 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-inspired carbon nitride mesoporous spheres for artificial photosynthesis: photocatalytic cofactor regeneration for sustainable enzymatic synthesis†
Jianhui
Huang
b,
Markus
Antonietti
a and
Jian
Liu
*a
aDepartment of Colloid Chemistry, Max-Planck Institute of Colloids and Interfaces, Research Campus Golm, 14476 Potsdam, Germany. E-mail: Jian.Liu@mpikg.mpg.de
bCollege of Environmental and Biological Engineering, Putian University, Putian 351100, P.R.China
First published on 27th March 2014
Abstract
Inspired by the photosynthetic thylakoid membrane of chloroplasts, we present here the rational design of mesoporous structured colloids made up of graphitic carbon nitride nanosheets and demonstrate their application in photocatalytic NADH regeneration for sustainable enzymatic synthesis.
Introduction
High efficiency light energy-to-chemical energy conversion is thought to be the ultimate goal in the field of sustainable energy, considering the sustainable and inexhaustible character of sunlight.1 In natural photosynthesis, green plants and algae have evolved into highly delicate, complex structures to convert light to chemical energy. Learning from nature might offer some possible key elements for realizing artificial photosynthesis.2–6 Inside the photosynthetic factories termed chloroplasts, a membrane-bound thylakoid is the site of the light-dependent reactions of photosynthesis. Following light harvesting by chlorophyll molecules, photoinduced electrons are passed through the electron transport chain to the enzyme ferredoxin–NADP (nicotinamide adenine dinucleotide phosphate) reductase, leading to the reduction of NAD(P) to NAD(P)H, completing the storage of light energy in one kind of “energy currency” of the cells (another kind is ATP). The newly formed NAD(P)H is used as a hydrogen source with high reducing power for the biosynthesis of carbohydrates from CO2 in the Calvin cycle of the dark reaction.7 NAD(P)+/NAD(P)H cofactors are also involved in many other enzymatic redox reactions of the metabolism. Recently, applying such enzymatic redox reactions for the synthesis of valuable and chiral compounds in vitro became highly attractive.8–10 However, high costs of enzyme-specific cofactors (NADH) hinder wider applications, and sustainable and economically acceptable regeneration methods for NADH are still eagerly demanded.11As in the photosynthetic light reactions, using light energy for the reduction of NAD+ to NADH is very promising and inherently sustainable, compared with the established but not optimal enzymatic regeneration method and electrochemical routes.12–16 By taking electrons from a light excited photocatalyst and coupling the protons to form the hydride electron pair, the NAD+ can be selectively reduced to 1,4-NADH in the presence of [Cp*Rh(bpy)H2O]2+ (abbreviated as M in the following context) acting as a hydride transfer reagent while triethanolamine (denoted as TEOA) is employed as a sacrificial electron donor.17 Despite previously reported works on the photocatalytic NADH regeneration, however, more facile and more efficient visible-light-driven photocatalytic systems are critically needed and are to be developed.18–23
Inspired by the folded, layered thylakoid membranes featuring very large surface area, which therefore could increase the amount of light absorbance, construction of a structural mimicry of the thylakoid membranes as a semiconductor object is promising to increase the efficiency of NADH regeneration.24 Graphitic carbon nitride (abbreviated as g-C3N4) is known as a layered material and found in recent years to possess visible-light-driven photocatalytic properties.25–32 Our previous work demonstrated that a synthetic carbon nitride photosynthetic structure onto diatom frustules could be used for photocatalytic NADH regeneration.33 However, the low surface area partly counterbalanced the benefits of refractive light harvesting brought about by the diatom structure.34 Higher efficiency could be achieved by coupling a higher specific surface area and strong light harvesting properties.35 Carbon nitride will act as the ultimate light-to-charge pair conversion site while a structured mesoporous sphere architecture could enhance the light intensity onto the active centers.36–38 The in situ regenerated NADH is then coupled with L-lactate dehydrogenase for sustainable photoenzymatic synthesis. In addition, as outlined in our previous report, direct transfer of a hydride electron pair from carbon nitride to NAD+ is also possible, and metal- and mediator free regeneration of NADH as the ultimate goal will also be investigated.39
Experimental section
Materials synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was added to the above SiO2 solution. The mixture was heated up to 100 °C and kept for 3 hours under refluxing to load PVP, and then cooled to room temperature. Under magnetic stirring, 37.5 mL of 0.1 g mL−1 sodium hydroxide aqueous solution was added to the as-cooled solution at room temperature. As more material is etched away from the silica particles, the solution becomes less and less opaque. The etching process could be monitored by measuring transmission of the etching solution at 700 nm using UV-Vis spectrometry. A mixture of 1 g of etched SiO2 mesoporous sphere and 5 g of cyanamide was degassed for 3 hours followed by sonication in water for 2 hours. After removal of surplus cyanamide by dissolving in water, the obtained white solid was transferred to a crucible with lid and heated under air at 2.3 °C min−1 up to 550 °C (4 hours) and kept at 550 °C for another 4 hours. The resultant yellow powder is treated with a 4 M NH4HF2 solution for 48 hours. The dispersion is then filtered and the yellow precipitate is copiously rinsed with deionized water and ethanol. Finally, the yellow powder is dried under a vacuum at 60 °C overnight.
000) was added to the above SiO2 solution. The mixture was heated up to 100 °C and kept for 3 hours under refluxing to load PVP, and then cooled to room temperature. Under magnetic stirring, 37.5 mL of 0.1 g mL−1 sodium hydroxide aqueous solution was added to the as-cooled solution at room temperature. As more material is etched away from the silica particles, the solution becomes less and less opaque. The etching process could be monitored by measuring transmission of the etching solution at 700 nm using UV-Vis spectrometry. A mixture of 1 g of etched SiO2 mesoporous sphere and 5 g of cyanamide was degassed for 3 hours followed by sonication in water for 2 hours. After removal of surplus cyanamide by dissolving in water, the obtained white solid was transferred to a crucible with lid and heated under air at 2.3 °C min−1 up to 550 °C (4 hours) and kept at 550 °C for another 4 hours. The resultant yellow powder is treated with a 4 M NH4HF2 solution for 48 hours. The dispersion is then filtered and the yellow precipitate is copiously rinsed with deionized water and ethanol. Finally, the yellow powder is dried under a vacuum at 60 °C overnight.
Characterization
XRD measurements were performed on a D8 Diffractometer from Bruker instruments (Cu Kα radiation, λ = 0.154 nm) equipped with a scintillation counter. N2 sorption experiments were done with a Quantachrome Autosorb-1 at liquid nitrogen temperature. TEM images were taken on Philips CM200 FEG (Field Emission Gun), operated at an acceleration voltage of 200 kV. SEM measurement was performed on a LEO 1550 Gemini instrument. The UV-Vis absorbance spectra were recorded on a T70 UV/Vis spectrophotometer. The FTIR spectrum was collected using a Varian 1000 FTIR spectrometer.NADH regeneration and photoenzymatic reaction
900 M−1 cm−1. NADH has peak absorption at 340 nm with an extinction coefficient of 6220 M−1 cm−1.
Results and discussion
Carbon nitride mesoporous spheres (abbreviated as CNMS) were synthesized by templating etched silica spheres, adopting a strategy termed “silica-etching chemistry”, as illustrated in Scheme 1.40 Briefly, the SiO2 sphere was first loaded with polyvinylpyrrolidone (PVP) for protecting an outside SiO2 shell, and a sodium hydroxide solution mildly etches the dense silica sphere towards a mesoporous system. These etched mesoporous structures then provide confined and necessary space for loading the cyanamide precursor for the subsequent in situ thermal condensation towards carbon nitride. At last, after removing the silica template by an aqueous NH4HF2 solution, a CNMS preserving the original sphere size, but with inverted inner structure could be obtained.26 In general, the moderation and control of the silica-etching chemistry is the determining factor for the successful construction of CNMS. The degree of etching can be facilely controlled by the etching time; an appropriate etching end point can be quantitatively determined by an increase of the original optical transmission at 700 nm wavelength up to 70%. The morphology evolution of a SiO2 sphere during different etching times was investigated, as illustrated in Fig. S1 (ESI†). Different etching degrees of SiO2 lead to entirely different morphologies of the obtained carbon nitride materials. As it is templated from the original SiO2 sphere or SiO2 sphere with low etching degree, only fragments and no defined particles could be obtained.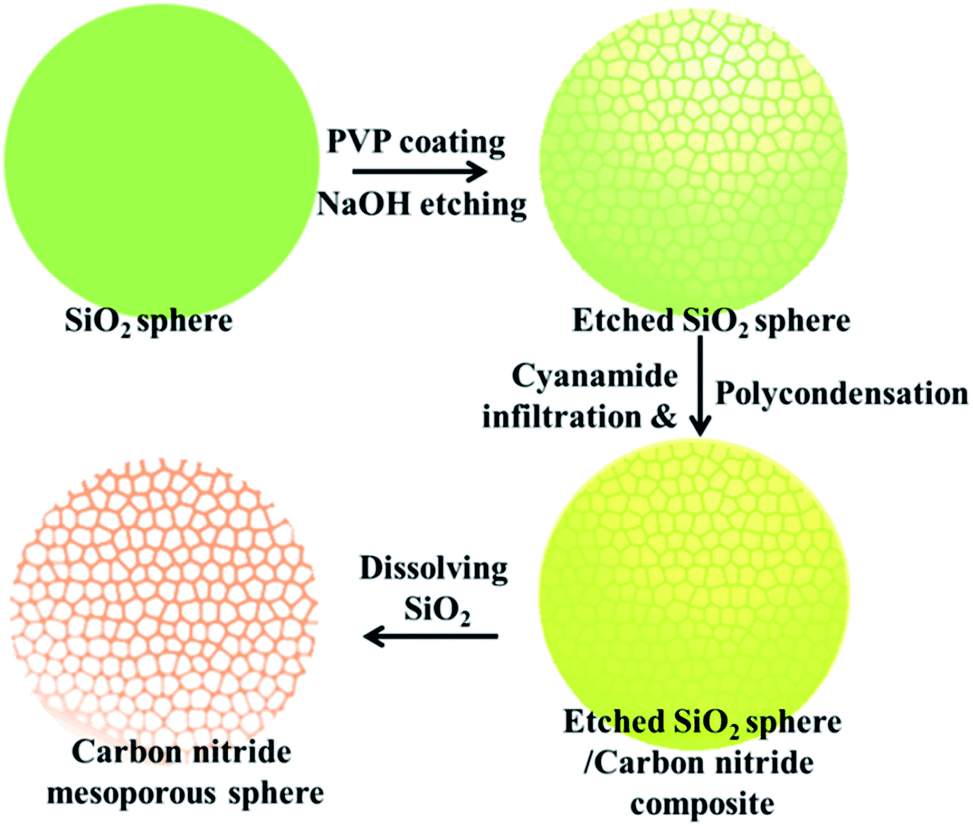 | ||
| Scheme 1 Schematic fabrication strategy for CNMS. The whole processes could be divided into four steps: (1) synthesis of a SiO2 sphere; (2) loading with PVP under refluxing conditions and mildly etching PVP coated SiO2 with NaOH to obtain mesoporous SiO2; (3) loading a cyanamide precursor into the porous space followed by in situ thermal condensation to carbon nitride; and (4) using an NH4HF2 aqueous solution to release CNMS. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images in low magnification for a large area overview of CNMS demonstrate the high homogeneity and uniformity of the obtained spherical material, as shown in Fig. 1a and b, and the local structure was evidenced by the TEM image shown in Fig. 1c. The CNMS was chosen to have a diameter around 420 nm to enable multiple internal light reflections and consequently higher light absorbance. The size matches with the peak wavelength of the blue LED light source which also is near the onset of optical absorbance of g-C3N4. The original SiO2 and etched SiO2 spheres are compared to those replicated structures in Fig. 1d & e, while the corresponding SEM images along with a large overview of CNMS are displayed in Fig. S2 of the ESI,† respectively. Throughout etching, the PVP protected SiO2 spheres do not decrease in diameter. The solid spheres turn mesoporous with the coarsened surface while still preserving the spherical morphology, indicating that the etching process is prohibited by the PVP protection of the shell. Compared with the sphere template, the size of obtained CNMS is a little smaller due to the shrinkage throughout thermal condensation. The typical material characterization results for CNMS are presented in Fig. S3 of the ESI.† The band at 810 cm−1 in the Fourier transform infrared spectroscopy (FTIR) spectra in Fig. S3a of the ESI† is ascribed to the s-triazine ring modes, while the bands at 1200–1600 cm−1 are characteristic of aromatic CN heterocycles. A powder X-ray diffraction (XRD) spectrum shows the two characteristic peaks of graphitic carbon nitride (Fig. S3b of the ESI†) at 13.0° and 27.4° which are ascribed to the in-plane repeat period and stacking of the conjugated aromatic system, respectively. N2 sorption measurement reveals that the CNMS is indeed mesoporous, with a specific surface area of 205 m2 g−1 (see Table 1, Fig. S3 and 4 of the ESI†).
 | ||
| Fig. 1 (a) SEM image of large area overview, illustrating the uniform and monodisperse properties of CNMS. (b) TEM image of CNMSs. (c) TEM image of a single CNMS particle, showing the porous property and nanosheet as the building blocks of CNMS. (d) and (e) show the TEM images of original solid SiO2 sphere and etched mesoporous SiO2 sphere, respectively. | ||
The (photo)electrochemical properties of CNMS accompanying two control samples (bulk g-C3N4 and mpg-C3N4) were investigated by electrochemical impedance spectroscopy (Fig. 2a). The results revealed semicircular Nyquist plots for CNMS, mpg-C3N4 and g-C3N4 with the trend of increasing diameter, which demonstrated that CNMS possessed an improved electronic conductivity already in the dark state as compared to the other morphologies. The photoinduced electron transfer processes were also investigated. The photocurrent measurements of the three materials cast on indium tin oxide glass were conducted in a 0.2 M Na2SO4 aqueous solution (Fig. 2b). Large enhancement of Iph for CNMS was indeed observed over an applied bias potential of 0.4 V versus Ag/AgCl, indicating strong light harvesting capability and faster transport of charged carriers.
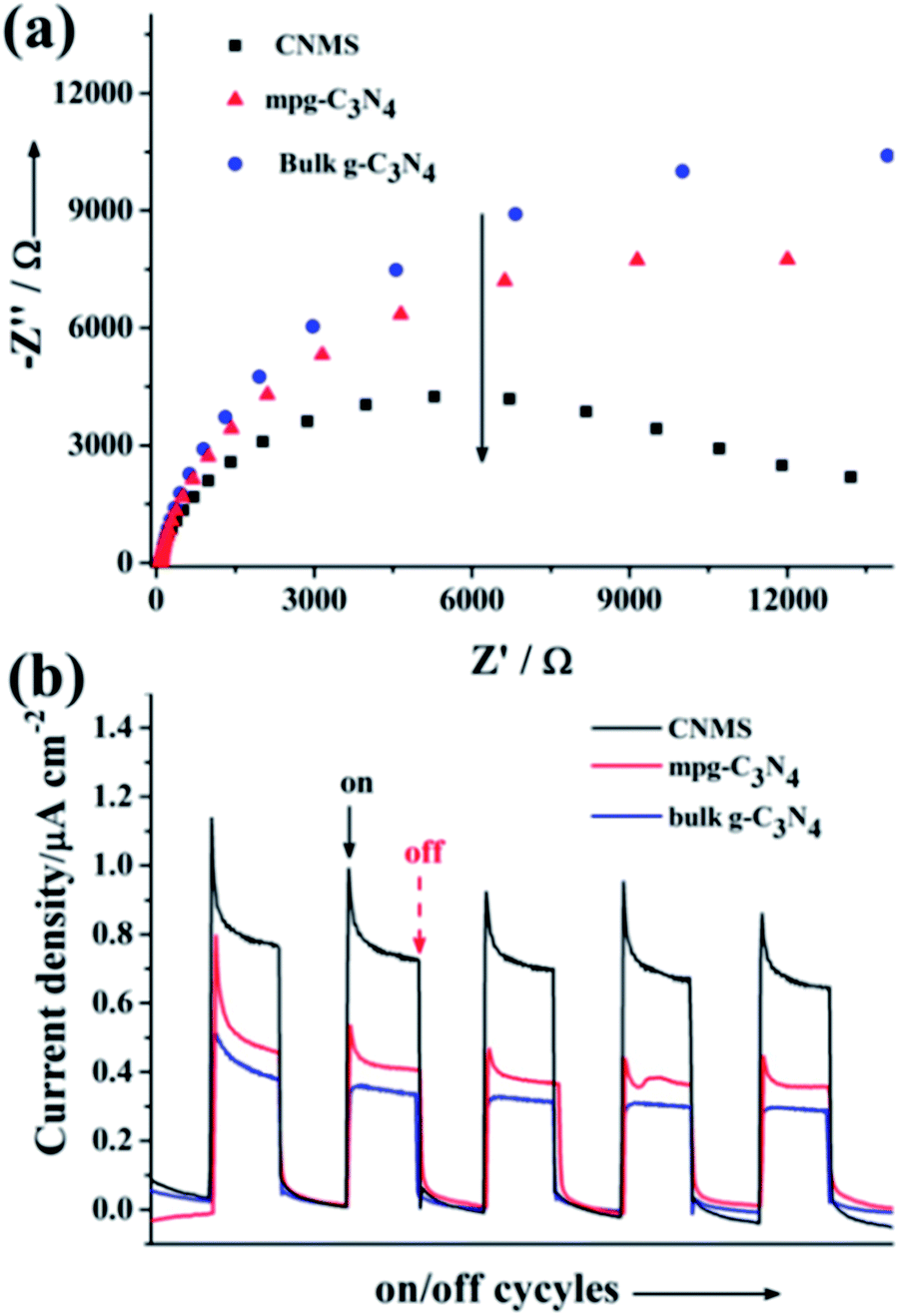 | ||
| Fig. 2 (a) Electrochemical impedance spectroscopy Nyquist plots for CNMS, mpg-C3N4 and g-C3N4, respectively. (b) The periodic on/off photocurrent output of CNMS, mpg-C3N4 and g-C3N4 cast on ITO glass at 0.4 V bias vs. Ag/AgCl in a 0.2 M Na2SO4 solution. | ||
Fig. 3a shows the diffuse reflectance spectra of the obtained materials, indicating typical semiconductor absorption behavior. The slight blue shift of CNMS in the optical absorption relative to bulk g-C3N4 might be ascribed to the smaller structural size of the nanosheet building block (see Fig. 1c and S5 of ESI†). The CNMS also exhibited a stronger absorption than two reference samples, especially in the 420–500 nm wavelength regions. A photoluminescence spectrum, coming from the recombination of free charge carriers, usually serves as a good technique for evaluating the processes of charge migration, transfer and separation. As illustrated in Fig. 3b, as compared to bulk g-C3N4, the substantially decreased fluorescence emission intensity of CNMS around 470 nm indicated the suppressed recombination rate of the photoinduced charge carriers, which is beneficial for further photocatalytic applications.26,41 The time-resolved photoluminescence spectrum was also employed to understand the photophysical behaviors of photoexcited charge carriers (Fig. S6 of the ESI†). The fluorescence intensities of all three samples decay exponentially. A lifetime of ∼6 ns was obtained for CNMS at room temperature (298 K), which gives an estimate of the time for charges to reach the surface stabilized sites and the related structural length scales of a few nanometers.
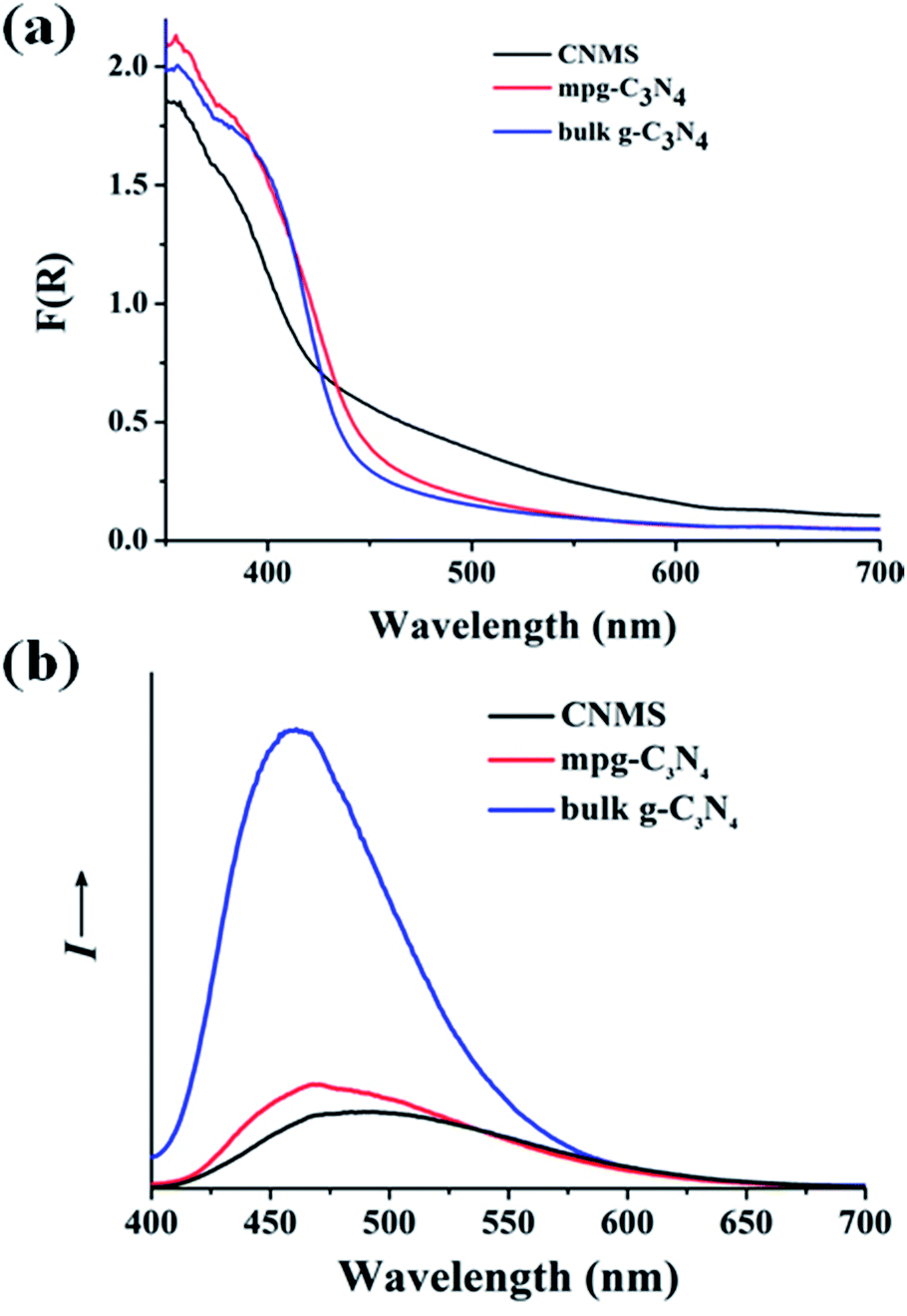 | ||
| Fig. 3 (a) Optical absorption of CNMS, mpg-C3N4 and g-C3N4 samples, respectively. (b) The photoluminescence spectra of CNMS, mpg-C3N4 and g-C3N4 samples. | ||
Based on the above photo- and electrochemical investigation of CNMS, the photocatalytic NADH regeneration was performed. Optimum reaction conditions were adopted from our previous work.33 A reaction solution with a pH value at 8 in a phosphate buffer solution with 15% TEOA was employed. M is a homogeneous redox catalyst acting as a hydride transfer reagent to shuffle hydride to the NAD+. An active carbon nitride content of 3 mg was maintained in all the experiments. The NADH regeneration was determined by measuring the increase of absorbance at 340 nm (with a molar extinction coefficient of 6220 M−1 cm−1) using the Lambert–Beer law, as illustrated in Fig. S7 of the ESI.†
The regeneration of NADH from NAD+ is summarized in the reaction equation in Scheme 2. The photocatalytic activity comparisons among CNMS, mpg-C3N4 and bulk g-C3N4 are presented in Fig. 4a. During the first illumination period, the conversion yields increase linearly. Under the same circumstances, typical well-known semiconductors such as TiO2 and ZnO were both inactive for NADH regeneration during the same illumination time. For comparison, no NADH regeneration took place in the dark. CNMS gives the best performance, and the NADH conversion yield reached 100% after only 30 min light illumination. mpg-C3N4 and g-C3N4 give 80% and 10% conversion yield within the same illumination period, respectively. Considering the even smaller surface area of CNMS compared to that of mpg-C3N4 (see Fig. S4† and Table 1), the higher activity can be ascribed to structural features which improve the light harvesting by inner reflections and field enhancement effects.42–44 High efficiency light harvesting by CNMS leads to more light-generated electron carriers, which are subsequently transferred to M to couple with protons, and then shuffled to NAD+ to create 1,4-NADH.
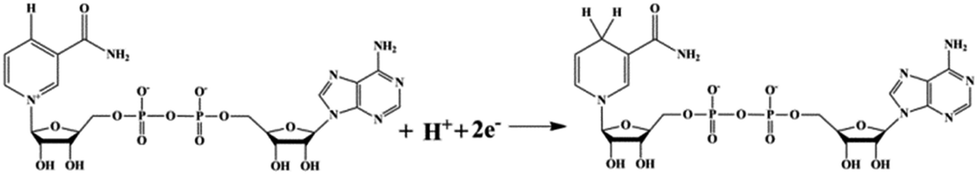 | ||
| Scheme 2 The summarized reaction equation shows the regeneration of NADH. | ||
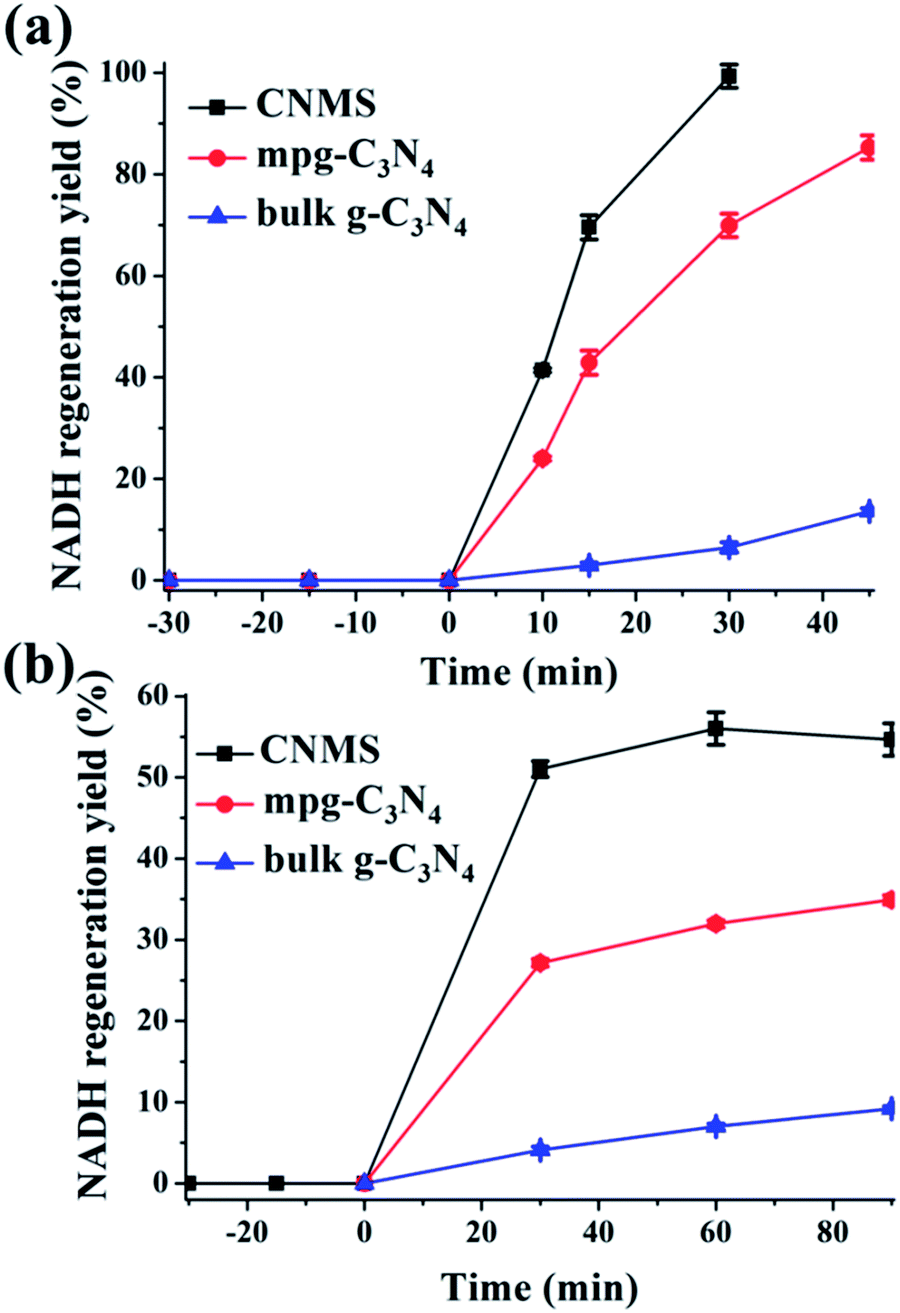 | ||
| Fig. 4 (a) Photocatalytic NADH regeneration kinetics in the presence of M (a) and mediator free (b) by CNMS, mpg-C3N4 and g-C3N4, respectively. For (a), mediator involved NADH regeneration: β-NAD+, 1 mM; M, 0.25 mM; TEOA, 15 w/v%; PBS buffer, 0.1 M, pH = 8; carbon nitride, 3 mg; for (b), mediator free NADH regeneration: β-NAD+, 1 mM; TEOA, 15 w/v%; PBS buffer, 0.1 M, pH = 10; carbon nitride, 3 mg. | ||
Even more striking, under alkaline reaction conditions (pH = 10) the CNMS regenerates NADH in 50% yield without an electron mediator under light after only 30 minutes (Fig. 4b). This again demonstrates the excellent performance of CNMS as a photocatalytic system for NADH regeneration. The direct hydride transfer between CNMS and NAD+ substrate is possibly promoted by the π–π stacking interaction between adenine subunit of β-NAD+ and the electron-rich, layered configurations of g-C3N4, as the building blocks of CNMS.45–47 For the mpg-C3N4, a 50% conversion yield was only achieved after two hours' illumination. After 30 min illumination, only 25% of conversion yield was reached. It is worth mentioning that regeneration of NADH in the mediator free system is accompanied by the loss of stereospecificity, and the 1,6- and 1,4-isomers are formed simultaneously.
The mechanisms of photocatalytic NADH regeneration process and the photosynthetic light reactions are illustrated in Fig. 5a & b, respectively. Under light excitation, CNMS generates electron–hole pairs. M can selectively regenerate 1,4-NADH by taking two electrons from the CNMS (−1.3 V vs. NHE, pH = 7) followed by coupling with one proton and regiospecific transfer to NAD+ (−0.3 V vs. NHE) (see H NMR of M in Fig. S8 of the ESI†). More specifically, the electrons are transferred to [Cp*Rh(bpy)(H2O)]2+, producing Cp*Rh(bpy), followed by coupling with one proton to form the hydriderhodium [Cp*Rh(bpy)(H)]+, which acts as a hydride transfer reagent towards NAD+. Due to the coordination of ring-slipped η3-[Cp*Rh(bpy)(H)]+ with the carbonyl-C-atom of oxidized NAD+, the enzymatically active 1,4-NADH isomer is exclusively generated. The holes (+1.4 V vs. NHE) take their electrons from the oxidation of TEOA (−0.84 V vs. NHE), producing glycolaldehyde and di(ethanol)amine.21 The g-C3N4 network featuring tri-s-triazine units as building blocks and the molecular structure of the NAD+ with the highlighted adenine subunit are presented in Fig. 5c & d, respectively. The π–π stacking interaction leads to the hydride transfer from g-C3N4 to NAD+ and the subsequent NADH regeneration but with poor selectivity for the enzymatically active 1,4-isomer. Upon taking the electron from CNMS, NAD+ could be reduced to NAD˙ and the free radical might undergo proton-assisted disproportionation to form 1,6-NADH. Only minor amounts of enzymatically active 1,4-NADH can be obtained via the isomerization by non-enzymatic transhydrogenation between NAD+ and 1,6-NADH.48,49 Due to the lack of substrate coordination as in the mediator involved system, the obtained mediator-free NADH regeneration is poor in selectivity. For overcoming the poor selectivity problem, experiments with metal-free biological mediators for NADH regeneration are currently in progress.50
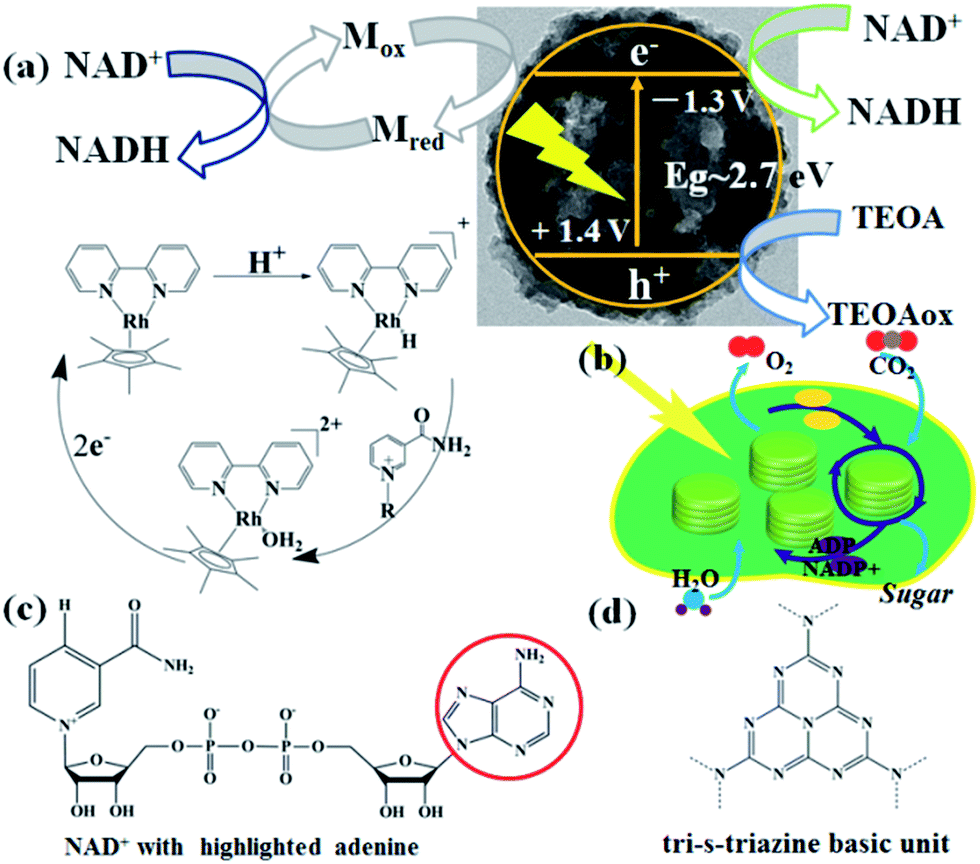 | ||
| Fig. 5 (a) Schematic illustration of photocatalytic NADH regeneration by CNMS, including mediator involved system and mediator free system, respectively. The regeneration of catalytically active [Cp*Rh(bpy)(H)]+ was also illustrated, which acts as a hydride transfer reagent towards NAD+. (b) The thylakoid structure and NADPH involved functions in photosynthetic chloroplasts under light irradiation. (c) The molecular structure of β-NAD+ with an adenine group highlighted in a red circle. (d) A perfect graphitic carbon nitride constructed from tri-s-triazine building blocks is illustrated. | ||
Based on the above NADH regeneration and its mechanism analysis, we performed the photoenzymatic synthesis of L-lactate assisted by L-lactate dehydrogenase (EC 1.1.1.27).51 The synthesis of L-lactate from pyruvate will provide a way to oxidize NADH to NAD+ for glycolysis when no oxygen is present. The enzymatic reaction is depicted in Fig. 6a.
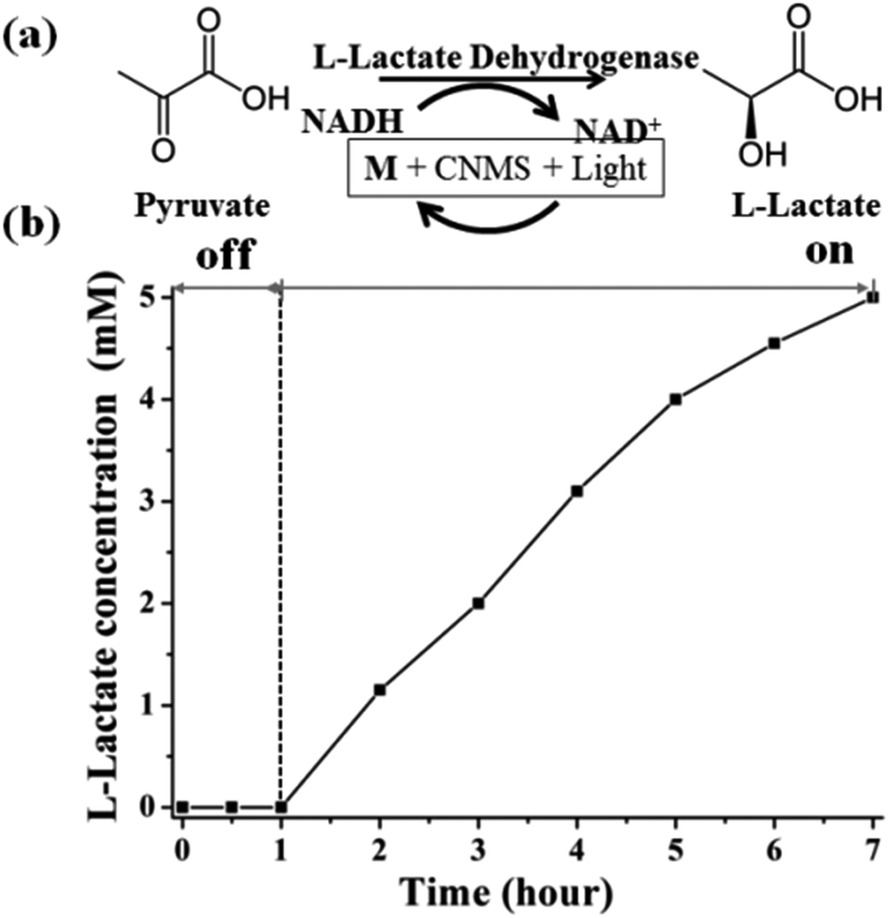 | ||
| Fig. 6 (a) The conversion between pyruvate and L-lactate by L-lactate dehydrogenase. (b) Photocatalytically propelled enzymatic synthesis of L-lactate by CNMS in the presence of M assisted by L-lactate dehydrogenase. Enzymatic reaction conditions: NAD+ (1 mM), M (0.25 mM), pyruvate (5 mM), L-lactate dehydrogenase (50 U), pH = 7.0 phosphate buffer with 15 w/v% TEOA, 3 mg CNMS. | ||
Firstly, the as-regenerated NADH is used to reduce pyruvate in the presence of L-lactate dehydrogenase. The NADH absorbance peak at 340 nm vanished within 5 minutes after addition of pyruvate solution and L-lactate dehydrogenase, which means that NADH is completely consumed for the L-lactate formation (Fig. S9a of ESI†). The two peaks around 300 nm are due to the characteristic absorbance of M in aqueous solution. Fig. S9b of the ESI† shows the pyruvate reduction to lactate by commercial NADH assisted by L-lactate dehydrogenase, also reflecting the complete consumption of NADH in the enzymatic reaction (Fig. S10 of the ESI†). The quantitative conversion demonstrates that the photocatalytically generated NADH is indeed in the active form (1,4-isomer), i.e. the artificial photosynthesis system produces a biologically accepted “currency”.
The sustainable synthesis of L-lactate was achieved by coupling the mediator involved NADH photogeneration system with a redox enzyme, as illustrated in Fig. 6b. The stoichiometric conversion of pyruvate to lactate was accomplished after 6 hours' light illumination, using continuous reaction conditions. During the light reaction, the in situ regenerated NADH was immediately consumed by L-lactate dehydrogenase for the synthesis of L-lactate from pyruvate, while the NAD+ could be repeatedly regenerated (the molar ratio of pyruvate to NAD+ was chosen to be 5, indicating that the NADH must be continuously regenerated for accomplishing the stoichiometric conversion). The yield was determined by the integration of NMR signals of the starting and ending reaction solutions, as shown in Fig. S11 of the ESI.† During the dark cycle, there is no L-lactate product detected. Complete conversion of pyruvate to L-lactate could be verified by the NMR spectra. Therefore, we can safely state that it is possible to realize the photoenzymatic synthesis of L-lactate from pyruvate in the presence of L-lactate dehydrogenase. Through a coupling with other enzymes, we can imagine synthesizing more valuable compounds, but also small fuel molecules such as methanol.52−56
Conclusions
In conclusion, we presented highly efficient cofactor regeneration systems which were synthesized by rational design and engineering of an existing photocatalytic system. Carbon nitride mesoporous spheres with a bioinspired inner structure were synthesized for the first time and employed as the catalyst for photocatalytic NADH regeneration in the presence of a hydride transfer reagent. The high surface area and strong light harvesting capability ensures that the CNMS possesses the highest activity for NADH regeneration in the examined series. In a mediator free system, the CNMS also gives the best performance for NADH regeneration but with poor 1,4 versus 1,6-selectivity. The in situ photocatalytically regenerated NADH could be coupled with L-lactate dehydrogenase for the synthesis of chiral L-lactate from pyruvate, and this reaction was run in a continuous fashion. This opens a general pathway for enzymatic synthesis of diverse chiral organic compounds, using ordinary light as the energy source. Owing to the facile synthesis and superior performance of CNMS, carbon nitride photocatalysis for NADH regeneration could turn compatible with the industrial demands for sustainable, high throughput synthesis. Furthermore, the silica-etching strategy for CNMS synthesis presented here could be extended to other nanomorphologies, the influence of which on reactivity stays unexplored here, as well as to other compositions of heterogeneous photocatalysts. The synthesis sequence developed here thereby might open further possibilities to engineer high efficiency artificial photosynthesis systems in the future.Acknowledgements
J. L. acknowledges the support from Alexander von Humboldt Foundation. J. H. acknowledges the support from National Natural Science Foundation of China (21103095) and the Excellent Young Scientific Research Talents in University of Fujian, China (JA12285). Dr Guo Liang Li and Dr Changqing Ye are acknowledged for helpful discussions and experimental assistance.Notes and references
- J. Barber, Chem. Soc. Rev., 2008, 38, 185 RSC.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767 CrossRef CAS PubMed.
- H. Li, P. H. Opgenorth, D. G. Wernick, S. Rogers, T.-Y. Wu, W. Higashide, P. Malati, Y.-X. Huo, K. M. Cho and J. C. Liao, Science, 2012, 335, 1596 CrossRef CAS PubMed.
- F. Wang, W. J. Liang, J. X. Jian, C. B. Li, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2013, 52, 8134 CrossRef CAS PubMed.
- S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283–2297 RSC.
- S. Fukuzumi, K. Doi, A. Itoh, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15572–15577 CrossRef CAS PubMed.
- M. L. Ghirardi, A. Dubini, J. Yu and P.-C. Maness, Chem. Soc. Rev., 2009, 38, 52 RSC.
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196 CrossRef CAS PubMed.
- P. Könst, H. Merkens, S. Kara, S. Kochius, A. Vogel, R. Zuhse, D. Holtmann, I. W. Arends and F. Hollmann, Angew. Chem., Int. Ed., 2012, 51, 9914 CrossRef PubMed.
- S. Staudt, E. Burda, C. Giese, C. A. Müller, J. Marienhagen, U. Schwaneberg, W. Hummel, K. Drauz and H. Gröger, Angew. Chem., Int. Ed., 2013, 52, 2359 CrossRef CAS PubMed.
- H. K. Chenault and G. M. Whitesides, Appl. Biochem. Biotechnol., 1987, 14, 147 CrossRef CAS.
- K. Seelbach, B. Riebel, W. Hummel, M. R. Kula, V. I. Tishkov, A. M. Egorov, C. Wandrey and U. Kragl, Tetrahedron Lett., 1996, 37, 1377 CrossRef CAS.
- A. Bardea, E. Katz, A. F. Bückmann and I. Willner, J. Am. Chem. Soc., 1997, 119, 9114 CrossRef CAS.
- H. C. Lo and R. H. Fish, Angew. Chem., Int. Ed., 2002, 41, 478 CrossRef CAS.
- R. Wichmann and D. Vasic-Racki, in Technology Transfer In Biotechnology, Springer, 2005, p. 225 Search PubMed.
- W. Ma, D.-W. Li, T. C. Sutherland, Y. Li, Y.-T. Long and H.-Y. Chen, J. Am. Chem. Soc., 2011, 133, 12366 CrossRef CAS PubMed.
- F. Hollmann, B. Witholt and A. Schmid, J. Mol. Catal. B: Enzym., 2002, 19, 167 CrossRef.
- Z. Jiang, C. Lü and H. Wu, Ind. Eng. Chem. Res., 2005, 44, 4165 CrossRef CAS.
- J. Ryu, S. H. Lee, D. H. Nam and C. B. Park, Adv. Mater., 2011, 23, 1883 CrossRef CAS PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367 CrossRef CAS PubMed.
- S. S. Bhoware, K. Y. Kim, J. A. Kim, Q. Wu and J. Kim, J. Phys. Chem. C, 2011, 115, 2553 CAS.
- S. Choudhury, J. O. Baeg, N. J. Park and R. K. Yadav, Angew. Chem., Int. Ed., 2012, 124, 11792 CrossRef.
- J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Angew. Chem., Int. Ed., 2012, 51, 517 CrossRef CAS PubMed.
- A. R. Parker and H. E. Townley, Nat. Nanotechnol., 2007, 2, 347 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2012, 135, 18 CrossRef PubMed.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642 CrossRef CAS PubMed.
- Y. S. Jun, J. Park, S. U. Lee, A. Thomas, W. H. Hong and G. D. Stucky, Angew. Chem., 2013, 125, 11289 CrossRef.
- Y. Zhang, Z. Schnepp, J. Cao, S. Ouyang, Y. Li, J. Ye and S. Liu, Sci. Rep., 2013, 3, 2163 Search PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486 CAS.
- J. Toster, K. S. Iyer, W. Xiang, F. Rosei, L. Spiccia and C. L. Raston, Nanoscale, 2013, 5, 873 RSC.
- J. Liu, J. Huang, D. Dontsova and M. Antonietti, RSC Adv., 2013, 3, 22988 RSC.
- Q. Yang, M. Li, J. Liu, W. Shen, C. Ye, X. Shi, L. Jiang and Y. Song, J. Mater. Chem. A, 2013, 1, 541 CAS.
- J. Liu, M. Li, J. Wang, Y. Song, L. Jiang, T. Murakami and A. Fujishima, Environ. Sci. Technol., 2009, 43, 9425 CrossRef CAS PubMed.
- J. Liu, G. Liu, M. Li, W. Shen, Z. Liu, J. Wang, J. Zhao, L. Jiang and Y. Song, Energy Environ. Sci., 2010, 3, 1503 CAS.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
- Q. Zhang, T. Zhang, J. Ge and Y. Yin, Nano Lett., 2008, 8, 2867 CrossRef CAS PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed.
- Z. Dong, X. Lai, J. E. Halpert, N. Yang, L. Yi, J. Zhai, D. Wang, Z. Tang and L. Jiang, Adv. Mater., 2012, 24, 1046 CrossRef CAS PubMed.
- H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406 CrossRef CAS PubMed.
- I. G. Yu, Y. J. Kim, H. J. Kim, C. Lee and W. I. Lee, J. Mater. Chem., 2011, 21, 532 RSC.
- H. Zhou, Z. Zhang, P. Yu, L. Su, T. Ohsaka and L. Mao, Langmuir, 2010, 26, 6028 CrossRef CAS PubMed.
- M. V. Jose, S. Marx, H. Murata, R. R. Koepsel and A. J. Russell, Carbon, 2012, 50, 4010 CrossRef CAS PubMed.
- J. Moiroux, P. J. Elving and J. Electroanal, J. Electroanal. Chem. Interfacial Electrochem., 1979, 102, 93 CrossRef CAS.
- P. J. Elving, W. T. Bresnahan, J. Moiroux and Z. Samec, Bioelectrochem. Bioenerg., 1982, 9, 365–378 CrossRef CAS.
- Y. Ohnishi, Y. Kikuchi and M. Kitami, Tetrahedron Lett., 1979, 32, 3005–3008 CrossRef.
- Y.-T. Long and H.-Y. Chen, J. Electroanal. Chem., 1997, 440, 239 CAS.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455 CrossRef CAS PubMed.
- R. Obert and B. C. Dave, J. Am. Chem. Soc., 1999, 121, 12192 CrossRef CAS.
- R. Cazelles, J. Drone, F. Fajula, O. Ersen, S. Moldovan and A. Galarneau, New J. Chem., 2013, 37, 3721 RSC.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132 CrossRef CAS PubMed.
- Q. Sun, Y. Jiang, Z. Jiang, L. Zhang, X. Sun and J. Li, Ind. Eng. Chem. Res., 2009, 48, 4210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, TEM images, N2 sorption isotherm plots, time-resolved photoluminescence, NMR spectra of CNMS can be found in the ESI. See DOI: 10.1039/c4ta00793j |
| This journal is © The Royal Society of Chemistry 2014 |