R. Suizu, K. Nomura, H. Yoshikawa, K. Awaga, Y. Noda, K. Kanai, Y. Ouchi, K. Seki and H. Ito, Angew. Chem., 2009, 121, 4082 CrossRef.
- E. Coronado and A. J. Epstein, J. Mater. Chem., 2009, 19, 1670 RSC; E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419 CrossRef CAS PubMed.
- G. Zoppellaro, A. Geies and M. Baumgarten, Eur. J. Org. Chem., 2008, 1431 CrossRef CAS; Y. Wang, L. Wang and L. Ma, J. Mol. Struct., 2008, 877, 138 CrossRef PubMed; H. Yamaguchi, S. Nagata, M. Tad, K. Iwase, T. Ono, S. Nishihara, Y. Hosokoshi, T. Shimokawa, H. Nojiri, A. Matsuo, K. Kindo and T. Kawakami, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 125120 CrossRef.
- E. Coronado, C. Gimenez-Saiz and C. J. Gomez-Garcıa, J. Mater. Chem., 2008, 18, 929 RSC; T. Yoshida and S. Kaizaki, Inorg. Chem., 1999, 38, 1054 CrossRef CAS PubMed; K. Yamaguchi, M. Okumura, J. Maki, T. Noro, H. Namimoto, M. Nakano, T. Fueno and K. Nakasuji, Chem. Phys. Lett., 1992, 190, 353 CrossRef.
- P. Siemsen and R. C. Livingston, Angew. Chem., Int. Ed., 2000, 39, 2632 CrossRef CAS.
- T. W. Greene, Protective groups in organic synthesis, 1991 Search PubMed.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Chem. Commun., 1980, 627 CAS.
- E. V. Tretyakov, S. Tolstikov and A. Mareev, Eur. J. Org. Chem., 2009, 78, 2548 CrossRef CAS; E. V. Tretyakov and V. I. Ovcharenko, Russ. Chem. Rev., 2009, 78, 971 CrossRef PubMed.
- G. R. Luckhurst and A. Hudson, Mol. Phys., 1967, 13, 409 CrossRef.
- L. Catala, J. L. Moigne, N. Gruber and P. Turek, Chem.–Eur. J., 2005, 11, 2440 CrossRef CAS PubMed; O. N. Chupakhin, I. A. Utepova, M. V. Varaksin and V. I. Ovcharenko, J. Org. Chem., 2009, 74, 2870 CrossRef PubMed; S. V. Klyatskaya, E. V. Tretyakov and S. F. Vasilevsky, Russ. Chem. Bull., Int. Ed., 2002, 51, 128 CrossRef.
- I. A. Grigor'ev, M. M. Mitasov, G. I. Shichikin and L. B. Volodarskii, Seriya Khimicheskaya, 1979, 11, 2606 Search PubMed; L. Rintoul, A. S. Micallef and S. E. Bottle, Spectrochim. Acta, Part A, 2008, 70, 713 CrossRef CAS PubMed.
- E. Mostovich, PhD thesis, Novosibirsk, 2011.
- T. Yoshida and S. Kaizaki, Inorg. Chem., 1999, 38, 1054 CrossRef CAS PubMed.
- A. Caneschi, D. Gatteschi and P. Rey, Prog. Inorg. Chem., 1991, 39, 331 CrossRef.
- M. Shoji, K. Koizumi, Y. Kitagawa, T. Kawakami, S. Yamanaka, M. Okumura and K. Yamaguchi, Chem. Phys. Lett., 2006, 432, 343 CrossRef CAS PubMed; T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223 CrossRef; K. Yamaguchi, F. Jensen, A. Dorigo and K. N. Houk, Chem. Phys. Lett., 1988, 149, 537 CrossRef.
- O. Kahn, Molecular Magnetism, VCH, Weinheim-New York, 1993 Search PubMed.
- B. Bleaney and D. K. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
- F. M. Romero, R. Ziessel, M. Bonnet, Y. Pontillon, E. Ressouche, J. Schweizer, B. Delley, A. Grand and C. Paulsen, J. Am. Chem. Soc., 2000, 122, 1298 CrossRef CAS.
- C. Rancurel, N. Daro, O. Benedi Borobia, E. Herdtweck and J.-P. Sutter, Eur. J. Org. Chem., 2003, 406, 167 CrossRef.
- K. Remović- Langer, E. Haussühl, L. Wiehl, B. Wolf, F. Sauli, N. Hasselmann, P. Kopietz and M. Lang, J. Phys.: Condens. Matter, 2009, 21, 185013 CrossRef PubMed.
- B. C. Watson, V. N. Kotov, M. W. Meise, D. W. Hall, G. E. Granroth, W. T. Montfrooij, S. E. Nagler, D. A. Jensen, R. Backov, M. A. Petruska, G. E. Fanucci and D. R. Talham, Phys. Rev. Lett., 2001, 86, 5168 CrossRef CAS.
- Ch. Wang, M. Kilitziraki, J. A. H. MacBride and I. D. W. Samuel, Adv. Mater., 2000, 12, 217 CrossRef CAS.
- G. Wulff, B. Heide and G. Helfmeier, J. Am. Chem. Soc., 1986, 108, 1089 CrossRef CAS.
- H. Lexy and T. Kauffmann, Chem. Ber., 1980, 113, 2749 CrossRef CAS.
- K. Maruyama and S. Kawabata, Bull. Chem. Soc. Jpn., 1989, 62, 3498 CrossRef CAS.
- A. Fürstner and Ch. Mathes, Org. Lett., 2001, 221 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–12 UV-Vis spectra of the nitronyl nitroxides 1–4c, IR spectra of dialdehyde 4a, imidazolidine 4b, biradicals 2c, 3c and 4c, EPR spectra of the biradical 3c, magnetic susceptibility data of compounds 1–3c, TOC graphic of 4c, respectively. CCDC 822066, 816633, 816635. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4tc00399c |
| ‡ Present address: Institute of Biochemistry, Ernst-Moritz-Arndt University Greifswald, Felix-Hausdorff-Straße 4, 17487 Greifswald, Germany. |
| This journal is © The Royal Society of Chemistry 2014 |