 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene oxide and Ag engulfed TiO2 nanotube arrays for enhanced electron mobility and visible-light-driven photocatalytic performance†
Lan Ching
Sim
a,
Kah Hon
Leong
a,
Shaliza
Ibrahim
a and
Pichiah
Saravanan
*ab
aEnvironmental Laboratory, Department of Civil Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: saravananpichiah@um.edu.my; Fax: +60 03 7967 5318; Tel: +60 03 7967 7678
bInstitute of Nanotechnology and Catalysis Research Center, University of Malaya, 50603, Kuala Lumpur, Malaysia. E-mail: saravananpichiah@um.edu.my; Fax: +60 03 7967 5318; Tel: +60 03 7967 7678
First published on 20th January 2014
Abstract
The visible-light-driven photocatalytic degradation of Methylene Blue (MB) and 2-chlorophenol (2-CP) were investigated using the composite of Ag nanoparticles (Ag NPs) and graphene oxide (GO) deposited over TiO2 nanotube arrays (TNTs). The resulting TNTs in the composite showed 100% anatase phase with no occurrence of the rutile phase. An implicit microscopic and spectroscopic technique (FESEM, HRTEM, FTIR and Raman analysis) confirmed the presence of Ag NPs and GO in the composite photocatalyst. It also exhibited an evident shift of the absorption edge in the visible range. The successful depositions of Ag contributed to improved photocatalytic activity in the visible spectrum owing to the existence of localized surface plasmon resonance (LSPR), and further the deposition of GO minimized the recombination of electron–hole pairs. The photocatalytic degradation of both MB and 2-CP followed pseudo-second order kinetics. In the primary run, both MB and 2-CP exhibited almost similar degradation efficiency of 68.3 and 66.8%, respectively. The reusability studies showed a deprived performance for MB degradation than that of 2-CP, due to chemisorption of MB. The prepared composite exhibited significantly larger enhancement in the photocatalytic oxidation of pollutants with greater electrons mobility to reactive sites of GO and Ag.
Introduction
Ever since the discovery of the photocatalytic splitting of water on titanium dioxide (TiO2) electrodes by Fujishima and Honda in 1972,1 TiO2 have attracted much attention for widespread environmental applications due to its non-toxicity, long-term stability, low cost, chemical inertness, easy availability and high photoactivity.2–4 Though TiO2 as nanoparticles have wider applications in energy and environmental domains, equivalently TiO2 nanotube arrays (TNTs) are also applied in similar domains for photodegradation of organic pollutants, carbon dioxide (CO2) reduction and dye-sensitized solar cells.5–9 TNTs possess a larger surface area, vectorial charge transfer, long term stability to photo- and chemical corrosion.10,11 However, the photocatalytic reactions of TNTs are limited by low absorption capability in the visible light region and a high recombination rate of photogenerated electron–hole pairs formed in photocatalytic activity. Consequently, significant efforts have been devoted to improve the photocatalytic efficiency of TNTs, such as doping metal ions,10,12,13 non-metal5,14 and coupling with semiconductor nanoparticles.15,16Noble metals, such as gold (Au) and silver (Ag), possess an additional ability to absorb visible light due to the existence of localized surface plasmon resonance (LSPR).17 Moreover, these metals function as electron donors to promote electron transfer from metal to TiO2 and act as electron traps in the metal–TiO2 nanostructures, minimizing the surface charge recombination in TiO2.18,19 Alternatively, conducting carbon materials are incorporated into TiO2 to promote electron transport. Graphene is a two-dimensional sp2-hybridized carbon nanosheet which possesses a high specific surface area with a large interface, high electron mobility and a tunable band gap.20 Metal or metal oxides (Ag, Au, TiO2 and SnO2) are combined with graphene and the resultant hybridized materials exhibit superior photocatalytic properties than the bulk metal or metal oxide.21,22 Nevertheless, hydrophobic graphene is not compatible with hydrophilic metal/metal oxides, which makes graphene difficult to deposit on the surface of metal/metal oxides.23
On the other hand, graphene oxide (GO) is a layer-structured graphite compound built up by hydrophilic, stacked graphene sheets bound to oxygen in the form of carboxyl, hydroxyl or epoxy groups.24,25 The better solubility of GO in water and other solvents allows to ease their deposition onto the surface of metal/metal oxides. Song et al. demonstrated Methylene Blue (MB) removal by GO–TNTs with a 15 times increase in the photoconversion efficiency.26 Gao et al. reported that GO–TiO2 hybrids exhibit higher adsorption ability for methyl orange (MO) than GO, and about ∼55% of MO was absorbed by the GO–TiO2 hybrids at the beginning of photodegradation.23 Utilising similar hybrid materials, Jiang et al. showed that the photo-oxidative degradation rate of MO and the photo-reductive conversion rate of Cr(VI) over the hybrids were as high as 7.4 and 5.4 times that over P25, respectively.27 These studies suggested that the enhancing effect of GO on the photocatalytic properties of TiO2 was attributed to a large surface area, adsorption capacity, and strong electron transfer ability of the GO in the hybrid materials.
Most of the findings on graphene-semiconductor composites are of nanoparticles while TNTs are scant. Indeed, TNTs can be combined with GO for practical application. In this work, we attempt to enhance photocatalytic performance of TNTs by using Ag and GO as an electron transfer channel or electron sink to reduce the recombination of photogenerated electron–hole pairs. Similarly, Tang et al. showed that the composite of Ag, reduced graphene oxide (RGO) and TNTs exhibited 2,4-dichlorophenoxyacetic acid removal efficiency of almost 100%, much higher than 49% over Ag–TNTs.28 The Ag particles were respectively deposited onto the surface of TNTs and RGO, forming Ag–TNTs and Ag–RGO–TNTs. However, it is inappropriate to compare the removal efficiency of both photocatalysts because the degradation mechanism might act differently according to the different location of Ag NPs. Many cases have mentioned that GO served as an electron sink to hinder electron–hole pairs recombination,23,26,29,30 but indeed the role of GO could change in different pollutant models. The present work offers several advantages over previously reported ones, including (1) deposition of Ag particles onto the surface of TNTs instead of GO to draw more conclusive results of comparison, (2) a low cost and a facile assembly method to deposit GO onto Ag–TNTs, (3) the photocatalytic activities were examined by comparing the photocatalytic degradation between Methylene Blue (MB) and 2-chlorophenol (2-CP), considering that the photocatalyst could respond differently to different types of pollutants.
Experimental
Preparation of GO
Graphite oxide was synthesized through a simplified Hummers method,31 3 g of natural graphite powder (99.99%, Sigma-Aldrich) were oxidized by a mixture of 400 mL of H2SO4 and 18 g of KMnO4. The mixture was stirred for three days to ensure complete oxidation of the graphite. Then, H2O2 solution was added to stop the oxidation process. The graphite oxide was washed with 1 M of HCl and DI water until a pH 4−5 was achieved. During the washing process, the graphite oxide underwent exfoliation to form GO gel. It was then vacuum dried at 60 °C for 24 h to obtain brownish GO solid.Preparation of Ag–TNTs and GO–Ag–TNTs
Ti foil (99.7%, Sigma-Aldrich) was first anodized in ethylene glycol (anhydrous, 99.8%) electrolyte containing 0.3 M ammonium fluoride (NH4F, 98%) and 2 vol% water (H2O) with graphite rod as the counter electrode under 50 V for 3 h. After annealing at 450 °C for 1 h, the anodized sample was sonicated for 30 min and then annealed for 2 h. Photodeposition of Ag on TNTs were carried out by dipping TNTs in an equal volume ratio of methanol–water mixture containing 1 mM of AgNO3. The surface of the TNTs was exposed to 400 W high pressure Hg lamp for 1 h under nitrogen atmosphere with sonication. The resulting product was designated as Ag–TNTs. The procedures for the preparation of GO–Ag–TNTs are illustrated in Fig. 1. Thus obtained Ag–TNTs were immersed in a 0.5 mg mL−1 aqueous GO suspension for 5 h and vacuum dried. The modified composite material was denoted as GO–Ag–TNTs.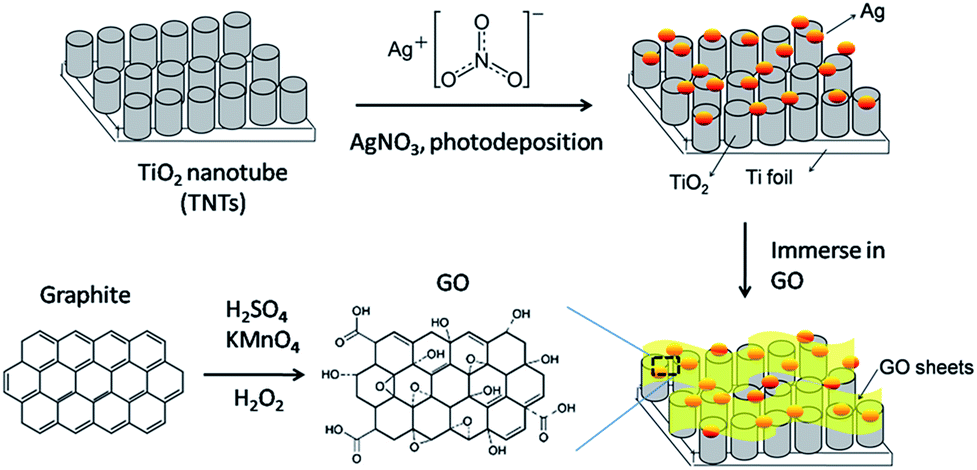 | ||
| Fig. 1 Schematic diagram of the procedures for preparation of GO–Ag–TNTs. | ||
Characterization
The phase composition of the synthesized photocatalysts was obtained using X-ray diffractometer (XRD, D8 Advance, Bruker) operated in the reflection mode with Cu Kα radiation (λ = 1.54 Å). The morphologies of samples were examined by a field emission scanning electron microscope (FESEM, SU8000, Hitachi) equipped with an EDS (energy dispersive X-ray spectroscopy) detector. The images were taken at an accelerating voltage of 20 kV. High resolution transmission electron microscope (HRTEM, JEM-2100F, Jeol) images were obtained at 200 kV. A Micro-PL/Raman spectroscope (Renishaw, inVia Raman Microscope) was used to acquire the Raman and photoluminescence (PL) spectra. Fourier transform infrared (FTIR) spectra were obtained on a Perkin Elmer Spectrum 400 spectrophotometer with scan range of 4000–450 cm−1. UV-vis diffuse reflectance spectra (UV-DRS) were measured using UV-vis spectrophotometer (UV-2600, Shimadzu) equipped with an integrating sphere attachment. The spectra were collected with BaSO4 as a reference. The surface chemical composition of the samples was analyzed by X-ray photoelectron spectroscopy (XPS, Omicron, Germany) with Al Kα radiation source.Photocatalytic experiment
The photocatalytic activities were evaluated based upon the removal of Methylene Blue (MB) and 2-chlorophenol (2-CP) in aqueous solutions. For comparison, the photocatalytic activities of TNTs, Ag–TNTs and GO–TNTs were also studied. The prepared photocatalysts were immersed in a glass beaker containing 100 mL aqueous solutions for MB (5 mg L−1) and 200 mL aqueous solutions for 2-CP (10 mg L−1), respectively. Prior to photodegradation, the solutions were magnetically stirred in the dark for 1 h to establish an adsorption–desorption equilibrium. A 500 W tungsten-halogen lamp was used as a visible light source, with any UV light below 400 nm being removed with a high-pass filter (FSQ-GG400, Newport Corp.). For the degradation of MB, the samples were collected at regular intervals, analyzed for residual MB concentration with visible spectrometer (Spectroquant® Pharo 100, Merck) at λmax = 664 nm. Similarly, 2-CP samples were withdrawn at regular intervals, centrifuged and analyzed for residual concentration with ultra-performance liquid chromatography (UPLC) (ACQUITY UPLC H Class, Waters) equipped with C18 column (50 mm × 2.1 mm × 1.7 μm). The mobile phase was acetonitrile (ACN) and water in the ratio 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 with a flow rate of 0.4 mL min−1. All photocatalytic experiments were carried out for duration of 6 h.
:40 with a flow rate of 0.4 mL min−1. All photocatalytic experiments were carried out for duration of 6 h.
Results and discussion
The FESEM images show that the synthesized TNTs are uniformly stacked in a tubular structure and vertically orientated with a tube diameter ranging from 100–120 nm and wall thickness of 15 nm. The cross-sectional image in Fig. 2a reveals that the tube length ranging from 8–9 μm. As shown in Fig. 2b, the photodeposited Ag particles have a wide range of sizes and shapes. After the photodeposition with a sonication process, the surface of TNTs is covered by Ag NPs with a near smaller average particle size of 100 nm (Fig. 2c). GO–Ag–TNTs sample was obtained with a sheet of GO coating the most surface of the TNTs, as shown in Fig. 2d. It is observed that GO has a flake-like structure with wrinkles and folds, which is consistent with earlier reports.21,32,33 This is a characteristic feature of GO when it is not conformally coated on the surface of TNTs.34 The inset in Fig. 2d is the corresponding EDX spectrum, confirming the presence of C, Ag, O and Ti in GO–Ag–TNTs. As illustrated in Fig. 2e, the Ag NPs were deposited onto the surface of the TNTs and even inside the tubes. The synthesized Ag and TiO2 can be clearly identified by the lattice fringes shown in the HRTEM image of GO–Ag–TNTs (Fig. 2f). The lattice fringes with 0.24 and 0.35 nm spacing are attributed to Ag (111) and anatase TiO2 (101) planes, respectively.35,36 | ||
| Fig. 2 FESEM images of the (a) cross-section of GO–Ag–TNTs, top view of Ag–TNTs (b) with no sonication, (c) with sonication, (d) top view of GO–Ag–TNTs. The inset of (d) is the EDX of GO–Ag–TNTs and (e–f) HRTEM images of GO–Ag–TNTs. | ||
Fig. 3 depicts the XRD of graphite, GO, TNTs, Ag–TNTs and GO–Ag–TNTs. Pure anatase TiO2 phase is observed in TNTs, Ag–TNTs and GO–Ag–TNTs. The two obvious peaks of tetragonal TiO2 anatase phase (JCPDS no. 21-1272) appeared at 25.3 and 48.0°, corresponding to (101) and (200) crystal planes, respectively. Additionally, Ag–TNTs and GO–Ag–TNTs show peaks at 38.1, 44.3, 64.4 and 77.4° which are assigned to the (111), (200), (220) and (311) planes of face centered cubic (FCC) Ag (JCPDS no. 65-2871). A (002) diffraction peak at 10.6° was observed for GO, indicating most of the natural graphite was oxidized into GO by expanding the d-spacing from 3.37 to 8.6 Å. This indicates the introduction of oxygen-containing groups on the GO sheets.21 However, there is no peak ascribed to GO that can be observed in the sample of GO–Ag–TNTs due to the low amount of GO which is below the detection limit of XRD.37,38 The average crystallite sizes of TiO2 anatase and Ag particles were calculated using the Scherrer equation:
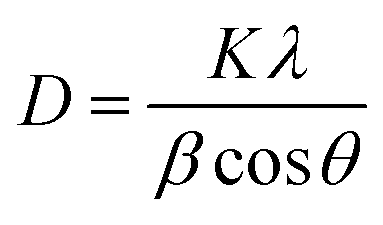 | (1) |
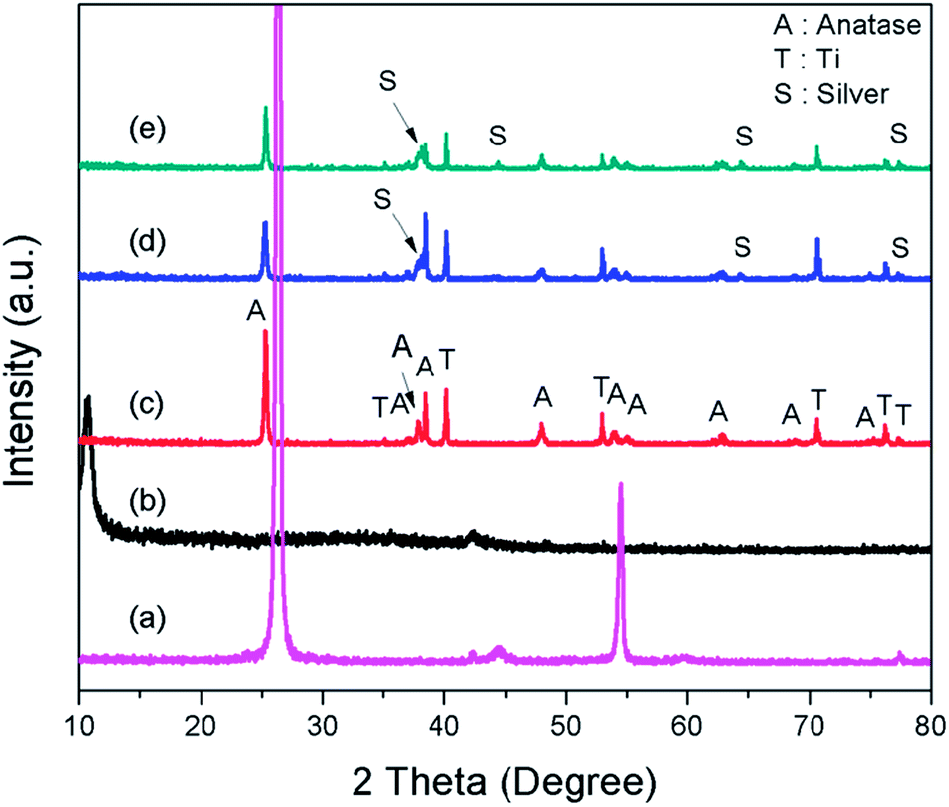 | ||
| Fig. 3 X-Ray diffraction patterns of (a) graphite (b) GO (c) TNTs (d) Ag–TNTs and (e) GO–Ag–TNTs. | ||
The UV-vis diffuse reflectance spectra (UV-DRS) of TNTs, Ag–TNTs and GO–Ag–TNTs are shown in Fig. 4. As expected, the sample of TNTs shows an absorption band lower than 380 nm (UV region) due to the charge transfer from O 2p valence band to Ti 3d conduction band.39 A broad absorption peak at approximately 460 nm is observed for Ag–TNTs, which is attributed to the surface plasmon absorption of Ag NPs.40 This clearly shows the presence of metallic Ag NPs on the surface of TNTs. In addition, GO–Ag–TNTs shows higher light absorption capacities in the entire visible region due to the presence of GO. Raman spectra of graphite, GO, TNTs, Ag–TNTs and GO–Ag–TNTs are depicted in Fig. 5. Four distinct Raman peaks of anatase TiO2 can be observed at 145 (Eg), 399 (B1g), 519 (A1g + B1g) and 639 cm−1 (Eg) for the samples of TNTs, Ag–TNTs and GO–Ag–TNTs. It further proved that all the combinations of synthesized samples resulted in 100% anatase phase. It is expected that GO and GO–Ag–TNTs have two peaks at around 1595 and 1350 cm−1, corresponding to the G- and D-bands, respectively. The G-band appearing around 1595 cm−1 is a significant characteristics of sp2 hybridized carbon materials, which can provide information on the in-plane vibration of sp2-bonded carbon domains,41,42 whereas the D-band appears at around 1350 cm−1 indicate the presence of sp3 defects within the hexagonal graphitic structure43 and can be associated with the amorphous carbon, or edges that break the symmetry and selection rule.44 Hence a smaller ID/IG peak intensity ratio of Raman spectra indicates lower defects of the graphitized structures. In comparison to Raman spectra of graphite, GO and GO–Ag–TNTs have broader G-band due to the enhanced isolated double bonds.45 While the D-band becomes sharper because of the increasing disorder with GO and GO–Ag–TNTs. After the oxidation of graphite, the ratio of ID/IG increases to 0.91, designating the formation of a large sp3 domain in the sample of GO. The D-band and G-band of GO–Ag–TNTs were roughly at similar positions to that of GO. However, the ratio of ID/IG for GO–Ag–TNTs is 0.95 which is slightly higher than GO, showing a marginal decline of the graphitic domains.
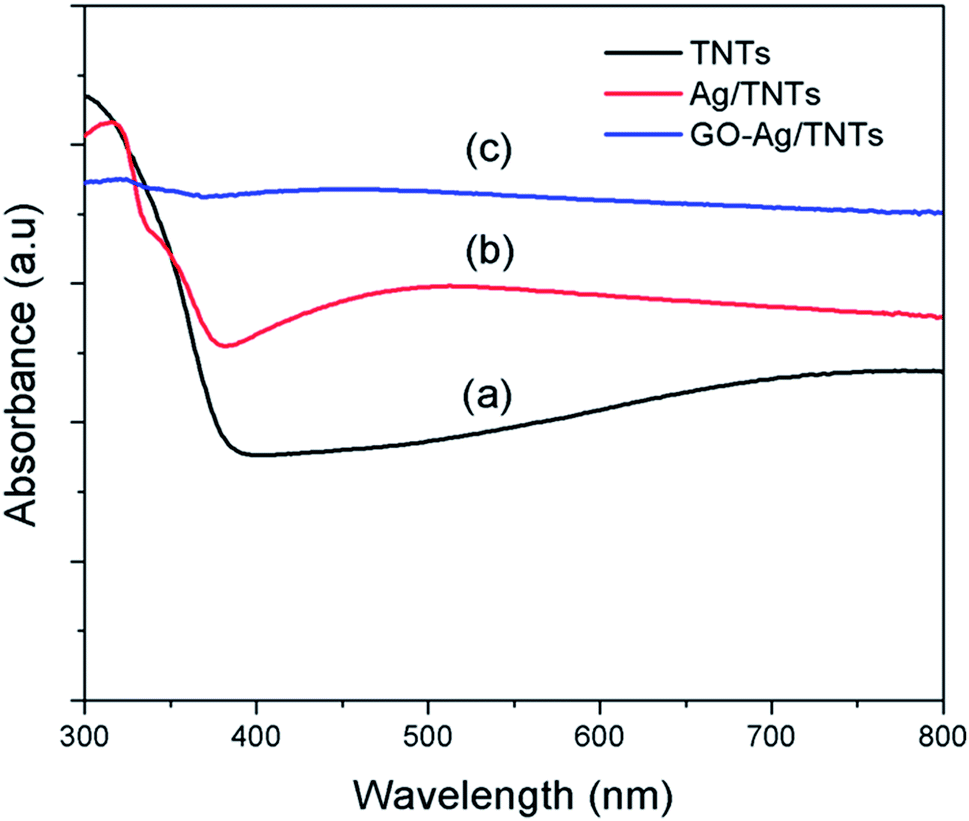 | ||
| Fig. 4 UV-visible absorption spectra of (a) TNTs (b) Ag–TNTs and (c) GO–Ag–TNTs. | ||
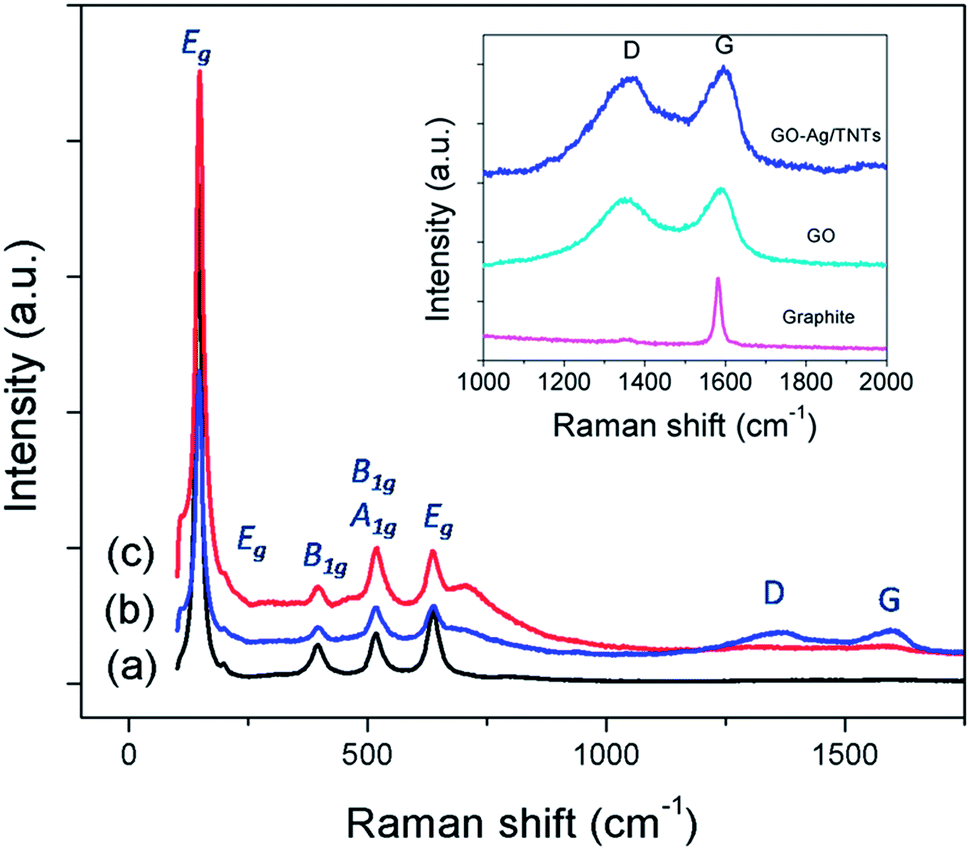 | ||
| Fig. 5 Raman spectra of (a) TNTs (b) GO–Ag–TNTs and (c) Ag–TNTs. The inset is the D and G band of graphite, GO and GO–Ag–TNTs. | ||
FTIR spectra were employed to characterize the carbon species in the prepared samples. Fig. 6 shows the FTIR spectra of GO, GO–Ag–TNTs, TNTs and Ag–TNTs. GO exhibits many strong absorption peaks corresponding to the stretching of the hydroxyl group (3300 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in carbonyl and carboxyl moieties (1720 cm−1), CC skeletal vibration bands from unoxidized graphitic domains or contribution from the stretching deformation vibration of intercalated water (1620 cm−1), carboxyl group (1375 cm−1), epoxide C–O–C or phenolic C–O–H stretching vibrations (1220 cm−1), and C–O stretching vibrations in the epoxy or alkoxy groups (1045 cm−1).35,46,47 For GO–Ag–TNTs, most of these groups are retained with a significant decrease in the peak intensity due to the lower GO dosage in the synthesis. The disappearance of C–O stretching band at 1220 cm−1 suggests that epoxide or phenolic groups in GO react with the surface hydroxyl groups of Ag–TNTs and finally form the Ti–O–C bonds in the GO–Ag–TNTs composite. The absorption peaks appear at 800 cm−1 can be assigned as a combination of Ti–O–Ti vibration in crystalline TiO2 and Ti–O–C vibration.48
O groups in carbonyl and carboxyl moieties (1720 cm−1), CC skeletal vibration bands from unoxidized graphitic domains or contribution from the stretching deformation vibration of intercalated water (1620 cm−1), carboxyl group (1375 cm−1), epoxide C–O–C or phenolic C–O–H stretching vibrations (1220 cm−1), and C–O stretching vibrations in the epoxy or alkoxy groups (1045 cm−1).35,46,47 For GO–Ag–TNTs, most of these groups are retained with a significant decrease in the peak intensity due to the lower GO dosage in the synthesis. The disappearance of C–O stretching band at 1220 cm−1 suggests that epoxide or phenolic groups in GO react with the surface hydroxyl groups of Ag–TNTs and finally form the Ti–O–C bonds in the GO–Ag–TNTs composite. The absorption peaks appear at 800 cm−1 can be assigned as a combination of Ti–O–Ti vibration in crystalline TiO2 and Ti–O–C vibration.48
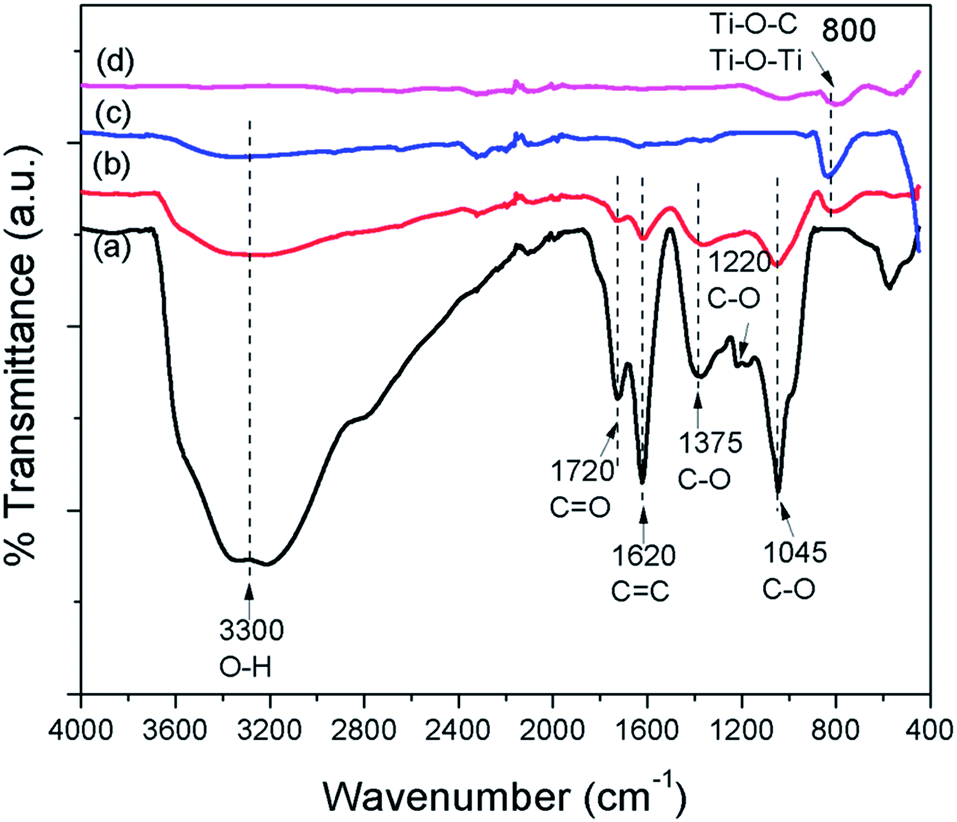 | ||
| Fig. 6 FTIR spectra of (a) GO (b) GO–Ag–TNTs (c) TNTs and (d) Ag–TNTs. | ||
The PL spectra in Fig. 7a were obtained to understand the emission mechanism of the prepared samples. PL emission intensity is related to the recombination rate of excited electron–hole pairs. Lower intensity indicates more excited electrons are transferred or trapped, and higher intensity means the faster the recombination rate. The emission peak of GO–Ag–TNTs and Ag–TNTs are obviously quenched as compared to that of TNTs. The quenching behavior revealed that both the GO and Ag trap electron or transfer electron to suppress electron–hole pairs recombination. The effective charge carrier separation could extend the reactive electron–hole pairs lifetimes and enhance the photocatalytic activity of GO–Ag–TNTs. High-resolution XPS was performed to determine the chemical composition and the oxidation state for GO–Ag–TNTs. As shown in Fig. 7b, there are two peaks observed at 459 eV (Ti 2p3/2) and 464.6 eV (Ti 2p1/2), both correspond to Ti4+ in pure anatase. The presence of Ag NPs can be detected at two peaks centered at 368.2 and 374.2 eV, which is assigned to Ag 3d5/2 and Ag 3d3/2, respectively (Fig. 7c). As shown in Fig. 7d, the C 1s XPS signals were deconvoluted into three components. The peak at 284.5 eV is assigned to the sp2 carbon atoms of GO. The peaks at higher binding energies are assigned to the oxygenated carbon species of GO, such as C–OH, CO and COOH.21,35 The contact between GO and TNTs can be proved by the presence of Ti–C (281 eV) and Ti–O–C (288.7 eV) signals. The former one is attributed to the formation of Ti–C bond in the interface between GO and TNTs. The coordination between carboxyl groups of GO and Ti(OH)x form Ti–O–C bond.49 The XPS results show that oxygenated groups of GO were retained in GO–Ag–TNTs and the formation of Ti–O–C bond, which is in good agreement with the FTIR results.
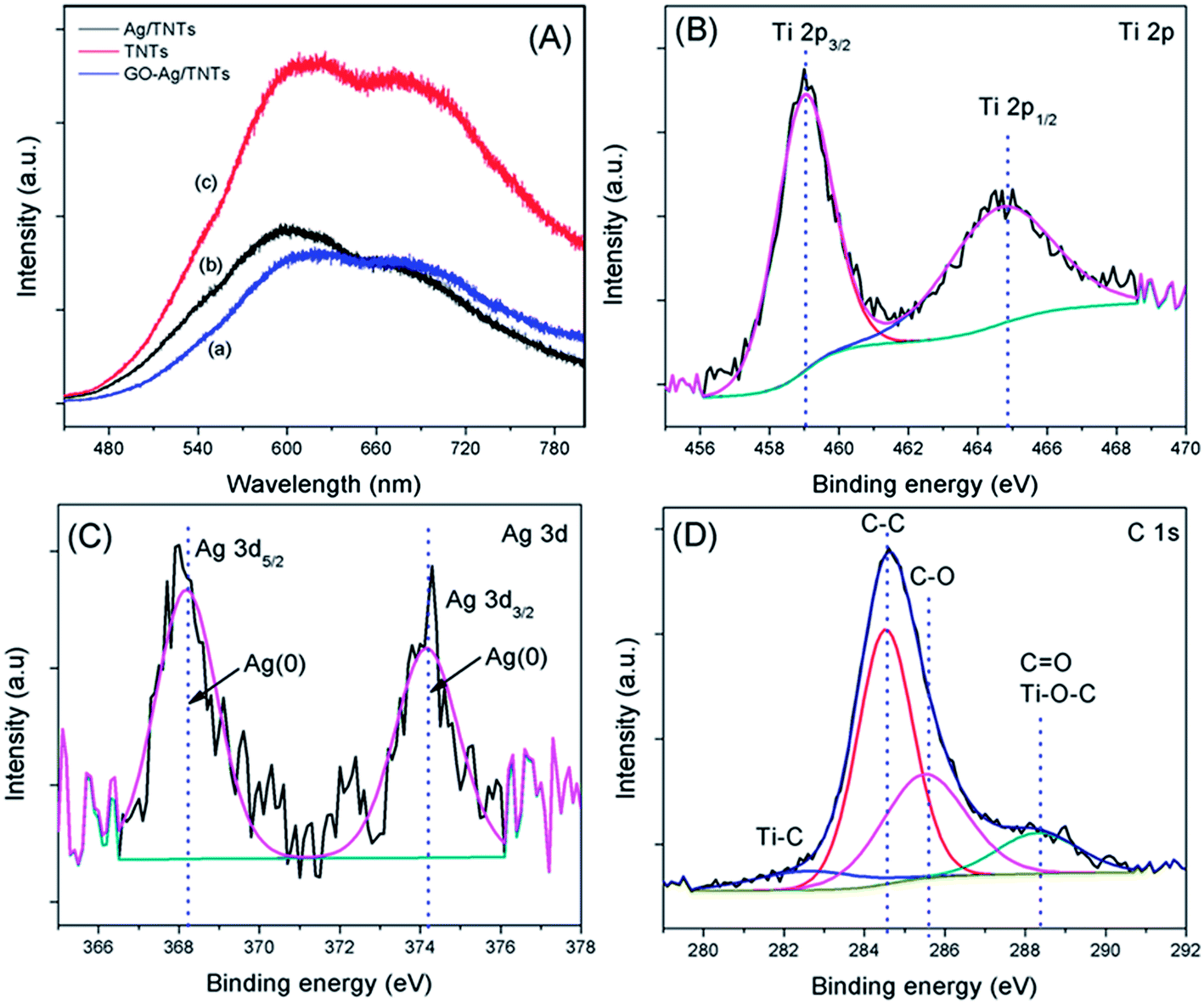 | ||
| Fig. 7 (a) Photoluminescence spectra of TNTs, Ag–TNTs and GO–Ag–TNTs. Core level XPS spectra of (b) Ti 2p (c) Ag 3d and (d) C 1s of GO–Ag–TNTs. | ||
The photocatalytic activity of the prepared GO–Ag–TNTs sample was evaluated by the degradation of MB and 2-CP under visible light irradiation as depicted in Fig. 8a and Fig. 8b. For an adsorption process in the dark, both of GO–TNTs and GO–Ag–TNTs exhibited adsorption capacity of almost 34% for MB and 12% for 2-CP, which is higher than the other samples. The reason for the high adsorption capacity of MB on the surface of GO is attributed to the strong π–π stacking interactions between the benzene rings of MB and the surface of GO.50 A significant decrease in the adsorption capacity of MB is observed for GO–Ag–TNTs after the first run, while it remains almost unchanged from the second to sixth run (Fig. 8c). It can be explained that the chemisorptions which is irreversible plays a dominant role at first. After many runs, there is almost only physical adsorption and therefore, MB adsorption rate is kept constant. On the other hand, physical adsorption is mainly involved in the adsorption of 2-CP since there is no significant loss in the adsorption capacity after many runs.
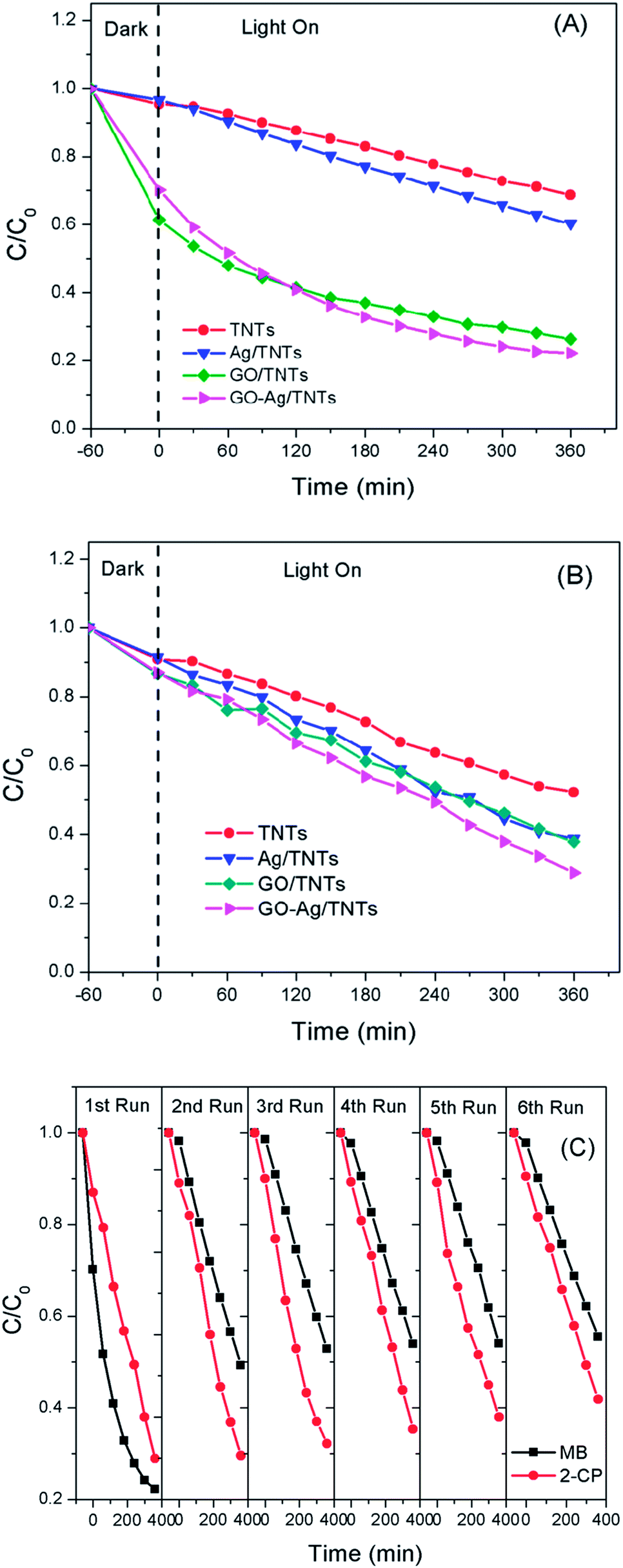 | ||
| Fig. 8 Photocatalytic degradation rates of (a) MB (b) 2-CP for TNTs, Ag–TNTs, GO–TNTs and GO–Ag–TNTs (c) recycled photocatalytic degradation rates of MB and 2-CP for GO–Ag–TNTs. | ||
The initial concentration (C0) is considered as the concentration of MB and 2-CP after adsorption–desorption equilibrium. As shown in Fig. 8a, the degradation efficiency of MB follows an order of GO–Ag–TNTs (68.3%) > GO–TNTs (57.2%) > Ag–TNTs (37.6%) > TNTs (27.9%). Fig. 8b shows the degradation efficiency of 2-CP follows an order of GO–Ag–TNTs (66.8%) > Ag–TNTs (57.7%) > GO–TNTs (56.2%) > TNTs (42.6%). These results exhibited that the degradation efficiency of both MB and 2-CP is comparable in the first run and also improved remarkably in the presence of GO, particularly with the coexistence of Ag and GO. In most cases, GO sheets were used as an electron sink to facilitate photogenerated electrons separation and store the separated electrons.51 The degradation mechanism of MB in Fig. 9a shows that GO can accumulate the electrons injected from the photogenerated MB because of the π-conjugated network and higher work function of GO than that of the excited MB. However, the injected electron could recombine with the surface adsorbed MB˙+ to lower the degradation efficiency. Besides that, the direct transfer of photogenerated electrons from GO to TNTs is restricted by the limited contact between GO and TNTs. Therefore, the photocatalytic activity of GO–TNTs is lower compared to that of GO–Ag–TNTs in the degradation of MB and 2-CP, respectively. Ag NPs were deposited onto the surface of TNTs prior to the decoration of GO to overcome these limitations. Ag NPs able to absorb visible light due to the existence of a localized surface plasmon resonance (LSPR),17 resulted in a better degradation efficiency for Ag–TNTs (37.6% for MB and 57.7% for 2-CP) compared to that of TNTs (27.9% for MB and 42.6% for 2-CP). The Ag NPs possess a higher work function (4.26 eV) than GO and also lying below the conduction band (CB) of TNTs (4.2 eV).52–54 Band gap energy (Eg) of GO is mainly formed by the anti-bonding π* orbital as a conduction band with a higher energy level and the O 2p orbital as a valence band.55,56 It has been reported that Ag+ can be reduced in graphene/TiO2 photocatalytic systems.57 This shows that the energy level of the anti-bonding π* orbital is higher than that of Ag NPs, and thus the electrons transfer from graphene to Ag+. In this case, it is appropriate to conclude that electrons can be injected from the excited GO to Ag NPs.58
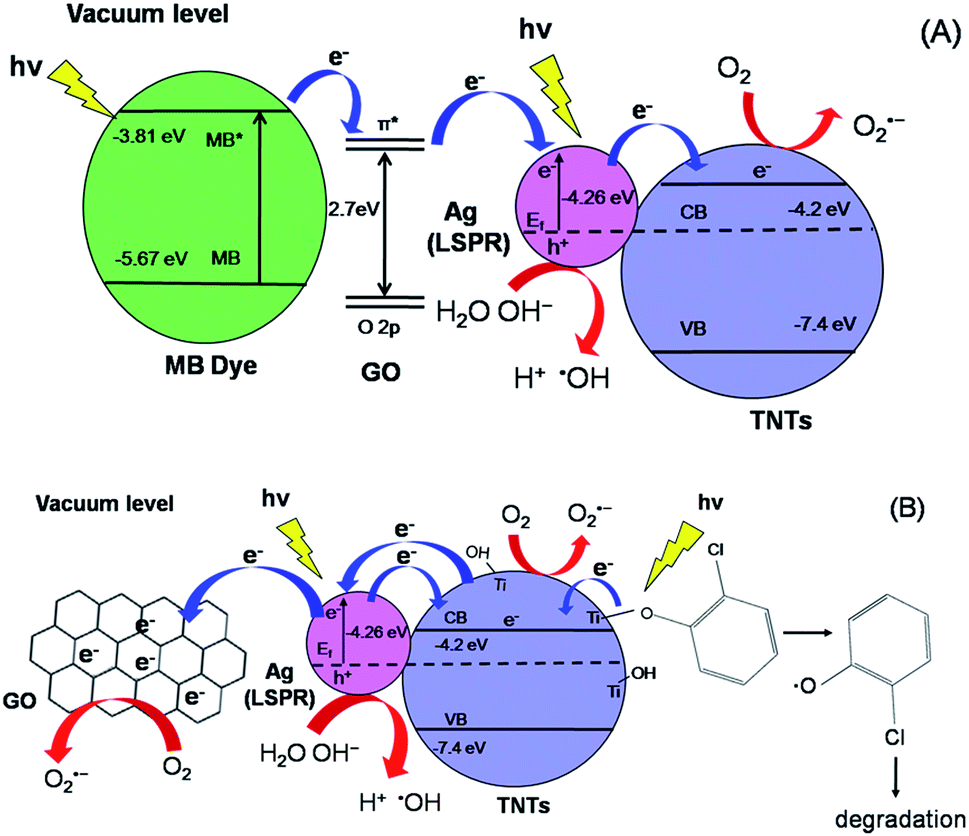 | ||
| Fig. 9 Schematic diagrams of electron transfer and degradation mechanism of (a) MB and (b) 2-CP. | ||
When Ag NPs and TNTs are in contact, a Schottky barrier is formed at the interface of Ag NPs and TNTs. Many groups have reported that Ag NPs can overcome the energy barrier at the interface of Ag–TiO2 upon LSPR-excitation to inject electrons from Ag NPs into the CB of TiO2 under the irradiation of visible light.59–64 Herein, the electrons generated by the LSPR effect in Ag NPs diffuse into the CB of TNTs. The TNTs function as an electron reservoir by capturing the electrons transferred from the GO and Ag to further increase the degradation efficiency of MB. GO served as an electron-accepting mediator between the MB and Ag NPs, which is consistent with the previous studies.65,66 Alternatively, the excited MB can also transfer electrons to TNTs and Ag due to its lower work function (3.81) than Ag (4.26) and lying above the conduction band of TNTs (4.2 eV). However, the electron transfer rate is slower because the deposited GO blocked the tube openings as visualised in FESEM, and perhaps decreased the effective area of Ag–TNTs for the electron transfer.
For the degradation of MB, GO–Ag–TNTs demonstrated a tremendous decreasing trend by 27% after the first run and followed by a significant loss of 36.7% after the sixth run (Fig. 8c). It can be speculated that the active sites of the GO can be undesirably occupied by the adsorbed MB through chemisorption which cannot be eluted, resulting in decreased photocatalytic activity after the first run. The involvement of some functional groups on the surface of GO in the adsorption of MB is shown in Fig. S1, ESI.† In contrast, GO–Ag–TNTs showed a greater stability in the reuse study of 2-CP with a total loss of 19.5% as the physically adsorbed 2-CP can be eluted and more active sites in GO are available for the degradation or charge transfer. It has been demonstrated previously that the 4-chlorophenol (4-CP) and other phenolic compounds can be degraded under visible irradiation due to the charge transfer surface complex formation between the phenolic compound and TNTs.67 Such a surface complex enables the excitation by visible light through ligand-to-metal charge transfer (LMCT) between the 2-CP (ligand) and the Ti4+ site on the surface.68,69 Since 2-CP is one of the phenolic compounds, the surface complex formation is taken into account when degrading 2-CP (Fig. 9b). This explains for higher degradation efficiency of 2-CP (42.6%) than MB (27.9%) for TNTs. The electrons are transferred from TNTs/2-CP surface complex to conduction band of TNTs. These electrons were subsequently injected to Ag NPs and finally to GO which served as an electron sink to facilitate the separation of the excited electrons. On the other hand, the LSPR effect in the Ag NPs provides electrons to the CB of TNTs.59–64 The electrons react with O2 to produce superoxide radical anion ˙O2−. While the photogenerated holes oxidize the organic molecule in MB or 2-CP to form R+, or react with OH− or H2O and then further oxidizing them into ˙OH radicals. The resulting ˙OH radicals are strong oxidizing agent to oxidize MB dye or 2-CP to end-products. The photocatalytic degradation of MB and 2-CP followed pseudo second-order reaction kinetics (Fig. S2 and S3, ESI†). The kinetic parameters of both pollutants are tabulated in Table S1 and S2, ESI,† respectively.
Conclusions
We have successfully deposited GO onto the surface of Ag–TNTs using a simple impregnation method to synthesize GO–Ag–TNTs. The hydrophilic behaviour of GO enabled its deposition onto the surface of Ag or TNTs. Ag NPs with average size of 100 nm were deposited onto the surface of TNTs and inside the tubes. A series of characterization works including FTIR, XPS and XRD confirmed the presence of oxygenated groups in GO after the oxidation of graphite. PL spectra clearly portrait the excellent electron–hole pairs separation performance of TiO2 rendered by both Ag and GO deposition. The prepared composite photocatalyst displayed superior photocatalytic activity under visible light irradiation. A duality contribution was unveiled by the GO where it acts not only as an electron sink for 2-CP degradation but also as an electron-accepting mediator for MB degradation. GO–Ag–TNTs well acted upon 2-CP than MB with higher repeatability and stability. The photocatalytic degradation mechanism clearly shows that every specific pollutant has a unique mechanism. Thus it presents a new insight on the utilization of Ag and GO for efficient visible light driven photocatalytic systems for pollutants removal.Acknowledgements
This work was supported by the University of Malaya Research Grant (RG167/12SUS) and a Postgraduate Research Grant (PPP) (PV106/2012A).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- G. K. Mor, H. E. Prakasam, O. K. Varghese and C. A. Grimes, Nano Lett., 2007, 7, 2356 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 18946 CAS.
- Y. K. Lai, J. Y. Huang, H. F. Zhang, V. P. Subramaniam, Y. X. Tang, D. G. Gong, L. Sundar, L. Sun, Z. Chen and C. J. Lin, J. Hazard. Mater., 2010, 184, 855 CrossRef CAS PubMed.
- N. Wang, X. Li, Y. Wang, X. Quan and G. Chen, Chem. Eng. J., 2009, 146, 30 CrossRef CAS PubMed.
- O. K. Varghese, M. Paulose, T. J. LaTempa and C. A. Grimes, Nano Lett., 2009, 9, 731 CrossRef CAS PubMed.
- D. B. Kuang, J. Brillet, P. Chen, M. Takata, S. Uchida, H. Miura, K. Sumioka, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2008, 2, 1113 CrossRef CAS PubMed.
- K. Shankar, G. K. Mor, H. E. Prakasam, S. Yoriya, M. Paulose, O. K. Varghese and C. A. Grimes, Nanotechnology, 2007, 18, 065707 CrossRef.
- X. He, Y. Cai, H. Zhang and C. Liang, J. Mater. Chem., 2011, 21, 475 RSC.
- K. S. Mun, S. D. Alvarez, W. Y. Choi and M. J. Sailor, ACS Nano, 2010, 4, 2070 CrossRef CAS PubMed.
- B. K. Vijayan, N. M. Dimitrijevic, J. Wu and K. A. Gray, J. Phys. Chem. C, 2010, 114, 21262 CAS.
- D. Feng, Z. Rui and H. Ji, Catal. Commun., 2011, 12, 1269 CrossRef CAS PubMed.
- G. Yan, M. Zhang, J. Hou and J. Yang, Mater. Chem. Phys., 2011, 129, 553 CrossRef CAS PubMed.
- L. R. Hou, C. Z. Yuan and Y. Peng, J. Hazard. Mater., 2007, 139, 310 CrossRef CAS PubMed.
- S. F. Chen, S. J. Zhang, W. Liu and W. Zhao, J. Hazard. Mater., 2008, 155, 320 CrossRef CAS PubMed.
- A. Zielińska-Jurek, E. Kowalska, J. W. Sobczak, W. Lisowski, B. Ohtani and A. Zaleska, Appl. Catal., B, 2011, 101, 504 CrossRef PubMed.
- S. Mubeen, G. Hernandez-Sosa, D. Moses, J. Lee and M. Moskovits, Nano Lett., 2011, 11, 5548 CrossRef CAS PubMed.
- N. Chandrasekharan and P. V. Kamat, J. Phys. Chem. B, 2000, 104, 10851 CrossRef CAS.
- Z. F. Liu, Q. Liu, Y. Huang, Y. F. Ma, S. G. Yin, X. Y. Zhang, W. Sun and Y. S. Chen, Adv. Mater., 2008, 20, 3924 CrossRef CAS.
- J. Zhang, Z. Xiong and X. S. Zhao, J. Mater. Chem., 2011, 21, 3634 RSC.
- X. J. Lv, S. X. Zhou, C. Zhang, H. X. Chang, Y. Chen and W. F. Fu, J. Mater. Chem., 2012, 22, 18542 RSC.
- Y. Gao, X. Pu, D. Zhang, G. Ding, X. Shao and J. Ma, Carbon, 2012, 50, 4093 CrossRef CAS PubMed.
- T. Szabó, Á. Veres, E. Cho, J. Khim, N. Varga and I. Dékány, Colloids Surf., A, 2013, 433, 230 CrossRef PubMed.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058 CrossRef CAS PubMed.
- P. Song, X. Zhang, M. Sun, X. Cui and Y. Lin, Nanoscale, 2012, 4, 1800 RSC.
- G. Jiang, Z. Lin, C. Chen, L. Zhu, Q. Chang, N. Wang, W. Wei and H. Tang, Carbon, 2011, 49, 2693 CrossRef CAS PubMed.
- Y. Tang, S. Luo, Y. Teng, C. Liu, X. Xu, X. Zhang and L. Chen, J. Hazard. Mater., 2012, 241, 323 CrossRef PubMed.
- X. Pu, D. Zhang, Y. Gao, X. Shao, G. Ding, S. Li and S. Zhao, J. Alloys Compd., 2013, 551, 382 CrossRef CAS PubMed.
- C. Y. Park, T. Ghosh, Z. D. Meng, U. Kefayat, N. Vikram and W. C. Oh, Chin. J. Catal., 2013, 34, 711 CrossRef CAS.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. Wang, X. Li, J. Chen and X. Tao, Chem. Eng. J., 2012, 198, 547 CrossRef PubMed.
- X. Y. Zhang, H. P. Li, X. L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801 RSC.
- D. K. Pandey, T. F. Chung, G. Prakash, R. Piner, Y. P. Chen and R. Reifenberger, Surf. Sci., 2011, 605, 1669 CrossRef CAS PubMed.
- S. S. M. S. Arif, K. Zhang, A. R. Park, K. S. Kim, N. G. Park, J. H. Park and P. J. Yoo, Nanoscale, 2013, 5, 5093 RSC.
- C. Ren, B. Yang, M. Wu, J. Xu, Z. Fu, Y. lv, T. Guo, Y. Zhao and C. Zhu, J. Hazard. Mater., 2010, 182, 123 CrossRef CAS PubMed.
- Y. Wang, Y. Tang, Y. Chen, Y. Li, X. Liu, S. Luo and C. Liu, J. Mater. Sci., 2013, 48, 6203 CrossRef CAS.
- W. S. Wang, D. H. Wang, W. G. Qu, L. Q. Lu and A. W. Xu, J. Phys. Chem. C, 2012, 116, 19893 CAS.
- A. Fuerte, M. D. Hernández-Alonso, A. J. Maira, A. Martínez-Arias, M. Fernández-García, J. C. Conesa, J. Soria and G. Munuera, J. Catal., 2002, 212, 1 CrossRef CAS.
- J. Liu, Y. Sun and Z. Li, CrystEngComm, 2012, 14, 1473 RSC.
- Z. Ni, Y. Wang, T. Yu and Z. Shen, Nano Res., 2008, 1, 273 CrossRef CAS PubMed.
- C. Chen, W. M. Cai, M. C. Long, B. X. Zhou, Y. H. Wu, D. Y. Wu and Y. J. Feng, ACS Nano, 2010, 4, 6425 CrossRef CAS PubMed.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238 CrossRef CAS PubMed.
- M. Long, Y. Qin, C. Chen, X. Guo, B. Tan and W. Cai, J. Phys. Chem. C, 2013, 117, 16734 CAS.
- P. Wang, Z. G. Liu, X. Chen, F. L. Meng, J. H. Liu and X. J. Huang, J. Mater. Chem. A, 2013, 1, 9189 CAS.
- Y. X. Xu, H. Bai, G. W. Lu, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS PubMed.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 3, 2653 CrossRef CAS PubMed.
- S. Sakthive and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef PubMed.
- O. Akhavana, M. Abdolahad, Y. Abdib and S. Mohajerzadeh, Carbon, 2009, 47, 3280 CrossRef PubMed.
- T. Wu, X. Cai, S. Tan, H. Li, J. Liu and W. Yang, Chem. Eng. J., 2011, 173, 144 CrossRef CAS PubMed.
- P. Wang, Y. Zhai, D. Wang and S. Dong, Nanoscale, 2011, 3, 1640 RSC.
- T. Yeh, F. Chan, C. Hsieh and H. Teng, J. Phys. Chem. C, 2011, 115, 22587 CAS.
- J. Hensel, G. Wang, Y. Li and J. Z. Zhang, Nano Lett., 2010, 10, 478 CrossRef CAS PubMed.
- D. Lin, H. Wu, R. Zhang and W. Pan, Chem. Mater., 2009, 21, 3479 CrossRef CAS.
- T. F. Yeh, J. M. Syu, C. Cheng, T. H. Chang and H. S. Teng, Adv. Funct. Mater., 2010, 20, 2255 CrossRef CAS.
- T. F. Yeh, J. Cihlar, C. Y. Chang, C. Cheng and H. S. Teng, Mater. Today, 2013, 16, 78 CrossRef CAS PubMed.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577 CrossRef CAS PubMed.
- G. H. Moon, H. I. Kim, Y. S. Shin and W. Y. Choi, RSC Adv., 2012, 2, 2205 RSC.
- W. Fan, S. Jewell, Y. She and M. K. H. Leung, Phys. Chem. Chem. Phys., 2014, 16, 676 RSC.
- Z. Chen, L. Fang, W. Dong, F. Zheng, M. Shen and J. Wang, J. Mater. Chem. A, 2014, 2, 824 CAS.
- C. Su, L. Liu, M. Zhang, Y. Zhang and C. Shao, CrystEngComm, 2012, 14, 3989 RSC.
- Y. He, P. Basnet, S. E. Hunyadi Murph and Y. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 11818 CAS.
- P. Wang, Y. Tang, Z. Dong, Z. Chen and T. T. Lim, J. Mater. Chem. A, 2013, 1, 4718 CAS.
- H. Eom, J. Y. Jung, Y. Shin, S. Kim, J. H. Choi, E. Lee, J. H. Jeong and I. Park, Nanoscale, 2014, 6, 226 RSC.
- S. Min and G. Lu, J. Phys. Chem. C, 2011, 115, 13938 CAS.
- Z. Mou, Y. Dong, S. Li, Y. Du, X. Wang, P. Yang and S. Wang, Int. J. Hydrogen Energy, 2011, 36, 8885 CrossRef CAS PubMed.
- S. H. Kim and W. Y. Choi, J. Phys. Chem. B, 2005, 109, 5143 CrossRef CAS PubMed.
- Y. H. Wang, K. Hang, N. A. Anderson and T. Q. Lian, J. Phys. Chem. B, 2003, 107, 9434 CrossRef CAS.
- T. Tachikawa, S. Tojo, M. Fujitsuka and T. Majima, Langmuir, 2004, 20, 2753 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra of GO–Ag–TNTs before and after the adsorption of MB, photocatalytic degradation kinetic parameters and kinetic plots of first order and second order. See DOI: 10.1039/c3ta14857b |
| This journal is © The Royal Society of Chemistry 2014 |