
Using common salt to impart pseudocapacitive functionalities to carbon nanofibers†
Richa
Singhal
and
Vibha
Kalra
*
Department of Chemical and Biological Engineering, Drexel University, 3141 Chestnut Street, Philadelphia, PA 19104, USA. E-mail: vk99@drexel.edu
First published on 20th October 2014
Abstract
A novel and simple method of incorporating pseudocapacitive surface functionalities on free-standing carbon nanofibers using common salt (sodium chloride) is presented. The blend of sodium chloride (NaCl) and polyacrylonitrile is electrospun together, followed by pyrolysis and mild acid treatment to obtain functionalized free-standing (binder-free) carbon nanofibers. The synthesized materials have a low surface area of only 24 m2 g−1, however the electrochemical studies show a five-fold increase in specific capacitance on incorporation of NaCl compared to that without NaCl. The XPS characterization demonstrates that the presence of NaCl leads to enhanced oxygen on the surface of carbon nanofibers, particularly in the form of carboxyl groups. These carboxyl groups then facilitate the adsorption of sulfur functional groups on acid treatment. A high specific capacitance of 204 F g−1, areal capacitance of 1.15 F cm−2, and volumetric capacitance of 63 F cm−3 in 1 M H2SO4 are obtained, which are attributed to the surface functional groups participating in the pseudocapacitive redox reactions. The fabricated nanofibers demonstrate good capacitance retention at high current densities and high cyclability.
Introduction
Supercapacitors, or ultracapacitors, with higher power density and longer cycle life than batteries, are an important class of energy storage devices, with growing range of applications including portable electronics, hybrid electric vehicles, and industrial power management.1–3 Supercapacitors store energy either using ion adsorption at the electrode/electrolyte interface (double layer capacitors) or using fast faradaic redox reactions between the electrolyte and the electrode (pseudocapacitors).1,4,5 The pseudocapacitors have gained attention due to higher achievable energy densities than double layer capacitors.6,7 The most studied pseudocapacitive materials are transition metal oxides such as RuO2, MnO2, etc.3,8,9 and conducting polymers such as polyaniline and polypyrrole.9,10 However, these materials present some important drawbacks such as they are expensive, operate in a low voltage window, cause decomposition of the electrolyte, and have poor stability.11–13 An alternative approach is the surface modification of carbon materials to include functional groups, which can participate in pseudo-faradaic charge transfer reactions and maintain the high cyclability of supercapacitors.14–16In the last decade, a number of studies have shown the high performance of heteroatom (nitrogen, oxygen, sulfur, etc.)-enriched carbons for supercapacitors. A recent study by Zhou et al.15 reported nitrogen and oxygen containing carbon nanotubes synthesized by KOH activation of polypyrrole nanotubes. These materials with a high surface area of 705.9 m2 g−1 demonstrated a high capacitance of 384.9 F g−1 at 0.5 A g−1 current density, attributed to both the high surface area and the interfacial functional groups. Chen et al.16 synthesized nitrogen-doped porous carbon nanofibers by carbonization of polypyrrole-coated carbon nanofibers. The composite nanofibers exhibited a surface area of 562.51 m2 g−1 and a specific capacitance of 202 F g−1 at a current density of 1 A g−1. However, their approach of incorporating nitrogen functional groups involves treatment of carbon nanofibers with polypyrrole by ultrasonicating carbon nanofibers with pyrrole, thereby losing the free-standing capability of nanofibers. Zhang et al.12 reported that the incorporation of nitrogen and sulfur into the carbon framework enhanced the capacitance as well as improved the conductivity of the ordered mesoporous carbons. Most studies on heteroatom-enriched materials employ porous carbons with a high surface area as supercapacitor electrodes and thus, the double layer capacitance contribution is significant and the contribution of pseudocapacitance from heteroatoms is not clear. In addition, for the incorporation of heteroatoms, these techniques utilize either activation processes which produce carbons with poor mechanical strength and lower electrical conductivity, costly templating methods, or complex treatment processes.14,17 Moreover, most of these techniques fabricate powder-based carbons, which need to be blended with insulating binders (such as PTFE, PVDF) to form electrodes. The incorporation of binders reduces the conductivity of the electrode and the effective available surface area in addition to adding dead weight to the device.18
In lieu of the issues presented above, we report a novel and simple technique for fabricating free-standing (binder-free) heteroatom-enriched carbon nanofibers with high supercapacitance using common salt (i.e. sodium chloride (NaCl)). To the best of our knowledge, this is the first time NaCl has been reported to enhance capacitance by modifying carbon surface functionalities, while still keeping intact the free-standing matrix of carbon nanofibers. Electrospinning is a simple and versatile technique for fabricating free-standing nanofiber mats that can be applied to polymers loaded with nanoparticles or active agents.19–24 This technique has been extensively used in the past to fabricate polyacrylonitrile (carbon precursor) nanofibers, which are then pyrolyzed in an inert environment to form carbon nanofibers (CNFs).18,21,25–27 Carbon nanofibers are of significant importance because of their properties such as high strength to weight ratios, excellent chemical resistance, and superior electrical and thermal conductivity.28,29 In this work, we demonstrated that blending NaCl with polyacrylonitrile in the electrospinning solution significantly enhances oxygen functional groups on the surface of resultant carbon nanofibers. In addition to providing pseudocapacitive properties, the oxygen functional groups also facilitated the adsorption of sulfur-functional groups during mild acid treatment (post carbonization) resulting in a five-fold improvement in specific capacitance (when compared to pure carbonized PAN nanofibers). The BET results indicated a low specific surface area (24 m2 g−1) in carbonized and treated PAN–NaCl nanofibers, thus allowing us to better understand the effect of surface functional groups on supercapacitor performance. A high specific gravimetric capacitance of 204 F g−1, areal capacitance of 1.15 F cm−2, and volumetric capacitance of 63 F cm−3 in 1 M H2SO4 are obtained. The electrodes demonstrated good capacitance retention at high current densities (57% at 20 A g−1) and high cyclability.
Results and discussion
Polyacrylonitrile–sodium chloride (PAN–NaCl) blend nanofibers with varying concentrations of NaCl (0–13.3 wt%) were electrospun. The nanofibers are denoted as CNF-X, where X is the weight percent of NaCl (rounded to the nearest integer) with respect to the total solid weight (PAN + NaCl) in the original electrospinning solution. Fig. 1 shows the scanning electron microscopy (SEM) images of CNF-13 at various stages i.e. as-made (after electrospinning), after stabilization, after carbonization, and after treatment. A smooth nanofibrous structure with very few beads was obtained on electrospinning, which was retained after the thermal and acid treatment processes. The carbonized CNF-13 (Fig. 1c) shows the appearance of nanocrystals (possibly NaCl) on the surface of nanofibers, which were removed on acid treatment. The SEM images of carbonized CNF-0, CNF-4, and CNF-7 are shown in Fig. S1 (ESI†). A similar morphology to that of CNF-13 was obtained except that the nanocrystal concentration on the surface of carbonized carbon nanofibers reduced with decreasing NaCl concentrations. The EDS elemental mapping of carbonized CNF-13 nanofibers showed a uniform distribution of sodium and chlorine on the surface of nanofibers (Fig. S2 in the ESI†). | ||
| Fig. 1 SEM images of (a) as-made, (b) stabilized, (c) carbonized, and (d) treated CNF-13 nanofibers. Scale bars are 500 nm. | ||
The cyclic voltammograms of treated CNF-0, CNF-4, CNF-7, and CNF-13 samples in a two-electrode cell at 5 mV s−1 scan rate are shown in Fig. 2. The specific capacitance increased with increasing NaCl concentrations and is the highest for CNF-13 nanofibers (193 F g−1). The specific capacitances of CNF-0, CNF-4, and CNF-7 were determined as 35.94, 70.2, and 111.6 F g−1, respectively. Thus, the specific capacitance is enhanced five-fold on adding 13.3 wt% NaCl to PAN (CNF-13 vs. CNF-0). It was observed that on further increasing the NaCl concentration beyond 13.3 wt%, no significant improvement in the specific capacitance of the prepared nanofibers was obtained because of settling of NaCl in the electrospinning solution. Hence, CNF-13 nanofibers were chosen for further study and are discussed below.
 | ||
| Fig. 2 Cyclic voltammograms of the treated carbon nanofibers in 1 M H2SO4 using a two-electrode test cell at a scan rate of 5 mV s−1. | ||
The specific surface area (SSA) and pore volume determined by BET analysis for CNF-13 and CNF-0 (control) nanofibers are listed in Table 1. The nitrogen adsorption–desorption isotherms of the carbonized and treated CNF-13 are shown in Fig. S2 (ESI†). The SSA slightly improves from 14 m2 g−1 for CNF-0 to 21 m2 g−1 for CNF-13 nanofibers. On treatment, the SSA is 24 m2 g−1. Since there is no significant improvement in the surface area due to NaCl, it shows that NaCl behaves differently from the previously reported zinc chloride30 and tin chloride.31 Both of them served as activating agents to create high surface area porous carbons. Based on the BET data, we can also conclude that the five-fold improvement in the capacitance of CNF-13 (vs. CNF-0) is not a result of the double layer capacitance. To better understand the effect of NaCl on capacitance, detailed chemical analysis was conducted as discussed below.
| Sample | BET SSA, m2 g−1 | Total pore volume, cm3 g−1 |
|---|---|---|
| Carbonized CNF-13 | 21 | 0.017 |
| Treated CNF-13 | 24 | 0.017 |
| Carbonized CNF-0 | 14 | 0.011 |
X-ray diffraction (XRD) was first used to identify the nanocrystals formed on the surface of carbonized nanofibers. Fig. 3 compares the XRD spectra of as-made, stabilized, carbonized, and treated CNF-13 samples. The XRD spectrum of as-made CNF-13 nanofibers shows a broad diffraction peak at 17°, which is commonly observed in the fiber diffraction patterns of PAN with hexagonal packing.32,33 On heat-treatment, the diffraction peak at 17° disappeared with the evolution of a broad peak at 2θ of ∼25° attributed to the (002) crystallographic plane of graphite crystallites. The broad peak is characteristic of the amorphous nature of carbon ascribed to the turbostratic structure, which are not as ordered as graphite.32 In addition to the carbon peak, XRD patterns display the major NaCl peaks at 2θ of 31.62°, 45.44°, 56.72°, 66.28°, and 75.52° corresponding to (200), (220), (222), (400), and (420) miller indices. These peaks disappear in the treated CNF-13 suggesting the removal of NaCl nanocrystals on acid treatment, as also seen via SEM (Fig. 1c and 1d). Based on the XRD and SEM data, it appears that NaCl does not undergo any chemical reaction during the stabilization and carbonization processes.
 | ||
| Fig. 3 XRD patterns of electrospun CNF-13 nanofibers at different stages of fabrication. | ||
X-ray photoelectron spectroscopy (XPS) was used to study the surface functional groups. In order to understand the role of NaCl and the effect of acid treatment on the fabricated nanofibers, the surface functional groups of four samples – carbonized CNF-13, treated CNF-13, carbonized CNF-0, and treated CNF-0 were compared. The surface elemental compositions and oxygen-to-carbon (O/C) and nitrogen-to-carbon (N/C) ratios from XPS data analysis of carbonized and treated CNF-13 and CNF-0 (as control) samples are summarized in Table 2. The elemental analysis shows over two times higher O/C content on the surface of carbonized CNF-13 (19.8%), compared to that of the control sample, carbonized CNF-0 (7.8%). To understand the origin of additional oxygen, we conducted XPS of CNF-13 and CNF-0 nanofibers after the air stabilization step, a process step prior to carbonization, where samples were exposed to oxygen. We found that both samples exhibited similar fractions of surface oxygen (14–17%). Given that the carbonization step occurs in an inert environment and there is no other source of oxygen, it is interesting to see significant enhancement of oxygen functional groups in the carbonized CNF-13 compared to carbonized CNF-0. We hypothesize that the following mechanism takes place during carbonization of CNF-13 nanofibers – sodium chloride reacts with the hydrogen radicals formed during the dehydrogenation reactions during carbonization to form the sodium metal as follows:
| NaCl + H* → Na + HCl | (1) |
| Carbonized CNF-0 (%) | Treated CNF-0 (%) | Carbonized CNF-13 (%) | Treated CNF-13 (%) | |
|---|---|---|---|---|
| O | 6.22 | 7.07 | 13.51 | 18.63 |
| C | 80.08 | 78.59 | 68.3 | 64.3 |
| N | 13.69 | 13.49 | 9.51 | 10.65 |
| Na | — | — | 4.93 | 0.31 |
| Cl | — | — | 3.75 | — |
| S | — | 0.84 | — | 6.11 |
| O/C | 7.8 | 9.0 | 19.8 | 29.0 |
| N/C | 17.1 | 17.2 | 13.9 | 16.6 |
The conversion is though not complete since we still detect sodium chloride after carbonization via XRD and XPS. The HCl gas diffuses into the nitrogen gas flowing through the furnace. The sodium atoms, thus, etch the carbon surface leaving reactive carbon sites. A portion of that sodium can reconvert into sodium chloride, since chlorine species is present in the gas. In addition, it is also expected that at ∼800 °C, sodium chloride melts and blocks the carbon surface including the remaining functionalized sites from the stabilization step (during stabilization, oxidation and cyclization reactions take place to form a ladder PAN structure34). Thus, complete carbonization of PAN is inhibited leaving the functional groups intact on carbon nanofibers that provide the pseudocapacitance.
On acidic treatment, the oxygen content is further enhanced from 13.51% to 18.63% for CNF-13 nanofibers due to surface oxidation by sulfuric acid. Almost all sodium and chlorine are removed from the surface of carbonized CNF-13 and 6.11% of sulfur is added on acidic treatment. Note the samples were thoroughly washed with DI water prior to any characterization, so we do not expect any residual H2SO4. The sulfur addition is, thus, due to the adsorption of sulfur functional groups on the surface of nanofibers. The XPS of treated CNF-0 nanofibers show only 0.84% of sulfur on the surface and no significant enhancement of surface oxygen was seen. This indicates that the surface of carbonized CNF-13 allowed for improved adsorption of oxygen and sulfur groups (compared to CNF-0). We further analyzed the surface species by deconvolution of various elemental peaks.
The deconvoluted C1s, O1s, and N1s peaks for carbonized and treated CNF-13 are shown in Fig. 4 and that of CNF-0 are shown in Fig. S3 (ESI†). The contribution from each species obtained after peak fitting is listed in Table 3. The deconvolution of C1s spectra yields four peaks: C-1 (284.6 eV) due to graphitic carbon (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) and/or –C–C–; C-2 (285.5–285.7 eV) due to carbon in CN, C–O (phenol, hydroxyl, and ether groups); C-3 (287.6–287.8 eV) due to CO bonds (carbonyl and quinone groups), and C-4 (288.4–289 eV) due to carbon in carboxyl or ester groups.35 On comparing the C1s peak distribution in Table 3, we note that while the carbonized CNF-0 sample shows no C-4 peak (carboxyl/ester), the carbonized CNF-13 sample exhibits a prominent C-4 peak showing the presence of a significant fraction (22%) of carboxyl and/or ester groups on its surface. This C-4 peak disappears after acid treatment as seen in the treated CNF-13 C1s peak distribution data. Fig. 5 shows a schematic showing the oxygen and nitrogen functional groups. The nitrogen containing functionalities are assigned to pyridinic (N-6, 398.1 ± 0.2 eV); pyrrolic/pyridone (N-5, 399.9 ± 0.3 eV), quaternary (N-Q, 400.9 ± 0.17 eV), and N-oxide (N-X, 402–404 eV) groups.36,37 N-6 and N-5 are the dominant peaks in both carbonized and treated CNF-0. Appearance of the N-X (N-oxide) peak is seen due to acid treatment in treated CNF-0. The N1s spectrum of CNF-13 shows all four peaks. N-6 is the dominant peak in carbonized CNF-13, however, on treatment, the contribution from N-6 peak reduces and N-5 peak increases, possibly due to the increase in pyridone nitrogen (N-5(1) in Fig. 5).36 The deconvolution of the O1s peak in carbonized and treated CNF-0 gives two peaks – O-1 at 531.40 eV assigned to oxygen in quinone and/or carbonyl groups, and O-2 at 532.5 eV assigned to oxygen in phenol/ether groups.37 However, the carbonized CNF-13 shows two additional peaks, O-3 at 533.4 eV assigned to oxygen in carboxyl groups and O-4 at 536.4 eV, corresponding to adsorbed oxygen or water. The treated CNF-13 shows only O-1 and O-2 peaks, with O-2 being the dominant one. The absence of O-3 peak indicates the elimination of carboxyl groups on acidic treatment, as also seen from the disappearance of the C-4 peak in the C1s spectrum of treated CNF-13. These observations suggest a reaction between sulfuric acid and carboxyl groups that also results in the adsorption of sulfur groups on the surface of treated CNF-13.38 The sulfur S2p XPS spectrum for treated CNF-13 is shown in Fig. 6. The deconvolution of the S2p peak gives two doublet peaks at 168 eV and 169 eV assigned to the –SO3H and –OSO3H groups, respectively.39 The sodium (Na1s) peak for carbonized CNF-13 occurs at 1072.3 eV corresponding to the Na–Cl bond.
C–) and/or –C–C–; C-2 (285.5–285.7 eV) due to carbon in CN, C–O (phenol, hydroxyl, and ether groups); C-3 (287.6–287.8 eV) due to CO bonds (carbonyl and quinone groups), and C-4 (288.4–289 eV) due to carbon in carboxyl or ester groups.35 On comparing the C1s peak distribution in Table 3, we note that while the carbonized CNF-0 sample shows no C-4 peak (carboxyl/ester), the carbonized CNF-13 sample exhibits a prominent C-4 peak showing the presence of a significant fraction (22%) of carboxyl and/or ester groups on its surface. This C-4 peak disappears after acid treatment as seen in the treated CNF-13 C1s peak distribution data. Fig. 5 shows a schematic showing the oxygen and nitrogen functional groups. The nitrogen containing functionalities are assigned to pyridinic (N-6, 398.1 ± 0.2 eV); pyrrolic/pyridone (N-5, 399.9 ± 0.3 eV), quaternary (N-Q, 400.9 ± 0.17 eV), and N-oxide (N-X, 402–404 eV) groups.36,37 N-6 and N-5 are the dominant peaks in both carbonized and treated CNF-0. Appearance of the N-X (N-oxide) peak is seen due to acid treatment in treated CNF-0. The N1s spectrum of CNF-13 shows all four peaks. N-6 is the dominant peak in carbonized CNF-13, however, on treatment, the contribution from N-6 peak reduces and N-5 peak increases, possibly due to the increase in pyridone nitrogen (N-5(1) in Fig. 5).36 The deconvolution of the O1s peak in carbonized and treated CNF-0 gives two peaks – O-1 at 531.40 eV assigned to oxygen in quinone and/or carbonyl groups, and O-2 at 532.5 eV assigned to oxygen in phenol/ether groups.37 However, the carbonized CNF-13 shows two additional peaks, O-3 at 533.4 eV assigned to oxygen in carboxyl groups and O-4 at 536.4 eV, corresponding to adsorbed oxygen or water. The treated CNF-13 shows only O-1 and O-2 peaks, with O-2 being the dominant one. The absence of O-3 peak indicates the elimination of carboxyl groups on acidic treatment, as also seen from the disappearance of the C-4 peak in the C1s spectrum of treated CNF-13. These observations suggest a reaction between sulfuric acid and carboxyl groups that also results in the adsorption of sulfur groups on the surface of treated CNF-13.38 The sulfur S2p XPS spectrum for treated CNF-13 is shown in Fig. 6. The deconvolution of the S2p peak gives two doublet peaks at 168 eV and 169 eV assigned to the –SO3H and –OSO3H groups, respectively.39 The sodium (Na1s) peak for carbonized CNF-13 occurs at 1072.3 eV corresponding to the Na–Cl bond.
 | ||
| Fig. 4 Deconvoluted C1s, N1s, and O1s XPS peaks for carbonized and treated CNF-13 samples. | ||
Sample![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
C1s peaks | N1s peaks | O1s peaks | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C-1a | C-2b | C-3c | C-4d | N-6e | N-5f | N-Qg | N-Xh | O-1i | O-2j | O-3k | O-4l | |
| a 284.6 eV. b 285.6 ± 0.12 eV. c 287.7 ± 0.1 eV. d 288.9 eV. e 398.1 ± 0.2 eV. f 399.9 ± 0.3 eV. g 400.9 ± 0.17 eV. h 402.5 ± 1.64 eV. i 531.4 ± 0.1 eV. j 532.3 ± 0.2 eV. k 533.4 eV. l 536.4 eV. | ||||||||||||
| Carbonized CNF-0 | 39.3 | 39.54 | 21.16 | — | 41.49 | 58.51 | — | — | 56.77 | 43.23 | — | — |
| Treated CNF-0 | 39.96 | 38.77 | 21.27 | — | 34.66 | 50.39 | — | 14.95 | 40.4 | 59.6 | — | — |
| Carbonized CNF-13 | 29.99 | 47.99 | — | 22.01 | 46.03 | 14.74 | 15.91 | 23.33 | — | 35.4 | 39.66 | 24.94 |
| Treated CNF-13 | 39.46 | 38.01 | 22.53 | — | 31.5 | 26.32 | 19.59 | 22.59 | 38.63 | 61.37 | — | — |
 | ||
| Fig. 5 Schematic diagram of the nitrogen and oxygen functionalities. | ||
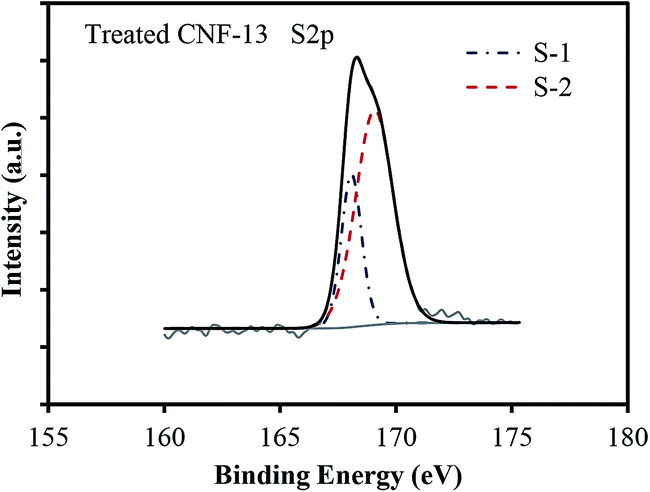 | ||
| Fig. 6 XPS S2p peak distribution for the treated CNF-13 sample. | ||
The results of XPS can be summarized as: (1) the oxygen on the surface of carbonized CNF-13 is almost double that of CNF-0. Thus, the presence of NaCl facilitated increased oxygen content on the surface of carbonized CNF-13. (2) The presence of NaCl modified the surface functional groups as is indicated by the presence of carboxyl groups and adsorbed oxygen, and the absence of carbonyl groups in carbonized CNF-13 (vs. carbonized CNF-0). (3) The sulfuric acid treatment significantly enhanced the oxygen content as well as incorporated sulfur by the adsorption of sulfonic and sulfate groups in CNF-13, but not in CNF-0. (4) The total nitrogen content on the surface of CNF-0 and CNF-13 is largely similar. The N1s spectrum of CNF-13 shows two additional peaks, N-Q and N-X, along with N-6 and N-5, which were not observed for carbonized CNF-0. It has been reported that while N-6 and N-5 nitrogen atoms mainly contribute to the pseudo-capacitive effect, N-Q and N-X groups improve the electron transfer through the carbon framework thereby providing better capacitance retention.17,40 Thus, we expect that the additional oxygen and sulfur functional groups are responsible for enhanced capacitance of CNF-13 nanofibers, while nitrogen functional groups improve the charge transfer leading to better capacitance retention.
The specific capacitance was determined from both two-electrode (practical performance measurement) and three-electrode cells (for studying faradaic reactions) and are compared in Fig. S4 (ESI†). The three-electrode specific capacitances were slightly higher than two-electrode capacitances at low scan rates (<50 mV s−1) but similar at high scan rates. The electrode loading was kept in the range from 8–11 mg cm−2 in accordance with the best practices for measuring capacitor performance reported by Ruoff and coworkers.41Fig. 7 shows the cyclic voltammetry (CV) curves for different scan rates varying from 5 mV s−1 to 500 mV s−1 for treated CNF-13 nanofibers in a three-electrode test cell. The CV curves show a near-rectangular shape even at high scan rates indicating good capacitive behavior. We do not see any redox peaks despite huge pseudocapacitive contributions indicating that charging and discharging occur at a pseudo-constant rate over the entire voltammetry cycles.42,43 Similar behavior has previously been reported for low-surface area, nitrogen-enriched carbons with high supercapacitance.14,17,44 A high specific capacitance of 193 F g−1 was obtained at a scan rate of 5 mV s−1 in a 1 V voltage window. As expected, the capacitance reduced with increasing scan rates due to mass transfer limitations at high scan rates. Nevertheless, we retain 66% of capacitance at 100 mV s−1 scan rate and 38.3% at 500 mV s−1 indicating high power handling capability due to the open pore structure (inter-fiber spacing) in nanofiber mats resulting in faster electrolyte/ion diffusion. The galvanostatic charge–discharge profiles at different current densities are shown in Fig. 8. A high specific capacitance of 204 F g−1 was obtained at 0.5 A g−1. We retain 57% of this capacitance (116 F g−1) even at a current density of 20 A g−1.
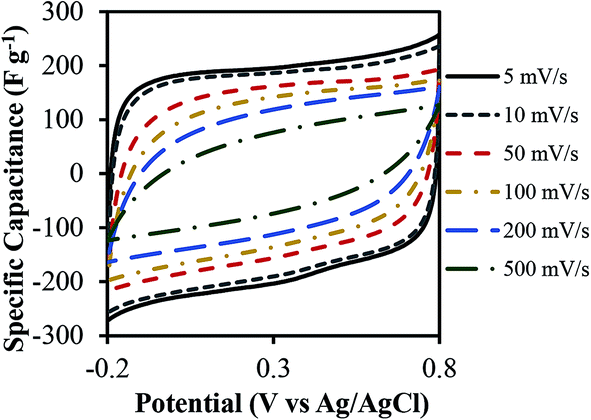 | ||
| Fig. 7 Cyclic voltammograms of the treated CNF-13 nanofibers in 1 M H2SO4 using a three-electrode test cell at different scan rates. | ||
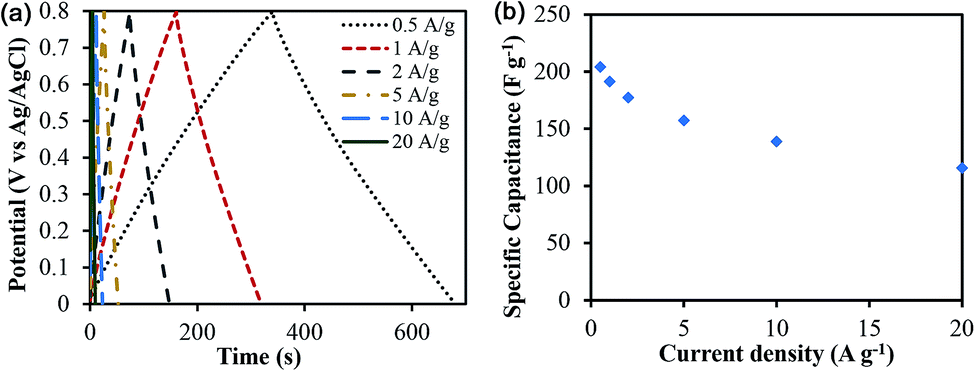 | ||
| Fig. 8 (a) Galvanostatic charge–discharge profiles of treated CNF-13 at different current densities, (b) specific capacitance as a function of current densities in 1 M H2SO4 electrolyte in a three-electrode test cell. | ||
As discussed above, the five-fold capacitance enhancement in CNF-13 (vs. CNF-0) is attributed to the additional oxygen and sulfur functional groups. The following continuous reversible oxidation/reduction reactions between hydroxyl, carbonyl and carboxyl groups are expected to occur in acidic medium:4
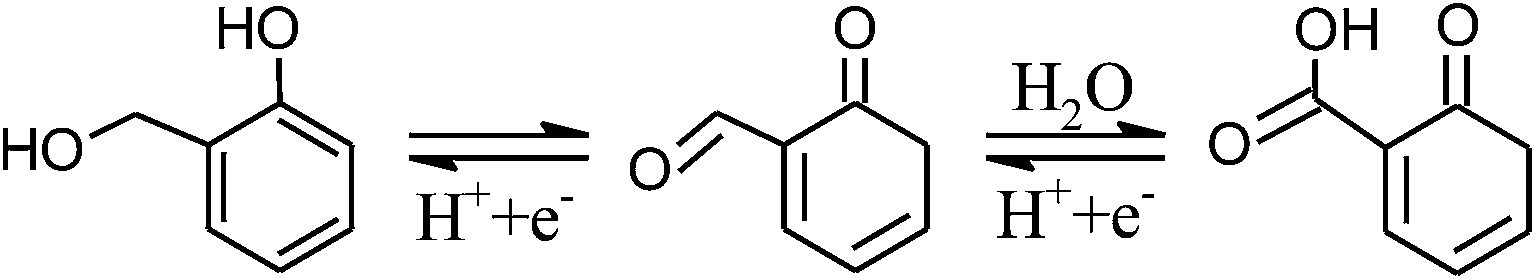 | (2) |
Thus, the transition between hydroxyl and carboxyl groups involves two electrons, thereby contributing to twice the charge storage. Kim et al.39 have shown that sulfuric acid treatment on PAN based hard carbons increases the reversible capacity by upto ∼20%. The sulfur functionalities contribute to pseudocapacitance by the redox reactions involving sulfone groups45 such as:
 | (3) |
 | (4) |
To further elucidate the high performance of the fabricated carbon nanofibers, the capacitance values reported in the literature for functionalized carbons have been compared with the present work (Table 4). It is to be noted that most studies had employed high surface area carbons (>200 m2 g−1); thus the contribution from the electric double layer capacitance is expected to be significant, and the contribution from the pseudocapacitive effect is not evident. Despite a low surface area of only 24 m2 g−1, the capacitance of our PAN–NaCl based carbon nanofibers is two-fold higher than that reported for carbon nanotube/PAN blend-based carbon nanofibers with 200 m2 g−1 surface area.46 Chen et al.16 recently reported the fabrication and electrochemical characterization of nitrogen-doped porous carbon nanofibers in supercapacitors. They followed a multistep synthesis procedure that involved coating of pre-formed carbon nanofibers with polypyrrole followed by re-carbonization to obtain N-doped porous carbon nanofibers with a surface area of >550 m2 g−1. They reported a capacitance of 202 F g−1 at 1 A g−1. The capacitance of the material fabricated in the present work is comparable despite the low surface area and with the additional advantages of a simpler fabrication procedure, inexpensive materials and potentially higher mechanical strength due to the low pore volume/surface area. Comparing with low surface area functionalized carbons reported in the literature, nitrogen-enriched carbons derived from melamine–mica composites14 with a surface area of 28 m2 g−1 have almost half the capacitance (115 F g−1) at an order of magnitude lower current density compared to our material.
| Carbon type | Surface area, m2 g−1 | Electrolyte | Scan rate/current density | Capacitance, F g−1 |
|---|---|---|---|---|
| (1) Carbonized carbon nanotube/PAN blend46 | 130 | 1 M H2SO4 | 5 mV s−1 | 100 |
| (2) Oxygen enriched seaweed biopolymer13 | 270 | 1 M H2SO4 | 2 mV s−1 | 198 |
| (3) Nitrogen-enriched carbon from silk fibroins47 | 2600 | 1 M TEABF4/PC | 1 mV s−1 | 52 |
| (4) Nitrogen-enriched carbons derived from melamine–mica composites14 | 28 | 1 M H2SO4 | 0.05 A g−1 | 115 |
| (5) Nitrogen- and oxygen-containing microporous activated carbon17 | 829 | 1 M H2SO4 | 0.05 A g−1 | ∼200 |
| (6) Oxygen-containing electrochemically modified graphite electrode4 | 6 | 2.3 M H2SO4 | 50 mV s−1 | 179.7 |
| (7) Nitrogen-doped carbon nanotubes48 | 200 | 1 M H2SO4 | 5 mV s−1 | 190.8 |
| (8) Nitrogen-doped carbon monolith36 | 679 | 6 M KOH | 1 mV s−1 | 246 |
| (9) Nitrogen-doped porous carbon nanofibers16 | 562.51 | 6 M KOH | 1 A g−1 | 202 |
| (10) Sulfur-doped activated carbons45 | 3010 | 0.5 M H2SO4 | 1 mV s−1 | 200 |
| (11) Nitrogen- and oxygen-containing activated carbon nanotubes15 | 706 | 1 M H2SO4 | 0.5 A g−1 | 384.9 |
| (12) Nitrogen-doped activated porous carbons49 | 1463 | 1 M H2SO4 | 0.1 A g−1 | 363 |
| Present work | 24 | 1 M H2SO4 | 5 mV s−1 | 193 |
| 0.5 A g−1 | 204 |
Fig. 9 compares the Nyquist plots of treated CNF-13 and CNF-0 nanofibers. At high frequencies (Fig. 9, inset), while the CNF-0 nanofibers do not show any semi-circle, CNF-13 shows a clear semi-circle associated with the charge transfer process arising from pseudocapacitance. The semicircular part in the high frequency region represents the electron-transfer-limiting process with its effective diameter equal to the faradaic charge transfer resistance.50 Furthermore, in the low frequency region, the straight line corresponding to the diffusion limiting process is less inclined for CNF-13 suggesting more capacitive behavior compared to CNF-0 nanofibers.
 | ||
| Fig. 9 Nyquist impedance plots of CNF-0 and CNF-13 measured in a three-electrode test cell in 1 M H2SO4 electrolyte. | ||
The cyclability is an important parameter for the practical application of supercapacitors. The cycle life of the treated CNF-13 nanofibers was examined at the scan rate of 100 mV s−1 for 1000 charge–discharge cycles in a three-electrode test cell (Fig. 10). The specific capacitance was 130.5 F g−1 after 1000 cycles. Thus, there was no change in specific capacitance even after 1000 cycles. This shows the good stability and high cyclability of the fabricated materials.
 | ||
| Fig. 10 Cycling performance of treated CNF-13 at a scan rate of 100 mV s−1 in 1 M H2SO4 in a three electrode test cell. | ||
Achieving high gravimetric capacitance is not sufficient for the practical application of nanomaterials as electrodes for supercapacitors. Good areal (per unit geometric area) and volumetric (per unit volume) capacitance is also necessary to compete with existing technology. Thus, the volumetric and areal capacitance was also determined for our materials in a two-electrode test cell. The areal capacitance of 1.15 F cm−2 was obtained. The volumetric capacitance of 52.33 F cm−3 was obtained for 240 μm thick treated CNF-13 electrodes, which is similar to that of a ∼300 μm electrode made from porous carbon powders (∼50 F cm−3).51,52 Since, the carbon nanofiber mats are compressible, the volumetric capacitance can be further increased. Hence, the PAN–NaCl electrospun fibers were compressed in a hydraulic press by applying a pressure of 1500 lbs at 60 °C for 1 min, followed by the regular stabilization, carbonization, and acid treatment processes. A higher volumetric capacitance of 63.03 F cm−3 for the 198 μm electrode for compressed CNF-13 nanofibers was obtained.
Experimental
Materials
Polyacrylonitrile (PAN, MW = 150000, Sigma-Aldrich, USA) and N,N-dimethylformamide (DMF, Sigma-Aldrich, USA) were used without any purification. Sodium chloride (Sigma-Aldrich, USA) was ground into fine powder and dried overnight under vacuum to remove any absorbed water.
Synthesis of carbon nanofibers
PAN (10 wt%) and NaCl (4, 6.7 & 13.3 wt%) were added to DMF and stirred at room temperature for 4–5 hours. Nanofibers were electrospun at room temperature with a relative humidity below 20% using a 20 gauge stainless steel needle (Hamilton Co.). The distance between the needle tip and the grounded collector (aluminum foil) was 15 cm, and the applied voltage of 13–14 kV was used to obtain a stable Taylor cone. The electrospinning was carried out in batches of 2 mL solution to minimize settling of sodium chloride in the syringe while electrospinning. The electrospun nanofibers were stabilized in air by heating to 280 °C at a rate of 5 °C min−1 for 5 hours. The stabilized nanofibers were then pyrolyzed (carbonized) under a nitrogen flow of 400 mL min−1 in a horizontal tube furnace at a heating rate of 2 °C min−1 to the temperature of 800 °C and held for 1 h. The carbonized NaCl–PAN nanofibers were then treated with 1 M H2SO4 solution and then thoroughly washed with distilled water to remove excess sulfuric acid.Characterization
The morphology and microstructure of the samples were characterized using a Zeiss Supra 50VP scanning electron microscope. A Quadrasorb (Quantachrome Instruments, USA) was used to conduct gas sorption measurements. Samples were tested at 77 K (liquid nitrogen baths were used as coolant) isotherms, using the nitrogen gas as the adsorbate. The Brunauer–Emmett–Teller (BET) formula was used to calculate specific surface areas in the P/Po range between 0.05 and 0.10. For XRD analysis, the samples were characterized using a Rigaku SmartLab X-ray diffractometer in Bragg–Bentano mode (Cu Kα radiation, voltage 40 kV, current 30 mA, scanning range 10–80° and step size 0.02°). XPS measurements were performed on a Physical Electronics VersaProbe 5000 spectrometer using a monochromated Al Kα excitation source. The binding energy scales were calibrated with respect to the C1s peak position (284.6 eV). The quantitative analysis was performed using CASAXPS software after Shirley background subtraction. The mixed 30% Gaussian–Lorentzian line shapes were used for peak fitting.Electrochemical measurements
The carbon nanofiber mats were directly used as binder-free electrodes for electrochemical measurements. The capacitive performances of the samples were investigated by using cyclic voltammetry (CV), galvanostatic charge–discharge, and electrochemical impedance spectroscopy (EIS) techniques, which were performed using a Gamry Reference 3000 potentiostat. For two-electrode analysis, a symmetric cell was assembled by placing two 3–4 mg each, 3/8′′ diameter discs of the fabricated carbon nanofibers separated by a Whatman glass fiber separator, sandwiched between two graphite current collectors one on each side in a swagelok cell. The carbon nanofiber electrodes were immersed in the aqueous electrolyte, 1 M H2SO4 overnight before measurements to ensure complete wetting of carbon electrodes. For three electrode analysis, an aqueous Ag/AgCl electrode was used as the reference electrode in the two-electrode test-cell. Thus, in a three-electrode cell, the potential difference is measured with reference to Ag/AgCl. The schematic of the two-electrode and three-electrode setups is shown in Fig. S5 of the ESI.†The specific capacitance per unit mass of one electrode in a two-electrode setup was calculated according to the equation: C = ∫2IΔt/(mΔV), where I is the response current (A), Δt is the discharge time (s), m is the average mass of the carbon nanofiber electrodes (g), and ΔV is the voltage window. For a three-electrode capacitance measurement, the above equation was modified as: C = ∫IΔt/(mΔV).41 The galvanostatic charge–discharge tests were performed at current densities ranging from 0.5 to 20 A g−1. The specific capacitance per electrode from galvanostatic charge–discharge experiments was calculated from the discharge curves after three charge–discharge cycles. EIS measurements were carried out by applying a sine wave with an amplitude of 10 mV over the frequency range of 100 kHz to 0.1 Hz.
Conclusions
Heteroatom-enriched free-standing (binder-free) carbon nanofibers for supercapacitors have been successfully synthesized using a simple fabrication technique. The surface area of the fabricated materials was very low (<25 m2 g−1), which allowed us to better understand the effect of surface functional groups. A high specific capacitance of 204 F g−1, areal capacitance of 1.15 F cm−2 and volumetric capacitance of 63.03 F cm−3 were obtained in 1 M H2SO4, which are comparable to carbons with a high surface area (∼2000 m2 g−1). Such a high capacitance in the reported materials is attributed to the surface functional groups participating in pseudocapacitive redox reactions. Electrospun PAN–NaCl nanofibers on carbonization showed higher oxygen content mainly in the form of carboxyl groups than those without NaCl. On mild acid treatment, these carboxyl groups react with sulfuric acid leading to adsorption of sulfate and sulfonate functional groups, which further add to the capacitance. Hence, NaCl played a major role in modifying the carbon surface to incorporate oxygen and sulfur functionalities. The fabricated fibers showed good capacitance retention at high current densities (57% at 20 A g−1). Moreover, no capacitance fade was observed after 1000 cycles showing good stability and high cyclability of the material. The method demonstrated in this work to generate pseudocapacitive surface functionalities can potentially be integrated with our previously published work on creating high surface area porous carbon nanofibers18 to generate heteroatom enriched-high surface area carbon nanofibers with further enhanced specific capacitance.Acknowledgements
This work is supported by the National Science Foundation under grant number CBET-1236466 and CBET-1150528. The authors would like to thank Boris Dyatkin and Prof. Yury Gogotsi for porosimetry measurements. The authors are grateful to Centralized Research Facility of Drexel University for instrumentation support.Notes and references
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed
.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC
- X. Fan, Y. Lu, H. Xu, X. Kong and J. Wang, J. Mater. Chem., 2011, 21, 18753–18760 RSC
- A. Pandolfo and A. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS PubMed
- Y. Hou, L. Chen, P. Liu, J. Kang, T. Fujita and C. Mingwei, J. Mater. Chem. A, 2014, 2, 10910–10916 CAS
- R. A. Fisher, M. R. Watt and W. Jud Ready, ECS J. Solid State Sci. Technol., 2013, 2, M3170–M3177 CrossRef CAS PubMed
- J. Zang, S.-J. Bao, C. M. Li, H. Bian, X. Cui, Q. Bao, C. Q. Sun, J. Guo and K. Lian, J. Phys. Chem. C, 2008, 112, 14843–14847 CAS
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213–234 CrossRef CAS PubMed
- T. Liu, L. Finn, M. Yu, H. Wang, T. Zhai, X. Lu, Y. Tong and Y. Li, Nano Lett., 2014, 14, 2522–2527 CrossRef CAS PubMed
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS
- D. Zhang, L. Zheng, Y. Ma, L. Lei, Q. Li, Y. Li, H. Luo, H. Feng and Y. Hao, ACS Appl. Mater. Interfaces, 2014, 6, 2657–2665 CAS
- E. Raymundo-Piñero, F. Leroux and F. Béguin, Adv. Mater., 2006, 18, 1877–1882 CrossRef
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS
- Z. Zhou, Z. Zhang, H. Peng, Y. Qin, G. Li and K. Chen, RSC Adv., 2014, 4, 5524–5530 RSC
- L.-F. Chen, X.-D. Zhang, H.-W. Liang, M. Kong, Q.-F. Guan, P. Chen, Z.-Y. Wu and S.-H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS
- C. Tran and V. Kalra, J. Power Sources, 2013, 235, 289–296 CrossRef CAS PubMed
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed
- T. Wang and S. Kumar, J. Appl. Polym. Sci., 2006, 102, 1023–1029 CrossRef CAS
- C. Tran and V. Kalra, Soft Matter, 2013, 9, 846–852 RSC
- V. Kalra, J. Lee, J. H. Lee, S. G. Lee, M. Marquez, U. Wiesner and Y. L. Joo, Small, 2008, 4, 2067–2073 CrossRef CAS PubMed
- A. Hu, C. Curran, C. Tran, A. Kapllani and V. Kalra, J. Nanosci. Nanotechnol., 2014, 14, 5501–5507 CrossRef CAS PubMed
- A. Kapllani, C. Tran and V. Kalra, Soft Matter, 2013, 9, 11014 RSC
- Z. Zhou, C. Lai, L. Zhang, Y. Qian, H. Hou, D. H. Reneker and H. Fong, Polymer, 2009, 50, 2999–3006 CrossRef CAS PubMed
- C. Tran and V. Kalra, ECS Trans., 2013, 53, 35–41 CrossRef CAS PubMed
- F. W. Richey, C. Tran, V. Kalra and Y. A. Elabd, J. Phys. Chem. C, 2014, 118, 21846 CAS
- L. Zhang, A. Aboagye, A. Kelkar, C. Lai and H. Fong, J. Mater. Sci., 2014, 49, 463–480 CrossRef CAS
- X. Mao, T. A. Hatton and G. C. Rutledge, Curr. Org. Chem., 2013, 17, 1390–1401 CrossRef CAS
- C. Kim, B. T. Ngoc, K. S. Yang, M. Kojima, Y. A. Kim, Y. J. Kim, M. Endo and S. C. Yang, Adv. Mater., 2007, 19, 2341–2346 CrossRef CAS
- G.-H. An and H.-J. Ahn, Carbon, 2013, 65, 87–96 CrossRef CAS PubMed
- Y. Yang, F. Simeon, T. A. Hatton and G. C. Rutledge, J. Appl. Polym. Sci., 2012, 124, 3861–3870 CrossRef CAS
- I. Karacan and G. Erdoğan, Fibers Polym., 2012, 13, 855–863 CrossRef CAS
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421–1432 CrossRef CAS PubMed
- J.-H. Zhou, Z.-J. Sui, J. Zhu, P. Li, D. Chen, Y.-C. Dai and W.-K. Yuan, Carbon, 2007, 45, 785–796 CrossRef CAS PubMed
- D.-W. Wang, F. Li, L.-C. Yin, X. Lu, Z.-G. Chen, I. R. Gentle, G. Q. Lu and H.-M. Cheng, Chem.–Eur. J., 2012, 18, 5345–5351 CrossRef CAS PubMed
- D. Hulicova-Jurcakova, E. Fiset, G. Q. M. Lu and T. J. Bandosz, ChemSusChem, 2012, 5, 2188–2199 CrossRef CAS PubMed
- C. M. Welch and H. A. Smith, J. Am. Chem. Soc., 1953, 75, 1412–1415 CrossRef CAS
- Y. J. Kim, H. J. Lee, S. W. Lee, B. W. Cho and C. R. Park, Carbon, 2005, 43, 163–169 CrossRef CAS PubMed
- D.-D. Zhou, W.-Y. Li, X.-L. Dong, Y.-G. Wang, C.-X. Wang and Y.-Y. Xia, J. Mater. Chem. A, 2013, 1, 8488–8496 CAS
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 CAS
- X. Lang, A. Hirata, T. Fujita and M. Chen, Nat. Nanotechnol., 2011, 6, 232–236 CrossRef CAS PubMed
- L. Yang, S. Cheng, Y. Ding, X. Zhu, Z. L. Wang and M. Liu, Nano Lett., 2011, 12, 321–325 CrossRef PubMed
- D. Hulicova, J. Yamashita, Y. Soneda, H. Hatori and M. Kodama, Chem. Mater., 2005, 17, 1241–1247 CrossRef CAS
- W. Gu, M. Sevilla, A. Magasinski, A. B. Fuertes and G. Yushin, Energy Environ. Sci., 2013, 6, 2465–2476 CAS
- F. Béguin, K. Szostak, G. Lota and E. Frackowiak, Adv. Mater., 2005, 17, 2380–2384 CrossRef
- Y. J. Kim, Y. Abe, T. Yanagiura, K. C. Park, M. Shimizu, T. Iwazaki, S. Nakagawa, M. Endo and M. S. Dresselhaus, Carbon, 2007, 45, 2116–2125 CrossRef CAS PubMed
- Y. S. Yun, H. H. Park and H.-J. Jin, Materials, 2012, 5, 1258–1266 CrossRef CAS PubMed
- M. Zhou, F. Pu, Z. Wang and S. Guan, Carbon, 2014, 68, 185–194 CrossRef CAS PubMed
- J. Yang and S. Gunasekaran, Carbon, 2013, 51, 36–44 CrossRef CAS PubMed
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480–483 CrossRef CAS PubMed
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P.-L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta05121a |
| This journal is © The Royal Society of Chemistry 2015 |