 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall-molecule azomethines: organic photovoltaics via Schiff base condensation chemistry†
M. L.
Petrus
ab,
R. K. M.
Bouwer
a,
U.
Lafont
a,
S.
Athanasopoulos
c,
N. C.
Greenham
c and
T. J.
Dingemans
*a
aDelft University of Technology, Faculty of Aerospace Engineering, Kluyverweg 1, 2629 HS Delft, The Netherlands. E-mail: t.j.dingemans@tudelft.nl
bDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX Eindhoven, The Netherlands
cUniversity of Cambridge, Optoelectronics Group, Cavendish Laboratory, J.J. Thomson Avenue, Cambridge, CB3 0HE, UK
First published on 8th May 2014
Abstract
Conjugated small-molecule azomethines for photovoltaic applications were prepared via Schiff base condensation chemistry. Bulk heterojunction (BHJ) devices exhibit efficiencies of 1.2% with MoOx as the hole-transporting layer. The versatility and simplicity of the chemistry is illustrated by preparing a photovoltaic device directly from the reaction mixture without any form of workup.
Most of the conjugated materials used in organic electronics are synthesized using time-consuming Suzuki-, Wittig-, or Heck-type coupling reactions. These chemistries often make use of expensive transition metal catalysts, require stringent reaction conditions, and extensive product purification.
In order to offer a more economic route towards organic photovoltaic materials we are currently exploring azomethines (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) as the conjugated linker unit. The azomethine bond is isoelectronic to the vinyl bond and possesses similar optoelectronic and thermal properties.1–4 Besides that, azomethines offer significant advantages over vinylenes as they can be prepared using Schiff base condensation chemistry under near ambient reaction conditions. Expensive catalysts are not required and water is the only by-product.5 The first photovoltaic devices based on poly(azomethine)s were published by Hindson et al. (PCE ∼ 0.12%)6 and Iwan et al. (PCE to ∼0.31%).7 The reported conjugated poly(azomethine)s are only moderately soluble and therefore difficult to process.8,9 Small-molecules, on the other hand, have the advantage of having a higher solubility, a well-defined molecular structure, and no batch-to-batch variations. Recent results have shown that conjugated small molecules can compete successfully with their polymeric counterparts.10,11
N–) as the conjugated linker unit. The azomethine bond is isoelectronic to the vinyl bond and possesses similar optoelectronic and thermal properties.1–4 Besides that, azomethines offer significant advantages over vinylenes as they can be prepared using Schiff base condensation chemistry under near ambient reaction conditions. Expensive catalysts are not required and water is the only by-product.5 The first photovoltaic devices based on poly(azomethine)s were published by Hindson et al. (PCE ∼ 0.12%)6 and Iwan et al. (PCE to ∼0.31%).7 The reported conjugated poly(azomethine)s are only moderately soluble and therefore difficult to process.8,9 Small-molecules, on the other hand, have the advantage of having a higher solubility, a well-defined molecular structure, and no batch-to-batch variations. Recent results have shown that conjugated small molecules can compete successfully with their polymeric counterparts.10,11
For this study we have synthesized two azomethine-based small molecules via a simple condensation reaction (Fig. 1).
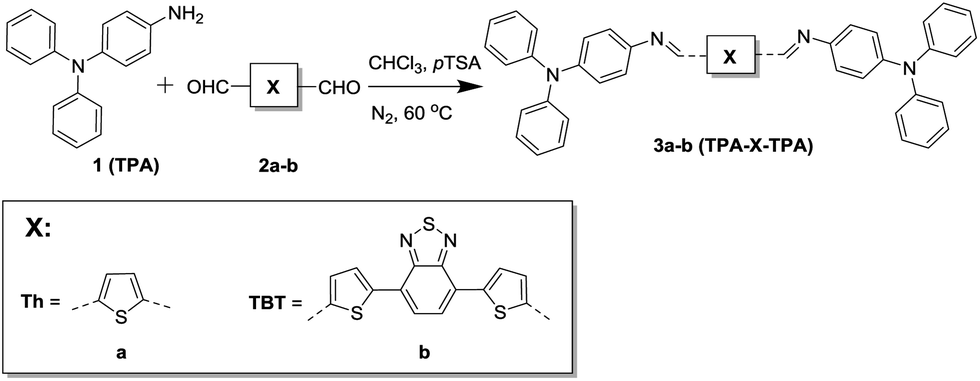 | ||
| Fig. 1 Synthesis and molecular structures of the small-molecule azomethines. | ||
4-Aminotriphenylamine (TPA, 1) was reacted with two conjugated dialdehydes. 2,5-Thiophenedicarbaldehyde (Th, 2a) was used since thiophenes have been studied extensively and possess good charge transport properties.12 Besides that, both materials are readily available from commercial sources. 4,7-Bis(5-formylthiophen-2-yl)-2,1,3-benzothiadiazole (TBT, 2b) was used because it is a strong electron-accepting moiety and this yields a bipolar donor–acceptor–donor molecule with a small bandgap and thus a larger overlap with the solar spectrum.13 In addition TBT is known to possess high mobilities and good film-forming abilities.14,15 The reaction was performed in chloroform and p-toluenesulfonic acid was used as a catalyst. The products (3a,b) were precipitated, and treated with diluted triethylamine to neutralize the protonated azomethine bond. The small molecules were obtained in good yields (>80%) and characterized using 1H and 13C NMR, FTIR, and mass spectrometry where possible. Detailed synthetic procedures and characterization of the small molecules and their intermediates are available in the ESI.†
The thermal properties of the new molecules were assessed using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Both small molecules show excellent thermal stabilities with degradation temperatures (Td5%) above 350 °C (ESI Fig. S1 and Table S1†). Comparing TPA-Th-TPA to its vinyl analogue, we found that the degradation temperature is approximately 40 °C higher,12 confirming the superior thermal stability of azomethines. DSC experiments showed that both TPA-Th-TPA and TPA-TBT-TPA exhibit a glass transition temperature (Tg) at 86 and 114 °C, respectively (ESI Fig. S2 and Table S1†). TPA-Th-TPA also showed a cold crystallization exotherm during the second heating at 143 °C. Both small-molecules melt above 200 °C and form isotropic melts, as was confirmed by hot-stage optical microscopy.
Absorption spectra of the small-molecules were recorded in solution (ESI Fig. S3†) and in the solid state (Fig. 2a) and the data are summarized in Table 1. The absorption peak around 310 nm can be ascribed to the triphenylamine moiety and is found for both TPA-X-TPA small-molecules. The push–pull character of TPA-TBT-TPA results in a large redshift (95 nm) of the absorption onset as compared to TPA-Th-TPA. In addition, TPA-TBT-TPA exhibits an extra absorption maximum at 400 nm, which was also observed in its vinyl analogue.15 The molecular extinction coefficients at the most red-shifted absorption maxima were calculated to be on the order of 104 M−1 cm−1. TPA-TBT-TPA displays the highest molar extinction coefficient (Table 1). The absorption spectra of the small-molecules in the solid state show no large shifts as compared to the solution spectra (Table 1). The bandgap of the small-molecules could be estimated from the onset of the absorption spectra. Comparing the absorption maxima with those of their vinyl analogues, a redshift for TPA-Th-TPA was observed (21 nm), while TPA-TBT-TPA shows a 22 nm blue shift.12,16 Halochromatic behaviour was observed when the azomethine solutions were acidified and is described in the ESI (Fig. S4†). Photoluminescence spectra were measured both in the solid state and in solution (ESI Fig. S5†). However, the photoluminescence is very weak, which indicates fast non-radiative decay channels.8,17
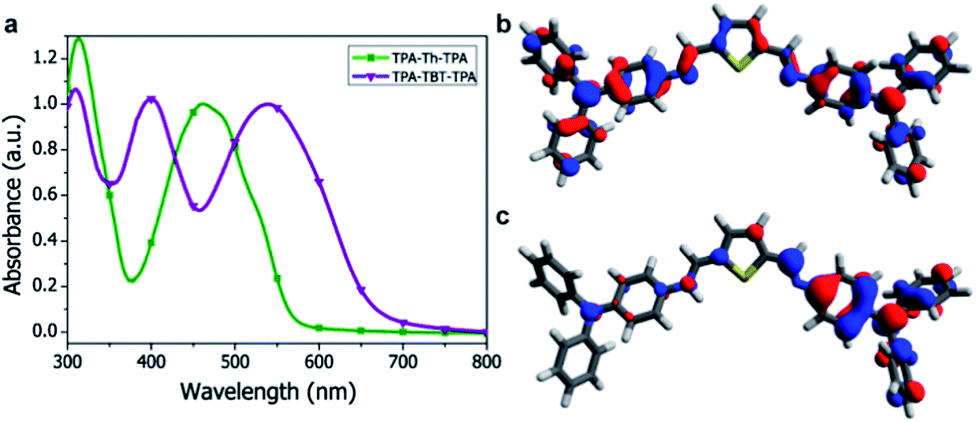 | ||
| Fig. 2 UV-Vis absorption spectrum and computational study of the small-molecules. (a) Normalized UV-vis absorption spectra of TPA-X-TPA small molecules in the solid state. (b) and (c) DFT-calculated (CAM-B3LYP/6-31G*) Kohn-Sham HOMO level for the TPA-Th-TPA molecule: (b) Shows the small-molecule in the neutral state where the HOMO is delocalized over the entire molecule. (c) Protonation of the azomethine nitrogen-atom interrupts conjugation and enforces localization of the HOMO to the non-protonated part of the molecule. | ||
| Compound | UV-vis solutiona | UV-vis filmb | ||||||
|---|---|---|---|---|---|---|---|---|
| λ max [nm] | ε [104 M−1 cm−1] | λ max [nm] | λ onset [nm] | E g [eV] | E oxi.onset [V] | HOMOd [eV] | LUMOe [eV] | |
| a Measured in dichloromethane. b Spincasted from chloroform. c Estimated from the onset of the absorption spectrum of the film, 1240/λonset. d Determined by cyclic voltammetry, with Fc/Fc+ at 4.8 eV below vacuum. e Estimated by subtracting the bandgaps from the HOMO energy levels. | ||||||||
| TPA-Th-TPA | 306, 467 | 3.6 | 314, 463 | 565 | 2.19 | 0.85 | −5.32 | −3.13 |
| TPA-TBT-TPA | 306, 389, 520 | 5.6 | 310, 400, 538 | 660 | 1.88 | 0.79 | −5.26 | −3.36 |
The electrochemical properties of the small-molecules in solution were investigated using cyclic voltammetry. Contrary to the azomethines reported by Gąsiorowski et al.,18 we found that oxidation of our azomethines is reversible (ESI Fig. S7†). However, when applying a higher potential (>1.2 V) the small-molecules are oxidized to their 2+ form, and this second oxidation appears to be irreversible. It is well-known from literature that oxidation of triphenylamines results in the formation of a cationic radical, which can dimerise via the para-position.19,20 The highest occupied molecular orbital (HOMO) energy levels of the small-molecules were determined by measuring the oxidation onset of the first oxidation and were found to be around −5.3 eV below vacuum (ESI Fig. S8†). The electron-donating triphenylamine moiety of the molecule is dominating the contributions to the HOMO energy level, explaining the similar HOMO energy levels (Table 1). The relatively deep HOMO energy levels are expected to result in a fairly high Voc and good oxidative stability. The lowest unoccupied molecular orbital (LUMO) energy levels were estimated by combining the optical bandgap with the obtained HOMO energy level. The LUMO energy level, which is dominated by contributions from the electron accepting part of the molecules, was significantly lower for TPA-TBT-TPA as compared to TPA-Th-TPA due to the strong acceptor unit (Table 1 and ESI Fig. S8†). The azomethine moiety slightly deepens the HOMO energy level compared to the vinyl and fully aromatic analogues, which are about 0.1–0.3 eV higher.12,15 This is attributed to the electron-withdrawing nature of the azomethine functionality.
PEDOT:PSS, which is most commonly used as the anode layer in OPVs, is acidic and could potentially protonate the azomethine functionality of our small-molecule azomethines.21 Density functional theory (DFT) calculations were carried out to study the effect of protonation of the azomethine linker and are described in the ESI.†22 The calculations indicate that protonation breaks the symmetry and enforces localization of the HOMO, which results in a lowering of the HOMO energy by approximately ∼2 eV (Fig. 2 and ESI S9–S12†) while the LUMO level shows a downward shift in energy by more than 3 eV. This suggests that upon protonation of the azomethine linker at the PEDOT:PSS interface, energy barrier states for holes and well states for electrons are formed. This can be detrimental for hole extraction and can also lead to enhanced recombination.
As protonation could limit the device performance, we compared the photovoltaic device characteristics of a conventional device (ITO/PEDOT:PSS/TPA-TBT-TPA:[60]PCBM/LiF/Al) with a device where the acidic PEDOT:PSS was replaced by neutral molybdenum oxide (MoOx) (ESI Table S3†). Replacing PEDOT:PSS with MoOx increases the efficiency from 0.35% to 0.42%, which could be attributed to an increase in voltage from 0.45 V to 0.68 V (ESI Table S3†). Also the lower work function of MoOx (between 5.3 and 5.7 eV while PEDOT:PSS is approximately 5.0 eV) could explain the higher voltage as a result of better energy alignment and higher driving force.23 Annealing at 70 °C further improved the device performance (ESI Table S4†) up to 0.65%; annealing at a temperature of 100 °C or higher destroyed the devices. The use of [70]PCBM resulted in an additional increase in efficiency up to 1.15% (ESI Tables S5, S6 and Fig. S14†), which is ascribed to the absorption of [70]PCBM, resulting in higher currents. The influence of the hole-blocking layer, annealing temperature and time, and type of fullerene were investigated for TPA-TBT-TPA and the optimal conditions were also used to prepare the TPA-Th-TPA devices (Table 2, Fig. 3a).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)/LiF/Al configuration after annealing at 70 °C
:2)/LiF/Al configuration after annealing at 70 °C
| Compound | J sc [mA cm−2] | V oc [V] | FF [%] | PCE [%] |
|---|---|---|---|---|
| a J sc was calculated by integrating the EQE spectrum with the AM1.5G spectrum under bias. b After annealing for 30 minutes. c After annealing for 5 minutes. | ||||
| TPA-Th-TPA | 3.73 | 0.83 | 39 | 1.21 |
| TPA-TBT-TPA | 4.50 | 0.75 | 34 | 1.15 |
| TPA-Th-TPA (one-pot)b | 2.60 | 0.75 | 31 | 0.60 |
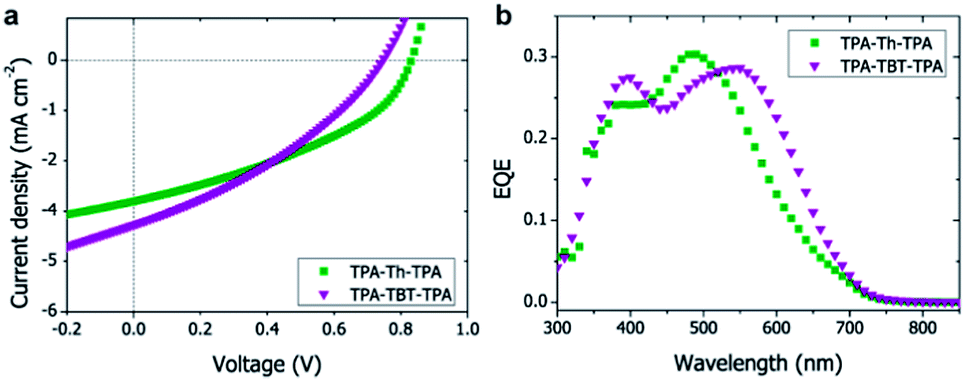 | ||
| Fig. 3 Device performance of small-molecule BHJ solar cells. (a) J–V characteristics and (b) EQE spectra of small-molecule bulk heterojunction solar cells based on TPA-X-TPA:[70]PCBM using MoOx as hole blocking layer after annealing. | ||
As expected from the absorption spectra, TPA-TBT-TPA, which has the smallest bandgap and thus the best overlap with the solar spectrum, shows a higher current. However, because of the slightly higher open-circuit voltage and fill factor (FF) TPA-Th-TPA shows the best power conversion efficiency (PCE). The high open-circuit voltages up to 0.83 V are in agreement with the fairly low-lying HOMO energy levels. All devices show a strong voltage dependence, i.e. significantly higher currents were obtained under reverse bias. This indicates that charge carriers are formed, but are not extracted under working conditions, which can be explained by a low charge carrier mobility and/or a poor morphology. The poor fill factors also hint towards unbalanced charge transport, which is likely attributed to the limited mobility of azomethines when processed under these conditions. The vinyl analogues of TPA-Th-TPA and TPA-TBT-TPA have been published by other groups and show PCEs of 0.34% and 0.26%, respectively after optimization,12,15,16 which is significantly lower than 1.21% and 1.15% found for the azomethine analogues published herein. Also a TPA-TBT-TPA analogue where the TPA moieties are directly connected to the TBT core without a vinyl or azomethine bond has been published and reached a PCE of 1.3% after extensive device optimization using [70]PCBM, which is only slightly higher compared to the azomethine we report.
The external quantum efficiency (EQE) spectra of the devices coincide with the peaks in the absorption spectra of the small-molecules (Fig. 3b), indicating that excitons created in the small-molecules are separated into charges at the interface with PCBM. Using [70]PCBM instead of [60]PCBM resulted in slight broadening of the absorption spectrum and an overall increase of the EQE to 30% (ESI Fig. S17†). The absorption maximum around 310 nm was not observed in the EQE spectrum, which is explained by the absorption of the ITO substrate and MoOx.24
The film morphology of the small-molecule:[70]PCBM active layers was investigated using transmission electron microscopy (TEM). Both active layers show an amorphous film with PCBM islands on the order of 2–3 nm (ESI Fig. S16†). These domains are relatively small; the use of a different solvent or a co-solvent might induce a stronger phase separation, which is expected to improve the device performance.
Condensation chemistry is very convenient for the synthesis of conjugated small-molecule azomethines. Besides the molecular components and a suitable solvent no other reagents or catalysts are required and water is the only side product. To demonstrate the simplicity and ease of this chemistry, we prepared a device by spincoating the reaction mixture without any form of workup or purification. For this, 2,5-thiophenedicarbaldehyde (2a, Th) and two equivalents of 4-aminotriphenylamine (1, TPA) were stirred overnight in dry chloroform at 60 °C. The next day the resulting deep orange solution was diluted to the required concentration and [70]PCBM was added prior to spincoating the active layer. The device showed a PCE of 0.60%, demonstrating that “one-pot” synthesized azomethines can be used as donor material in organic photovoltaic devices (Table 2 and ESI Fig. S15†). The lower efficiency might be explained by the presence of unreacted functional groups, which are known to act as charge traps, and/or protonation of the azomethine bond. The EQE spectrum of the “one-pot” synthesized TPA-Th-TPA device coincides with that of the devices prepared using the conventional method, indicating that in both cases charge transfer from TPA-Th-TPA was obtained (ESI Fig. S18†).
In conclusion, two conjugated small-molecule azomethines were synthesized via Schiff base condensation chemistry. The small-molecules both exhibit reversible oxidation behaviour, a fairly deep lying HOMO energy level, and a bandgap between 1.9 and 2.2 eV. Photovoltaic devices based on MoOx as the hole-transporting layer performed better when compared to PEDOT:PSS, which is mainly attributed to an increased voltage. Both small-molecules showed efficiencies around 1.2% with [70]PCBM as the electron acceptor. TPA-Th-TPA performed slightly better (PCE of 1.21% with a Jsc of 3.7 mA cm−2, Voc of 0.83 V, and FF of 39%) after post annealing. Both azomethine-based devices outperform their vinyl analogues, indicating that azomethines are promising candidates for organic photovoltaic applications. We also demonstrated that this chemistry enables the fabrication of OPV devices directly from the reaction mixture without any product workup. These devices showed a PCE of 0.6%. Because azomethine chemistry is easy, clean and proceeds under near ambient conditions we believe that this approach has the ability to reduce materials and production costs of organic photovoltaic devices.
Acknowledgements
This research forms part of the research program of the Dutch Polymer Institute (DPI), project #717. The work in Cambridge was supported by the Engineering and Physical Sciences Research Council (grant number EP/G060738/1). We acknowledge the EPSRC UK National Service for Computational Chemistry Software (NSCCS) for access to the Columbus Cluster. We acknowledge Prof. René Janssen, Dr Martijn Wienk, and Dr Dhritiman Gupta of the Eindhoven University of Technology for access to their device fabrication and characterization facilities. We also acknowledge Prof. Laurens Siebbeles for access to the photoluminescence equipment.Notes and references
- A. Bolduc, A. Al Ouahabi, C. Mallet and W. G. Skene, J. Org. Chem., 2013, 78, 9258 CrossRef CAS PubMed.
- P. W. Morgan, S. L. Kwolek and T. C. Pletcher, Macromolecules, 1987, 20, 729 CrossRef CAS.
- A. S. Perez Guarin, M. Bourgeaux, S. Dufresne and W. G. Skene, J. Org. Chem., 2007, 72, 2631 CrossRef PubMed.
- L. Sicard, D. Navarathne, T. Skalski and W. G. Skene, Adv. Funct. Mater., 2013, 23, 3549 CrossRef CAS.
- H. Schiff, Justus Liebigs Ann. Chem., 1864, 131, 118 CrossRef.
- J. C. Hindson, B. Ulgut, R. H. Friend, N. C. Greenham, B. Norder, A. Kotlewski and T. J. Dingemans, J. Mater. Chem., 2010, 20, 937 RSC.
- A. Iwan, M. Palewicz, A. Chuchmała, L. Gorecki, A. Sikora, B. Mazurek and G. Pasciak, Synth. Met., 2012, 162, 143 CrossRef CAS PubMed.
- M. L. Petrus, R. K. M. Bouwer, U. Lafont, D. H. K. Murthy, R. J. P. Kist, M. L. Böhm, Y. Olivier, T. J. Savenije, L. D. A. Siebbeles, N. C. Greenham and T. J. Dingemans, Polym. Chem., 2013, 4, 4182 RSC.
- H. Niu, H. Kang, J. Cai, C. Wang, X. Bai and W. Wang, Polym. Chem., 2011, 2, 2804 RSC.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44 CrossRef CAS PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020 CrossRef CAS PubMed.
- J. Kwon, W. Lee, J. Kim, S. Noh, C. Lee and J.-I. Hong, New J. Chem., 2010, 34, 744 RSC.
- H. Zhou, L. Yang, S. Stoneking and W. You, ACS Appl. Mater. Interfaces, 2010, 2, 1377 CAS.
- M. Zhang, Y. Sun, X. Guo, C. Cui, Y. He and Y. Li, Macromolecules, 2011, 44, 7625 CrossRef CAS.
- Q. He, C. He, Y. Sun, H. Wu, Y. Li and F. Bai, Thin Solid Films, 2008, 516, 5935 CrossRef CAS PubMed.
- C. He, Q. He, Y. He, Y. Li, F. Bai, C. Yang, Y. Ding, L. Wang and J. Ye, Sol. Energy Mater. Sol. Cells, 2006, 90, 1815 CrossRef CAS PubMed.
- M. Bourgeaux and W. G. Skene, Macromolecules, 2007, 40, 1792 CrossRef CAS.
- J. Gąsiorowski, E. D. Głowacki, B. Hajduk, M. Siwy, M. Chwastek-ogierman, J. Weszka, H. Neugebauer and N. S. Sariciftci, J. Phys. Org. Chem., 2013, 117, 2584 Search PubMed.
- E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 178, 3498 CrossRef.
- H.-J. Yen and G.-S. Liou, Org. Electron., 2010, 11, 299 CrossRef CAS PubMed.
- E. L. Ratcliff, R. C. Bakus II, G. C. Welch, T. S. van der Poll, A. Garcia, S. R. Cowan, B. a. MacLeod, D. S. Ginley, G. C. Bazan and D. C. Olson, J. Mater. Chem. C, 2013, 1, 6223 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010.
- A. Garcia, G. C. Welch, E. L. Ratcliff, D. S. Ginley, G. C. Bazan and D. C. Olson, Adv. Mater., 2012, 24, 5368 CrossRef CAS PubMed.
- Y. Sun, C. J. Takacs, S. R. Cowan, J. H. Seo, X. Gong, A. Roy and A. J. Heeger, Adv. Mater., 2011, 23, 2226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, materials and methods, computational study and figures and tables as referred to in the text. See DOI: 10.1039/c4ta01629g |
| This journal is © The Royal Society of Chemistry 2014 |