
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-aqueous microgel particles: synthesis, properties and applications
J. A.
Bonham
a,
M. A.
Faers
b and
J. S.
van Duijneveldt
a
aUniversity of Bristol, School of Chemistry, UK
bBayer CropScience AG, Germany
First published on 20th October 2014
Abstract
Microgels are cross-linked polymer latex particles that can form stable colloidal dispersions. Their typical sizes range from 10 to 1000 nm and they can swell in response to their external environment (pH, temperature and solvency). This swelling behaviour is central to many potential applications for microgels. The existing literature is dominated by studies of the properties of aqueous microgel dispersions. In contrast, this review focusses on the development of microgel particles in non-aqueous systems, looking at the challenges of studying these particles as well as their swelling behaviour. The five main mechanisms of producing microgel particles will be discussed and examples of materials used for microgels that swell in non-aqueous solvents will be given. Finally some examples of applications for non-aqueous microgels are given.
1 Introduction
Microgels were first synthesised in 1935 by Staudinger and Husemann.1 The term microgel was not used until 1949![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 and since then microgels have been defined as cross-linked polymer latex particles, in the size range of 10–1000 nm, that form stable colloidal dispersions (Fig. 1). It is important to note that in the literature, a number of different terms are used for microgel, including microsphere,3 microbead,4 nanogel,5 and minigel for larger particles.6 For simplicity in this review we will only use the term microgel.
2 and since then microgels have been defined as cross-linked polymer latex particles, in the size range of 10–1000 nm, that form stable colloidal dispersions (Fig. 1). It is important to note that in the literature, a number of different terms are used for microgel, including microsphere,3 microbead,4 nanogel,5 and minigel for larger particles.6 For simplicity in this review we will only use the term microgel.
 | ||
| Fig. 1 A colloidal dispersion of microgel particles. | ||
Microgels have the ability to swell and de-swell in response to changes in the external environment, e.g. pH, temperature and solvency (see Fig. 2). The swelling force is balanced by the elastic forces of the cross-links, which hold the particle together and enable the particle to maintain its shape. This ability for microgels to change size depending on their environment is of particular interest in research and there are many different external stimuli that have been reported in the literature to influence the particle swelling in aqueous solvents, including temperature,7 pH8 and ionic strength.9 This multi responsive nature of microgels makes them ideal for a vast range of applications including photochemistry,10 biomedical applications,11 optical switches or sensors12 and in coating technologies.9
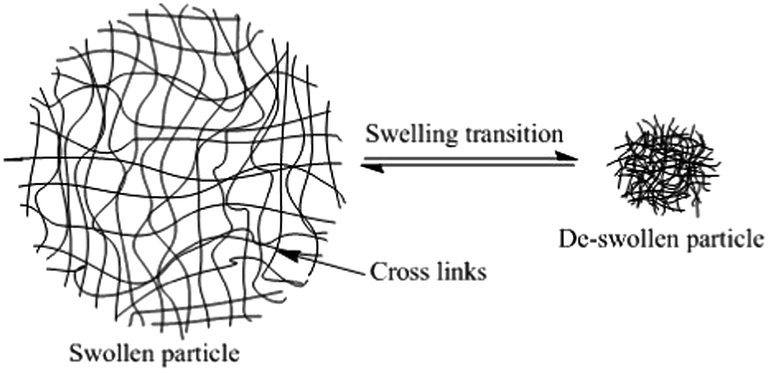 | ||
| Fig. 2 A swollen and de-swollen microgel particle. | ||
The majority of the development work in this field has focussed on aqueous microgels, where polymer latex particles are swollen and de-swollen in water in response to temperature and pH, for example poly(N-isopropylacrylamide) (p(NIPAM)) which has a known lower critical solution temperature (LCST) of 35 °C.7 These aqueous microgels have a number of different applications including in cosmetics,13 pharmaceuticals,11,13,14 catalysts,15 photochemistry10 and in optical switches or sensors.12
However, there is significantly less literature on the development of non-aqueous microgels, where microgel particles are swollen and de-swollen in organic solvents, such as toluene and benzene. Non-aqueous solvents are often preferred, or even essential for both academic and industrial research, for example to aid with chemical stability or performance. From an academic point of view, some model systems, for example model hard spheres, are easier to design in non-aqueous solvents. Non-aqueous solvents also offer opportunities to design colloidal systems, with properties such as refractive index and density matching which are important for scattering and imaging studies.16
Importantly, microgels in non-aqueous solvents have a number of differences to their water based counterparts and they face a number of challenges in their synthesis, stability and swelling. This review will discuss these differences and challenges as well as some of the specific applications of non-aqueous microgels.
2 Synthesis and materials
There are two ways of approaching the synthesis of non-aqueous microgels. The first way is to synthesise the microgel particles in water using emulsion polymerisation (EP),17 surfactant free emulsion polymerisation (SFEP) or microemulsion polymerisation (MEP), and then transfer the particles to an organic medium at the end of the reaction.18 This can be done in a number of different ways including: freeze drying,19 rotary evaporating or washing with an appropriate solvent (e.g. methanol).20 This method could in theory be applied to any type of microgel particle, however, this review will focus on literature where the particles have been transferred into organic solvents.The second way to produce microgel particles in non-aqueous solvents is to carry out the reaction directly in non-aqueous solvents via dispersion polymerisation (DP)18 or precipitation polymerisation (PP). However, making the particles directly in non-aqueous solvents presents some challenges as it is unknown if the particles will be swollen or de-swollen in the non-aqueous solvent during synthesis, and well-defined spherical particles do not always result.
The basic principles of these reaction mechanisms are shown in Table 1. There are also a number of other less common methods of microgel formation that will not be covered in this review, such as solution polymerisation,21 seeded polymerisation and combined mechanisms.22 Some advantages and disadvantages of the various polymerisation methods discussed are given in Table 2 and Table 3 gives an overview of the non-aqueous microgel synthesis.
| Namea | Initial systemb | Particle size/nm | Stabilisation | Ref. |
|---|---|---|---|---|
| a EP = emulsion polymerisation, SFEP = surfactant free emulsion polymerisation, DP = dispersion polymerisation, MEP = microemulsion polymerisation, PP = precipitation polymerization. b O/W E = oil-in-water emulsion, A = aqueous solution, O = organic solution, O/W ME = oil-in-water micro-emulsion, M = marginal solvents. | ||||
| EP | O/W E | 50–1000 | Surfactants | 17, 23 and 24 |
| SFEP | O/W E | 50–1000 | Charges on initiators | 19, 22 and 25 |
| DP | A or O | 1000–15000 |
Electrostatically (aqueous), sterically (non-aqueous) | 3, 9, 22, 26 and 27 |
| MEP | O/W ME | 10–100 | Surfactants | 20, 21, 28 and 29 |
| PP | M | 50–1000 | Low monomer content | 30–33 |
| Namea | Advantages | Disadvantages |
|---|---|---|
| a For abbreviations see Table 1. | ||
| EP | Easy to produce non-aqueous microgels | Surfactant needs removing after the reaction, non-uniform cross-linking occurs, can get core–shell morphologies |
| SFEP | No surfactant needed | Non-uniform cross-linking, difficult to do in aqueous medium |
| DP | Can be done in an organic or aqueous medium, the extent of reaction depends on the concentration of monomer | Cross-linking needs to be initiated after the particles have formed |
| MEP | Small particles produced, high yield achieved | Often needs a cosurfactant, have to remove surfactants after the reaction |
| PP | One step reaction, quick | Low yield due to low monomer content, need to find a good solvent |
| Monomera | Cross-linkera | Cross-link densityb mol% | Methodc | Initial solventa,d | Other solventsa,d | Diameter | Ref. |
|---|---|---|---|---|---|---|---|
| a For abbreviations see Table 4 and Fig. 6. b Expressed as a percentage of total monomer. c For abbreviations see Table 1, SP = seeded polymerization. d / = a mixture of more than one solvent. e For a full list of solvents used see ref. 37. f Comonomers were added at 10 wt% relative to NIPAM. | |||||||
| S | DVB | 7–70 | PP | AN, Tl | — | 1000–2500 nm | 3 |
| MEMA | EGDMA | 0.6–5 | DP | Hx | W | 120–720 nm | 9 |
| S | DIB | 0.02–5 | MEP | W | Tl | 39–168 nm | 20 |
| S | DVB | 0–55 | EP, DP | MEK/H | — | 1800–4600 nm | 34 |
| S | DVB | 0.09–3 | DP | W | EB | 403–1150 nm | 18 |
| MMA | EGDMA | 0.3–1.4 | SFEP, DP | W/A | — | 40–502 nm | 22 |
| S | DVB | 0–0.03 | EP | W | MEK, B | — | 23 |
| NIPAM, AA, BA, VL, VP, VBf | BIS | 6–7 | SFEP, DP | W | Hx, M | 300–500 nm | 19 |
| MMA, GMA, HEMA | EGDMA | 1–10 | MEP | W | BA | 40–100 nm | 29 |
| MA | DVB | 3–22 | PP | MEK/H | — | 100–500 nm | 30 |
| S | PETEA | 14–78 | PP | E | — | 580–750 nm | 31 |
| AC | DVB | 6–80 | PP | AN | — | 2300–3100 nm | 33 |
| MAA, EGMM | EGDMA | 15–75 | PP | MEK, H | — | 450–2090 nm | 35 |
| S, FS, MMA, CHMA | DIB | 0.6–5 | MEP | W | Tl | 9–106 nm | 36 |
| BMA, MMA, HEMA | AMA | 1–1.3 | EP | W | A, MiPKe | 109–235 nm | 37 |
| PS | DIB | 10–50 | EP, SFEP | W | EN, oDC | 56–185 nm | 38 |
2.1 Emulsion, surfactant free emulsion and microemulsion polymerisation
EP, SFEP and MEP are all polymerisation techniques used to produce microgels in non-aqueous solvents where the particles are originally made in water and are then transferred to organic solvents. MEP is one of the most common ways to produce microgels and produces particles in high yields. MEP also enables the particle size to be controlled and can make microgels between 5–100 nm.21The mechanism for these three types of polymerisation are very similar. In EP free radicals initiate chain polymerisation of monomers in the presence of an aqueous solution of surfactants. The surfactants create micelles in the aqueous phase which act as micro-reactors, limiting the size of the resultant microgel particles39 (Fig. 3). According to Harkins24,39 the mechanism can be split into three stages: particle formation, propagation and termination.
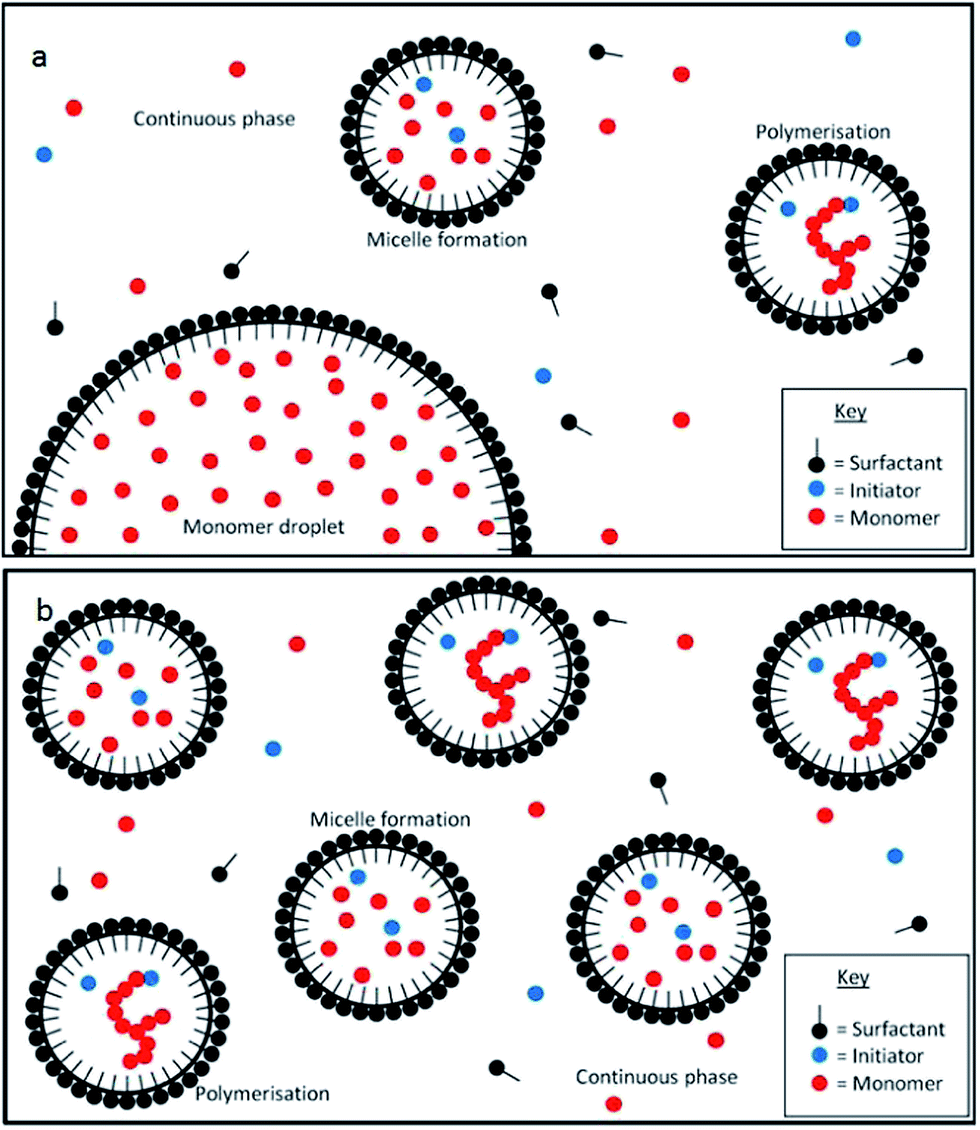 | ||
| Fig. 3 The mechanisms of emulsion polymerisation (a) and microemulsion polymerisation (b). | ||
There are a number of examples of non-aqueous microgels made via EP in the literature. In 1952 Cragg and Manson used EP to make PS and DVB microgels in aqueous solution and then transfer them to non-aqueous solvents, for example benzene or MEK. These particles were transferred to organic solvents via a solvent wash (Section 2.1.1). Machotova et al. used EP to make acrylic microgels in water and then transferred them to organic solvents, including acetone, MEK and MiPK (see ref. 37 for a full list). Whilst EP can successfully be used to produce microgels of various monomers in a number of different solvents, more recently EP has been largely replaced with SFEP and DP.
SFEP has a very similar mechanism to EP, however there is no surfactant added to the reaction mixture and the stabilisation is achieved from charges on the initiator molecules. This makes SFEP more difficult to carry out in non-aqueous solutions as charges in non-aqueous solvents are often very weak, there are a few examples though where SFEP has been used to make NIPAM microgels with additional co-monomers that swell in non-aqueous solvents. Camli et al. used SFEP to create PMMA microgels cross-linked with EGDMA in a mixture of water and acetone. Increasing the acetone content initially made the particle size increase as a reduction in dielectric constant reduced the particle stability during nucleation. An acetone content >20% made the particle size decrease due to increased monomer solubility that dominated the dielectric constant effect.22 Nur et al. used SFEP to produce p(NIPAM) microgels co-polymerised with a number of different monomers including: AA, BA, VP, and VL, the particles were re-dispersed in hexane and methanol.19
In MEP the droplets at the beginning of the reaction are much smaller than EP or SFEP and there are no monomer droplets present, but only micelles.21 In order to make these droplets small enough, the oil–water interfacial tension (γ) needs to be reduced to a few mN m−1. This is not always possible using only one surfactant due to the onset of micellisation and consequently a co-surfactant, or an alcohol, is often needed. Fig. 3 illustrates the mechanisms of MEP and EP.
Whilst MEP is one of the most common methods of producing microgels, there are still conflicting reports on its mechanism. One questionable characteristic of a microemulsion is its fragility. If a microemulsion is unstable towards slight thermodynamic changes, then the system may not remain in the same phase during the whole reaction process. Such systems are said to be fragile and consequently, the system is not always a microemulsion during polymerisation.40 In contrast, sturdy microemulsions would maintain their place in the phase diagram throughout the whole reaction and are important for successful MEP synthesis.
Antonietti et al. used MEP to produce PS and DIB microgel particles in toluene with a size of around 100 nm. Nie et al. used MEP to prepare a number of different microgels and then transferred them to non-aqueous solutions and determined their swelling ratio. They used MMA with a cross-linker of EGDMA as well as two different functional monomers, GMA and HEMA. Increasing the HEMA content increased particle aggregation due H-bonds between the particles. They also looked at a number of different surfactants and co-surfactant systems, including sodium dodecyl sulfate (SDS) and sodium bis(2-ethylhexyl) sulfosuccinate (AOT), they found that increasing the surfactant content increased the particle diameter and swelling.29
Whilst it can be seen that EP, SFEP and MEP can all be used to produce microgels in non-aqueous solvents, none of these mechanism are without limitations and it is important that the appropriate mechanism is chosen for any given microgel preparation.
The first method to transfer particles to organic solvents is freeze drying, where the particles are frozen and then subjected to a reduced pressure, causing any frozen water in the particles to sublime. Once the particles are in this dried state they can then be re-dispersed in a variety of organic solvents. For example Nur et al. used freeze drying to dry p(NIPAM) microgels that were co-polymerised with AA, BA, VP and CL and re-disperse them in hexane and methanol.19 Likewise Saunders and Vincent freeze dried PS and DVB microgels and re-dispersed them in ethylbenzene. These particles were also centrifuged five times to remove any un-cross-linked polystyrene.41
The second method to transfer microgels to organic solvents is to use a solvent wash. Here the particles are washed with an excess solvent that will cause them to coagulate. They can then be filtered and re-washed. The particles can then be dried down in a vacuum oven to ensure that all the water has been removed. Once in a dried state the particles can then be re-dispersed in a number of solvents. Cragg and Manson used this method on PS and DVB microgels, where excess ethanol was used to wash the particles.23 Antonietti et al. precipitated out PS and DVB microgels in an excess of hot methanol, the particles were then re-dispersed in THF. This process was repeated three times before the particles were dried down in a vacuum oven to form a white powder.36
The major drawback with both of these methods is that the particles need to be fully dried down before they are re-dispersed in non-aqueous solvents. This can be problematic as often the particles can irreversibly aggregate during the drying or alternatively, water may still be present inside the particles, both of which prevent re-dispersion. A further problem arises if the surfactant has not been fully removed. Vigorous shaking can be used to break up any aggregates that form during the drying process.19 For highly cross-linked microgels, shaking is not always sufficient to break up these aggregates and these particles may not re-disperse in non-aqueous solvents.23 Allowing the microgels to equilibrate for at least 24 hours before any swelling measurements are taken is important to ensure the particles are fully dispersed and swollen;19 centrifuging has also been used to ensure the microgels can be well re-dispersed.41 Nonetheless, often the drying and re-dispersing process can be unpredictable, causing additional challenges in the synthesis of non-aqueous microgels.
2.2 Dispersion and precipitation polymerisation
DP and PP are both ways of making microgel particles directly in non-aqueous solvents. DP is named after the milky white dispersion that is formed at the end of the reaction and can produce particles in the size range of 1000–15000 nm;3,27 DP can also be carried out in aqueous mixtures. DP was originally developed in the 1960's and made large scale production of microgels much more attainable. This led to the increased interest in these particles in the 1990's.26
DP consists of two stages, nucleation and growth. In nucleation radicals are initiated and then form oligomeric radicals via propagation. When the concentration of these radicals reaches a critical level, they form micelles. These micelles are stabilised electrostatically and solubilise the monomer; henceforth the inside of the micelles becomes the locus of polymerisation. The particle number and size distribution are determined in this first step. The second stage is the growth of the particles, which is a much longer and more robust stage.26 As the particles form, a “milky” dispersion results and the reaction only terminates when all the monomer has been consumed. The remaining radicals then favour reaction with each other and the reaction ends. However, because DP is often carried out in organic media, electrostatic stabilisation is not effective due to the low dielectric constant.27 Therefore, in order to stabilise non-aqueous DP suspensions, soluble polymer chains are grafted onto the microgels to provide steric stability to the particles by preventing coagulation.
Polymer grafting can often lead to flocculation or an increased particle size distribution when the cross-linker is added. Thus, the cross-linking needs to take place after the particle has formed, otherwise the grafted polymer responsible for the particle's stabilisation will be incorporated into the interior of the cross-linked polymer and the particles will coagulate. Alternatively, adding the cross-linker in stages or carefully choosing the solvent are ways to overcome this problem and produce cross-linked microgels by DP.26
Butun et al. used DP to produce multi-responsive MEMA and EGDMA microgels in n-heptane. The particles were sterically stabilised and ranged between 120–720 nm in diameter.9 Kobayashi and Senna used DP to produce core–shell microgels where the core was a poly(styrene) latex with ethanol and methoxyethanol as the solvent and poly(acrylamide) as the stabiliser. Seeded polymerisation was then used to create a poly(styrene) and divinylbenzene cross-linked shell. When these particles were swollen in toluene or MEK, it was seen that increasing the DVB content decreased the swelling ratio.42
DP can also be carried out in an aqueous medium. One of the most documented cases of preparing a PS and DVB latex in water is to use aqueous DP in the absence of an emulsifier. However, difficulties can arise in this mechanism when the latexes are transformed to non-aqueous solutions due to polar groups on the surface of the particles.18
In PP, the microgel polymerisation is carried out in a marginal solvent, such that the monomer is soluble in the solvent, but the polymeric material formed is insoluble and consequently precipitates out of the solution. This mechanism does not require use of external stabilisers to prevent the particles from aggregating. Instead, the stabilisation comes from a low monomer content in the reaction mixture. One further advantage of this mechanism is that it can be performed in one single step and therefore is quicker than some of the other methods described above.
Jiang et al. used PP to produce monodisperse cross-linked microgels. Ethanol was used as the solvent with a co-monomer mix of S and PETEA. Particles were produced in a size range of 580–750 nm,31 but only when the monomer content was between 1 and 2 vol%. At any higher concentrations a coagulum was formed and at lower concentrations no product was formed at all.31 Frank et al. looked at the different morphologies of particles produced from PP of DVB and MA as the MEK volume fraction increased. They concluded that microgels were produced when MEK was between 70–80 vol%. From these studies they determined a mechanism for the production of microgels using electron microscopy, viscosity and light scattering, Fig. 4, and distinguished how they are different from microspheres.30
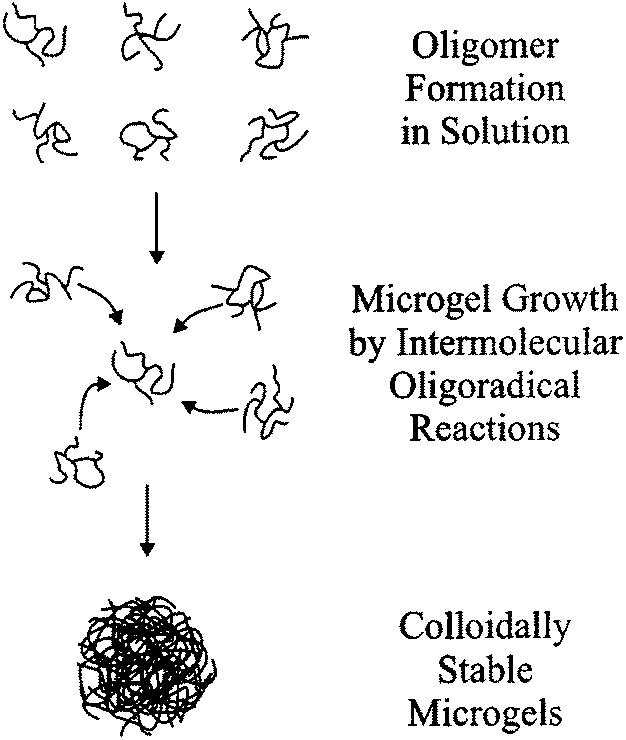 | ||
| Fig. 4 The PP mechanism of microgel formation in 80% MEK proposed by Frank et al. (reprinted with permission from ref. 30. Copyright (2002) American Chemical Society). | ||
Cui et al. made highly cross-linked PS and DVB microgels in AN in a size range of 1000–2500 nm and they showed that increasing the monomer content increased the size of the particles produced.3 Cui et al. produced particles in a blend of AN and Tl and found that increasing the Tl content increased the particle size. When the Tl content was over 50 vol%, a macrogel was formed, due to the increase in solvency of the solvent mixture with increasing Tl content.3 Li and Stover made highly cross-linked microgels using DVB in AN. They also looked at using blends of solvents and found that only mixtures of AN and n-butanol or n-propanol led to microgels and all other solvents formed either: gels, coagulum, or non-spherical particles. Once the particles were made Li and Stover found the particles did not swell in any common solvent.32
Downey et al. formed DVB and 4-methyl-styrene microgels via PP using solvent mixtures of MEK and heptane, the morphologies formed due to the difference in solvency were discussed in some detail and are given in Section 4. Goh and Stover look at PP in mixtures of MEK and heptane and how the solvency affected the morphology of the particles; they found that decreasing the solvency caused the particles to change from microgels to microspheres.35 They used polar monomers such as MAA and EGMM and an EGDMA cross-linker and found that microgels were only formed when the EGDMA content was between 4 and 25 vol%.35 They also found that varying the ratio of acid:ethylene oxide groups had a significant impact on the morphology of the particles formed.35
A wide variety of examples of PP can be found in the literature and often the solvency of the system used greatly affects the particles that are formed. Often a decrease in solvency leads to more coagulation in the system, which can prevent microgels forming. Additionally the monomer content can affect the size of the particles and often a high monomer content will prevent any particles from forming. The exact details of each polymerisation depend on the system used and each polymerisation can be specifically tailored to match the desired properties of the particles.
2.3 Materials used
Table 4 and Fig. 6 show the variety of materials that have been used to make non-aqueous microgels. The functionalities of the monomer and cross-linker have a major influence on the particle's properties as does the solvent the particle is both made in and swollen in. Additionally, the initiators used, as well as any surfactants or stabilisers used in the synthesis affect the particle's properties.| Abbreviations | Solvents |
|---|---|
| A | Acetone |
| AN | Acetonitrile |
| B | Benzene |
| BA | Butyl acrylate |
| E | Ethanol |
| EB | Ethylbenzene |
| EN | 2-Ethyl naphthalene |
| H | Heptane |
| Hx | Hexane |
| oDC | ortho-Dichlorobenzene |
| M | Methanol |
| MEK | Methyl ethyl ketone |
| MiPK | 3-Methyl-2-butanone (isopropyl methyl ketone) |
| MOE | Methoxyethanol |
| Tl | Toluene |
| W | Water |
The choice of monomer in a microgel preparation is vital in determining the resultant particle's properties. The monomer determines the χ parameter, which has a major influence on the swelling ratio of the particles (Section 4.2) and can also add additional properties into the particles such as conductivity and functional groups which govern how the particle will respond to changes in the environment. One of the major advantages of microgel particles is the ease at which the properties can be altered using the monomers and co-monomers and consequently a huge library of microgels have been seen in the literature.
Styrene is one of the most common monomers used for non-aqueous microgels which could be due to its easy handling and ability to form spherical, amorphous particles.43 Additionally, there are a number of known solvents that will swell styrene and there is a wealth of information available in the literature. Styrene can be bought from a number of suppliers and often includes an inhibitor such as 4-tert-butylpyrocatechol which needs to be removed via purification. A number of other monomers have also been used successfully in non-aqueous microgel preparation.
Butun et al. used MEMA to make microgel particles that could swell in hexane and Camli et al. used MMA to make microgel particles that were swollen in a mixture of water and acetone.22 The addition of functional monomers, such as MMA, HEMA29 and MMA alter the surface properties of the particle and it has been shown that MMA can also act as a co-surfactant.44 Nur et al. made NIPAM microgels, which typically only swell in water, however with the addition of co-monomers, such as AA, BA and VL, the particles were shown to swell in methanol.19 It is also possible to use monomers to make microgels in organic medium conductive using poly(aniline) and dodecylbenzenesulfonic acid as monomers.45
The cross-linker in the particle is important, as the cross-link density controls the swelling ratio of the particles. There is less variety in the cross-linkers used and DVB is by far the most common cross-linker used to make non-aqueous particles. It is often purchased commercially as a mixture 55% DVB and 45% ethylstyrene. Other cross-linkers seen in the literature include EGDMA,34 DIB18 and BIS, which is commonly used in conjunction with NIPAM.19 Jiang et al. used PETEA as a cross-linker, which has four functional cross-linking sites so will be able to cross-link four different chains together at once.31
There are differences in the literature as to how much cross-linker is needed before a microgel will form. Jiang et al. noticed that when the XLD was less than 14 mol% relative to total monomer the yield of product made was very low.31 It is unusual that particles would not be made until 14 mol% as there are reports in the literature of particles being made with a XLD as low as 0.01 mol%;23 this difference may be due to the functionalities of the cross-linkers. Cragg and Manson claimed that microgel particles made with 0.0006 mol% DVB were very similar in both appearance and solubility to pure PS latex particles and that only when the XLD was increased to 0.01 mol% did the particles begin to act as microgels.23 Clarke and Vincent made PS and DVB microgels with 0.09 mol% DVB and Camli et al. made MMA microgels with 0.3 mol% EGDMA. The lowest possible XLD needed to form a microgel is reasonably low and of the order of 0.01 to 0.1 mol%, yet the exact number is a topic that is not agreed on in the literature and depends on, amongst other things, the functionality of the cross-linker and the mechanism of polymerisation.
The initiator used in a microgel preparation can affect the mechanism and properties of the particles, for example, it can affect the locus of polymerisation, the yield and the particle size. Additionally, the functionality of the initiator often influences the surface properties and charges on the particles which plays a large role in determining how well the particles can be re-dispersed in organic solvents. There are a number of initiators used in the literature and the majority of initiators used are anionic initiators, including AIBN, a hydrophobic initiator which forms radicals in the hydrophobic phase of EP.34,9 Potassium persulfate (KPS) is another common example of an anionic initiator. However, KPS is hydrophilic, and consequently the radicals formed from KPS are in the aqueous phase.23 Benzyl peroxide initiators have also been used.42
Nair used two different initiators, 4,4′-azobis(4-cyanovaleric acid) (ACVA) and KPS, in the preparation of acrylic microgels. The particles containing ACVA had weakly acidic surfaces and low surface charge densities, consequently they could be re-dispersed in a low dielectric solvent e.g. heptane, whereas particles made with KPS had strongly acidic surfaces with high charge surface densities which did not re-disperse in heptane.46
The solvent used during particle synthesis can affect the size, shape and stability of the particles that are made. This is especially true for polymerisation (PP) and dispersion polymerisation (DP) mechanisms, where the solvent plays a very important role in the mechanism of microgel formation. Downey et al. looked at how varying the solvency of DVB microgels affected the structure that was formed. MEK was used as the good solvent and heptane as the poor solvent with the resulting morphologies shown in Fig. 5. Microspheres were produced when the MEK content was between 33–20 vol%. When the MEK content was higher than 33 vol% a macrogel was formed, and when the MEK content was lower than 20 vol% a coagulum was formed.34
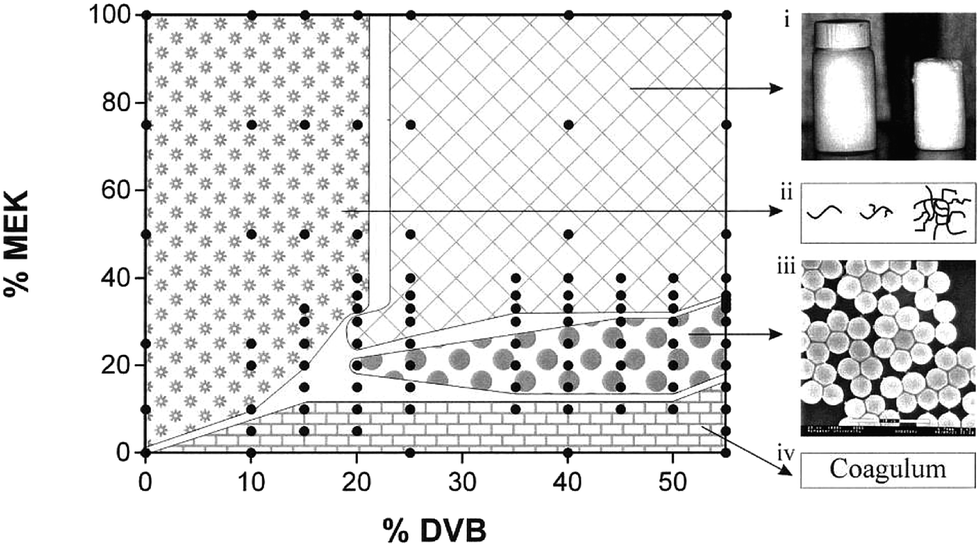 | ||
| Fig. 5 Morphology map for polymerisations reported by Downey et al. The 4 vol% monomer feed contained from 0 to 55% DVB. The solvent compositions for the polymerisations were varied from neat MEK to neat heptane. Four distinct phases are represented: (i) space-filling macrogels; (ii) soluble polymer; (iii) microgels; (iv) coagulum (reprinted with permission from ref. 34. Copyright (2001) American Chemical Society). | ||
Frank et al. also looked at varying the solvency during PP of DVB and alt-maleic anhydride microgels and showed that coagulum formed under the poorest solvency conditions and macrogels formed when the MEK content was highest (highest solvency).30 Therefore, the resulting morphology is a complex combination of the swelling and the inter-particle forces.
Consequently, it is possible to design microgel particles with the exact properties required by suitable choice of starting materials. The monomer will affect the swelling of the particles as well as any functional groups in the particles. The initiator can influence the surface properties of the particle which will govern how the particles respond to external stimuli and the cross-linker will control the cross-link density as well as the extent of homogeneous cross-linking within the particle.
3 Stability
The first question surrounding microgels in non-aqueous solvents is how they are stabilised to form colloidal dispersions. The stability of a colloidal suspension depends on three factors: the electrostatic repulsion, the van der Waals attraction and the steric repulsion.47In general the DLVO theory is used to describe the stability of a colloidal suspension as it describes the first two of these terms.48 In non-aqueous systems the scope for charge stabilisation is much more limited than in aqueous systems. Non-aqueous solvents have a low concentration of ions due to their low dielectric constants, although charged particles in non-aqueous solvents are not unheard of, with the first example seen in the 1950s.49 Charge control agents, such as surfactants have been used to successfully add charge to non-aqueous solvents49 and a number of techniques have been used to confirm the existence of long range electrostatic interactions in non-aqueous solvents, including atomic force microscopy, surface force analysis and optical tweezers.
Whilst it has been shown that charges can be important in non-aqueous solvents and often DLVO theory is applicable, in general non-aqueous colloids require steric stabilisation to form a dispersion. For microgels this steric repulsion is governed by the number of polymer chains that extend into the solvent from the particle itself, which is often determined by how homogeneously the microgel swells (Section 4.1).50 Often microgels have a “hairy layer” caused by inhomogeneous cross-linking, protruding into the solvent and resulting in a repulsive interaction between the particles due to a loss in entropy when the protruding polymer chains become compressed. A number of different surfactants and stabilisers have been used to aid the stability of non-aqueous microgel dispersions. For some mechanisms, such as microemulsion polymerisation (MEP), the choice and concentration of surfactant impacts the size, morphology and stability of the resultant particles.29,40 On the other hand, surfactant free emulsion polymerisation (SFEP) does not require any surfactants but relies on initiator molecules to stabilise dispersions.22
4 Swelling
Unlike conventional (un-cross-linked) latex particles, microgels can be swollen under suitable solvent conditions, yet retain their integrity. Swelling occurs when the conditions (for example, temperature) of the microgel particles change towards better solvent conditions for the polymer, causing the chains within the microgel to move away from each other. This “swelling force” is balanced by the elastic force of the cross-linking agents that hold the microgel particle together.41 Therefore, the particle does not de-form when exposed to a swelling force, but it holds its shape and simply swells.The most common way to swell non-aqueous microgel particles is to vary the solvency. Solvents that swell the microgel particles are said to be good solvents whereas solvents that collapse the particles are poor solvents. As discussed below, there is often a gradual change between these two conditions, with the theta point, where χ (the Flory–Huggins parameter) is 0.5, being the nominal boundary between the two regions. There are a number of non-aqueous solvents which have been shown to swell various microgels, and these are listed in Table 3. The initial solvent describes the solvent that the particles were synthesised in and other solvents describes the solvent(s) the particles were transferred to after synthesis (if applicable).
For convenience of synthesis, microgels are often made in poor solvents, where they are un-swollen, and then transferred into good solvents where the particles swell. For example, Saunders and Vincent made poly(styrene) and divinylbenzene (PS and DVB – see Table 4 and Fig. 6 for abbreviations throughout) microgels in water, freeze dried them and then transferred them to ethylbenzene, a good solvent for PS. A 4.5 fold increase in cross-linker concentration led to a more than 2 fold increase of the swelling ratio in ethylbenzene, as shown by electron microscopy and dynamic light scattering.41
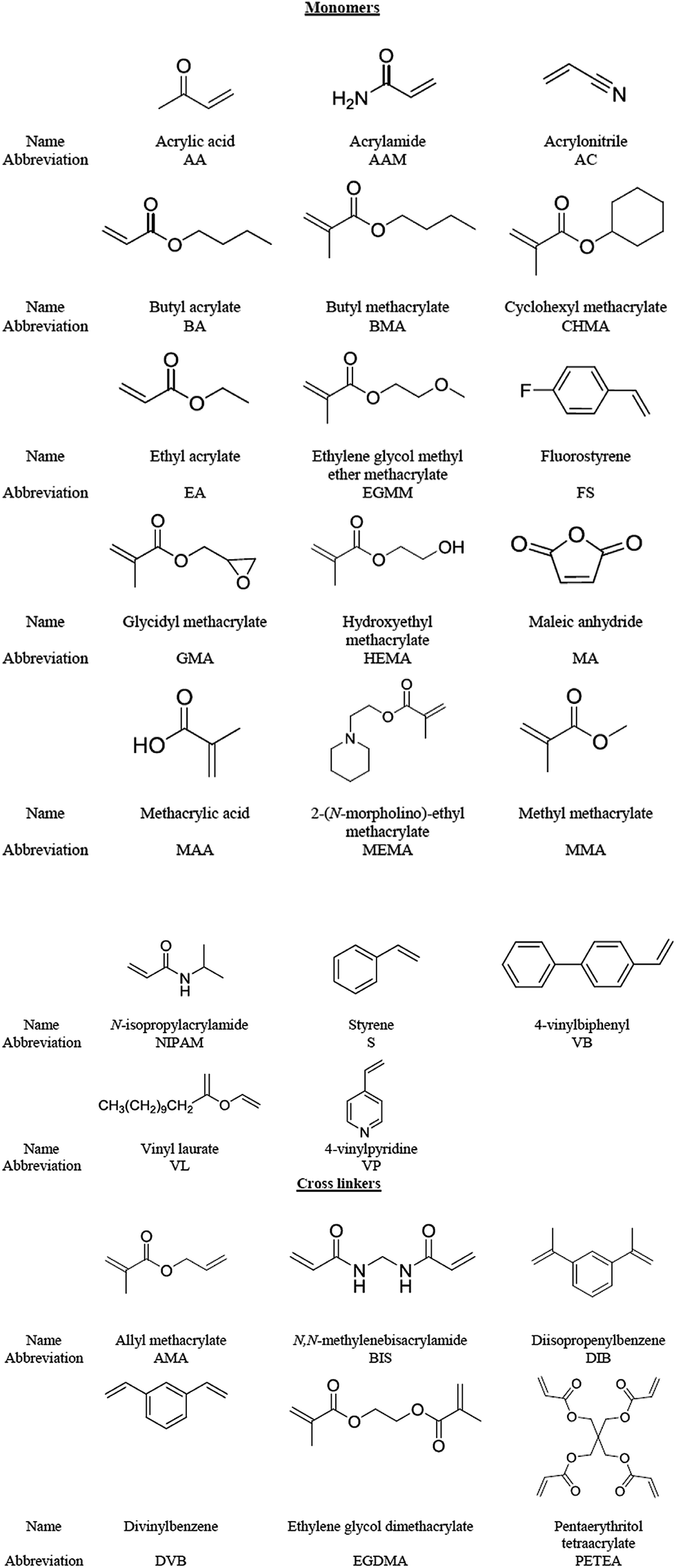 | ||
| Fig. 6 Examples of the different monomers and cross-linkers used in non-aqueous microgels, including their abbreviations and structures. | ||
Butun et al. made multi-responsive microgels particles using MEMA and EDGMA. They showed that the particle swelling was responsive to temperature, pH and ionic strength in aqueous media and that the particles also swelled in response to different solvents, demonstrating the possibilities to control the swelling in non-aqueous systems through the choice of solvents. Kobayashi and Senna swelled PS and DVB microgels in MEK and showed that increasing the DVB content reduced the swelling. At sufficiently large concentrations of DVB, there was a sufficient amount of cross-linker to prevent swelling altogether.42 Nur et al. made microgels using p(NIPAM) with different co-monomers and showed that the particles dispersed in methanol but precipitated when the particles were dissolved in hexane;19 they claim this is a result of the hydrophilic nature of the p(NIPAM)-based microgels.
Machotova et al. made acrylic microgels from MMA, BMA and HEMA co-monomers and an AMA cross-linker. The particles were swollen in a number of organic solvents, including acetone, MEK and MiPK (see ref. 37 for a full list). They found that acetone was the strongest swelling solvent and that MiPK was the weakest solvent, and they attributed this difference to the solubility parameters (δ), where higher δ values gave higher swelling ratios. The solubility parameters are one way to estimate the χ parameter for particles in solvent:51
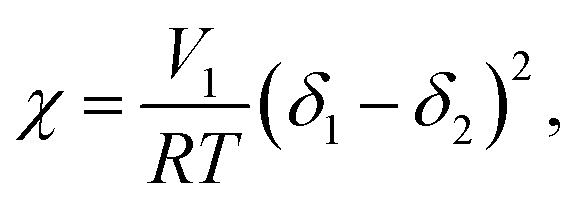 | (1) |
The degree of swelling of a microgel particle can also be affected by other components present in solution, especially polymers (as used in order to generate depletion interactions between the microgel particles52). The depletion interaction for hard particles by free polymer is well understood, however, for microgel particles where the particles are deformable and penetrable, the mechanism is not as well understood and often an osmotic de-swelling is observed.41,53 The chemical potential of the solvent surrounding the microgel particle must equal the chemical potential of the solvent inside the particle and one possible explanation for the de-swelling is that the increased osmotic pressure outside the microgel is counter-balanced by expulsion of solvent from inside the microgel. It can be seen that microgels in non-aqueous solvents have been shown to swell in response to solvency and osmotic pressure, however there are few reports of microgel particles in non-aqueous solvents swelling in response to other stimuli such as temperature. This is in contrast to microgels in aqueous solvents where changing the temperature of the solution can alter the size of the particles.
Swelling makes microgel particles uniquely valuable to many applications (Section 5). However, swelling also produces a number of challenges in determining and describing the properties of microgel particles. Firstly, the properties will depend on how swollen the particles are, which will change depending on the environment of the particles. Secondly, a microgel particle with a high swelling ratio will consist of a polymer network embedded in a large number of solvent molecules; thus microgels behave less like hard spheres as their swelling increases. Most commonly, the size of a swollen microgel is characterised by its hydrodynamic radius, as probed by dynamic light scattering.
The swelling of a microgel particle can be described by its swelling ratio, which can be expressed empirically as a ratio of hydrodynamic radii of swollen (rHs) and collapsed (rHc) particles, eqn (1). Equivalently, a volume swelling ratio can be used, eqn (2). In general (especially for aqueous microgels), particles may still contain solvent in the collapsed state. However, for a system of hydrophobic particles, such as PS microgels, it is assumed that particles in water are fully collapsed with no solvent present and that particles in non-aqueous solvents are swollen. With this in mind, Q will refer to the swelling ratio as defined below throughout the review.
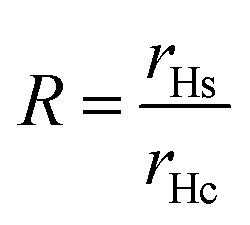 | (2) |
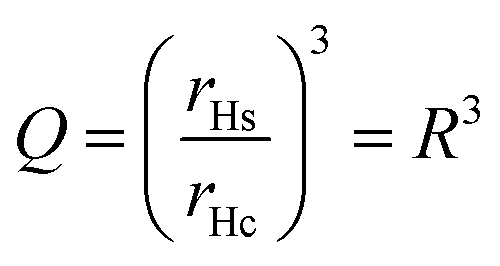 | (3) |
4.1 Is swelling homogeneous?
One of the major challenges for characterising and modelling microgel particles is that often the particle swelling is inhomogeneous. For particles in the collapsed state, a description assuming a homogeneous sphere should be appropriate. However, it is common that the cross-link density decreases with the distance from the particle centre, due to the difference in reaction kinetics between the cross-linker and monomers. Therefore, for the swollen state this assumption may not be appropriate.12One of the most common techniques used to analyse microgel particles is small angle neutron scattering (SANS) which allows the determination of a number of properties of microgels such as the molecular weight and particle size as well as more detailed structural properties such as the average chain conformation, the mesh size and the sizes of both the cores and shells of particles.54 Data analysis provides structural information on the particles and for microgels the model used in this analysis needs to take into account the properties of the specific microgel particle, such as the extent of inhomogeneous cross-linking. Furthermore, soft microgels can deform and penetrate at high concentrations and the structure factor S(q) which describes the particle interaction, needs to take this into consideration. For good quality scattering data, it is possible to obtain the radial segment density profile. If the particle is homogeneous, then this will be a box like density profile, however it is more likely that the cross-link density is inhomogeneous.14
Holley et al. used SANS to study PS and DVB microgels in deuterated toluene and THF and determined three models for the particles based on their cross-link densities (XLD) and synthesis routes. They found that particles made via a batch microemulsion preparation (MEP) (see Section 2.1 for details on synthesis routes) with a XLD <10 mol% had a “fuzzy gel” shape. These particles had a well defined core with homogeneous cross-linking, surrounded by an inter-facial layer made of dangling chains and loops. However, particles made via semi-batch MEP, where the monomer is added to the reaction continuously, were better modelled as smooth gels. These gels, with an XLD of 4.6 mol% had no significant “fuzzy layer”. Particles with a XLD >10 mol% and made by batch MEP were best modelled as dendritic gels, where there is no homogeneous core and a small mesh size (Fig. 7).54
 | ||
| Fig. 7 Different types of PS and DVB particles depending on their cross-link density and polymerisation technique. (a) Fuzzy gel, XLD = 0.8 mol% batch MEP, (b) smooth gel, XLD = 4.6 mol%, semi-batch MEP (c) dendritic gel, XLD = 11 mol% batch MEP (reprinted from Polymer, 55, D.W. Holley, M. Ruppel, J.W. Mays, V.S. Urban and D. Baskaran, Polystyrene nanoparticles with tunable interfaces and softness, 58–65, Copyright (2014), with permission from Elsevier). | ||
Bradley et al. used quantum dots (QDs) to investigate how the distribution of cross-links affects the swelling of PS microgel particles in chloroform. When the particles were only partly swollen the QDs only penetrated the periphery of the particle, with smaller pores in the centre of the particle preventing further penetration. At higher swelling ratios the QDs entered into the whole particles, showing that the spread of cross-links governs the swelling of the particles.55 Scheffold et al. argued that thermo-sensitive NIPAM microgels can be described as having a dense core surrounded by a brush like shell.12 These brushes form as a result of the inhomogeneous cross-linking of the particle and provide steric stability for the particles. Whilst Scheffold et al. used this theory on aqueous microgels it could still apply for non-aqueous microgels and therefore steric stability will always play a role in microgel particles irrespective of whether they are aqueous or non-aqueous. Antonietti et al. claim that PS and DIB microgel particles swell homogeneously.20 They use the P ratio:
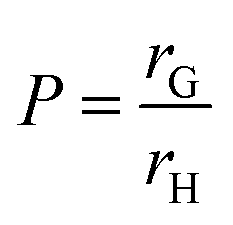 | (4) |
It can be seen that the extent of inhomogeneous cross-linking in a microgel depends on the polymerisation and the cross-link density of the particle and has a great influence on how the particles will swell. It is commonly expected that most particles will have some extent of inhomogeneous swelling, with the inner most regions of the microgel being less swollen than the outer regions. This makes modelling microgels difficult as their properties change not only between solvents but also within the particle.
4.2 Flory–Rehner theory
One way to describe the swelling of a microgel is to use the Flory–Rehner theory (FRT) which is a theoretical model used to describe the swelling in (macroscopic) polymer gels and is an extension of the phase theory for polymer solutions.51 It is the earliest model for the swelling of polymer gels and was first discussed in 1943 by Flory and Rehner;56 it is simple and applicable to many different situations57 and is often used to estimate the cross-link density of a polymeric system.58 Whilst it is originally designed for a macroscopic gel, the theory can apply for all polymer networks, including microgels. The FRT is not the only theory that can be used to describe the swelling of a gel, with other theories including the Flory–Wall theory20 and the rubber elasticity theory.The main limitations of the FRT are that it neglects a number of factors regarding the structure of the gel; as previously mentioned most microgels do not have homogeneous cross-linking, a factor which is not considered in the FRT. Changes in the entropy and/or enthalpy of mixing of the cross-linker are also not considered in the theory, which for large cross-linkers may be a poor assumption. Nonetheless, it is one of the oldest and most well known theories and it should be applicable in particular to microgels in non-aqueous solvents, as it does not take into account any electrostatic interactions or hydrogen bonding which are considered to be irrelevant for these systems.
The key result of the FRT is given in eqn (5):
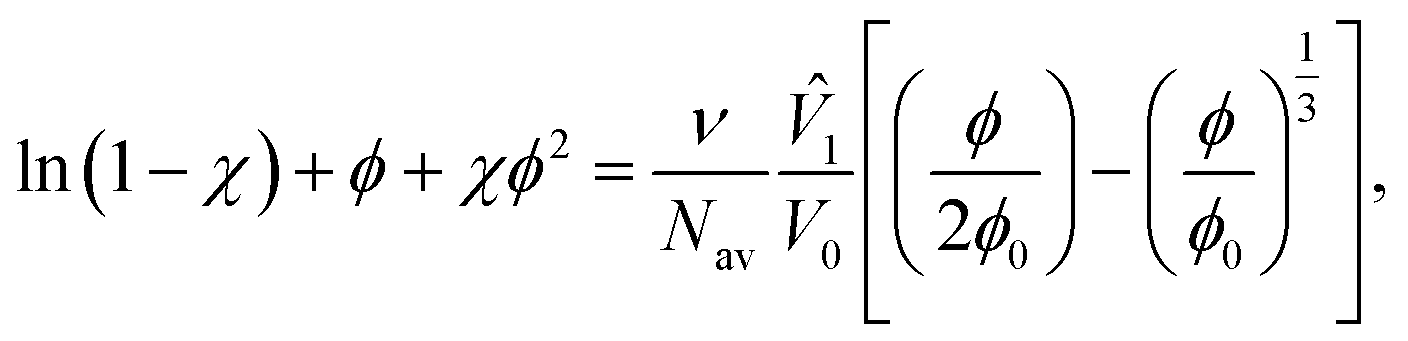 | (5) |
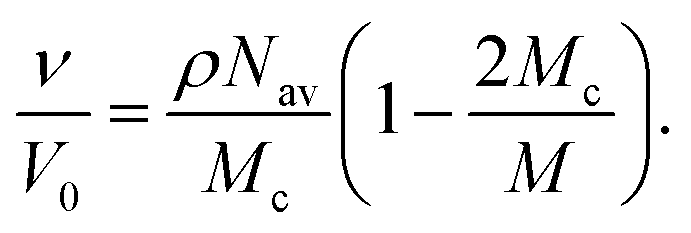 | (6) |
In these expressions, φ is the volume fraction of polymer in the swollen particle, φ0 is the volume fraction of polymer in the reference state and χ is the Flory–Huggins parameter, Mc is the average molar mass between cross-links and ![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) 1 is the solvent molar volume. Nav is Avagadro's number, V0 is the volume of the network in the reference state, M is the molar mass of the pre-networked chain, ρ is the density of the polymer and v is the number of elastically active strands. For a full derivation of the FRT see ref. 51.
1 is the solvent molar volume. Nav is Avagadro's number, V0 is the volume of the network in the reference state, M is the molar mass of the pre-networked chain, ρ is the density of the polymer and v is the number of elastically active strands. For a full derivation of the FRT see ref. 51.
The reference state from eqn (5) is a topic of debate in the literature with two main arguments being used to describe this state.57 Khokhlov gives a theoretical argument that the reference state is given at the θ condition,59 from a more practical point of view the reference state is given by the synthesis conditions.60 If we take the reference state to be that of the polymer in the synthesis conditions, and there is no solvent present in the collapsed particles, it can be said that φ0 = 1 and that Q = 1/φ. This would not be the case for hydrophilic particles, however, for PS and DVB microgels that are synthesised in water and transferred to non-aqueous solvents this assumption stands and will thus be used in the following discussion.
The FRT highlights three key parameters in the swelling behaviour of gels:
1. The average molar mass between cross-links, Mc.
2. The solvent molar volume, 1.
3. The solvent quality, expressed as the χ parameter.
Any variation of microgel size with temperature would mainly arise through a change of χ. Croll illustrates how these parameters influence the swelling ratio (see Fig. 8) by solving eqn (5) and calculating φ, the equilibrium volume fraction of polymer in the network. Experimentally, a large variation in χ such as the one depicted in Fig. 8 would be difficult to achieve in non-aqueous solvents, and solvent blends would be the best way to achieve such extreme values for χ.
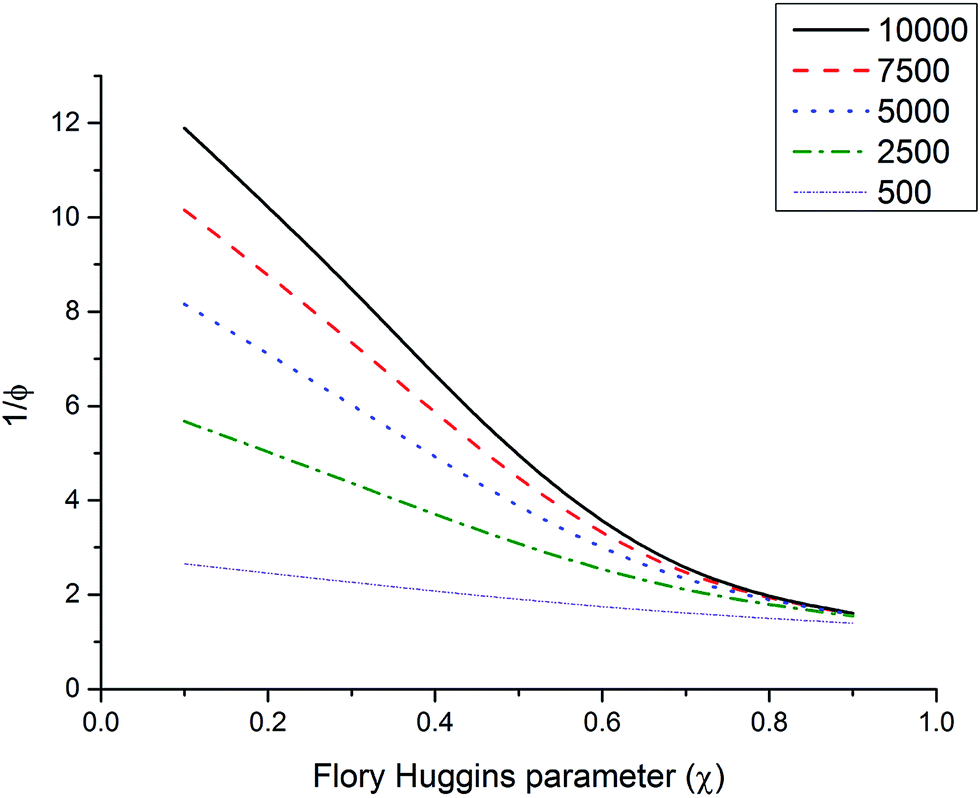 | ||
| Fig. 8 The equilibrium volume fraction of polymer in the network as a function of the Flory–Huggins parameter and molar mass between cross-links. ρ = 1.2 g cm−1 and 1 = 63 cm3 mol−1. Re-drawn from ref. 58. | ||
The FRT was originally designed for use on macroscopic polymer gels and there are many examples of this in the literature. However, there are few detailed results available on testing this theory for non-aqueous microgel systems.61 In order to test this theory for non-aqueous microgels, eqn (5) was used, using the same method as in ref. 58. The system studied was PS and DVB microgels in toluene at 25 °C. The calculations were compared to experimental swelling data of these systems from ref. 20 and 36 (Fig. 9). Mc was calculated using eqn (7).
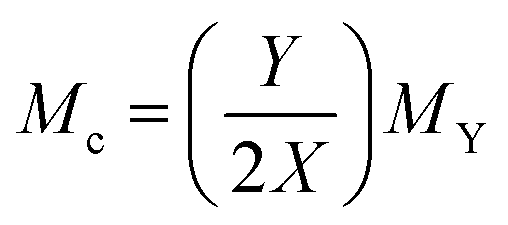 | (7) |
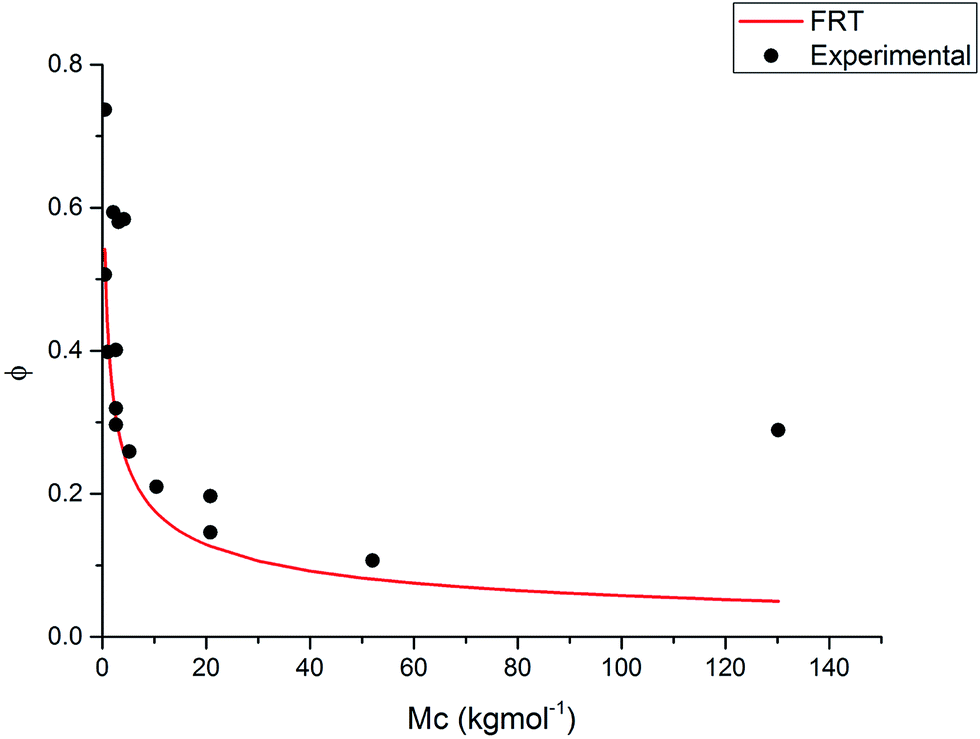 | ||
| Fig. 9 The equilibrium volume fraction of polymer in the network of PS and DVB microgels in toluene at 25 °C over a range of molar masses between cross-links cross (Mc). Experimental data taken from ref. 20 and 36 (•). The line shows the theoretical predictions. χ = 0.4 (ref. 62) and 1 = 105.91 cm3 mol−1. | ||
Where Y is the number of moles of monomer, X is the number of moles of cross-linker and MY is the molar mass of the monomer.
The theory predicts that as Mc increases the equilibrium volume fraction decreases and the majority of experimental data fit along this trend. In Fig. 9, the largest deviation from this prediction is at a very high Mc where the equilibrium volume fraction is much higher than predicted. At very low Mc values, the theory also deviates from the experimental data, with the experimental data having a higher equilibrium volume fraction than expected. Thus, for this case it can be said that the FRT is not applicable towards the extremities of Mc values. Consequently, more comparisons need to be made using a wider range of particles and solvents before any finalised statements about how applicable the FRT is to non-aqueous microgel systems can be made.
5 Applications of non-aqueous microgels
Microgels in non-aqueous solvents have been used for a variety of applications and their size and shape makes them particularly desirable. Microgels are highly adaptable and can be tuned to achieve particular properties that can be utilised in various applications. The most common application of non-aqueous microgels is in coatings technologies, but there is also a wide range of literature on a variety of additional applications of microgels in non-aqueous solvents, including rheology modifiers and model systems.5.1 Coatings technology
One of the first commercial uses for microgel particles was in both solvent and water based coatings where the majority of microgels are used to modify the rheological behaviour of paint formulations. Microgels are particularly adapted to this application as they are easily modified and applicable to a large number of coatings.63 Today microgels are often made of regulatory approved monomers and are thus easy to incorporate into formulations in accordance with legislations.63Highly cross-linked, viscous, non-swellable microgels make good binders in organic coatings, especially in high solids paint formulations.63 Ishikura et al. used microgels to produce novel binders for coatings containing linear polymers. Adding microgels to the coatings increased the maximum film thickness and improved sagging properties. Furthermore, the microgel influenced the rheology and tensile strength of the film properties.64
Highly swellable microgels are also incorporated into paints as rheology modifiers.63 They can give pseudoplastic behaviour, meaning they have a high low shear viscosity and a low high shear viscosity. Consequently, the paint can be stored without pigments settling to the bottom and directly sprayed without thinning down.63 Microgels are also used as fillers in paints to improve the film forming properties of the paint. Furthermore they have been reported to reinforce cured paint films and improve their mechanical properties63 as well as weather and shock resistance.65
5.2 Rheology modifiers
A very common application of microgels in non-aqueous solvents is as rheology modifiers, particularly in paints and coatings, Section 5.1. However, they are also used as additives in a number of other systems in order to customize the rheology. The unique swelling properties also allow microgels to be used for high performance needs such as engine and engineering oils where high temperature rheological modifications are essential.66When microgels are swollen they take up a sufficiently large amount of space and become closely packed. This results in strongly interacting particles that have an increased viscosity, where the shear viscosity of the system is dominated by the microgel swelling and effective volume fraction of the microgels.67 EA and MAA particles are often used to thicken non-aqueous systems, where the viscosity is governed by the particle size and the cross-link density. Aggregated particles, or non-spherical particles, also increase the viscosity of the system.47
Electro-rheology can be used to tune the rheology of a system using an electric field. The particles, in a non-conducting fluid, will align with an electric field, resulting in a loss of fluidity and an increase in rheology.47 Xie et al. studied electro-rheology of an MMA colloidal suspensions in a number of non-aqueous media including paraffin oil and poly(diethylsiloxane).68 Combining such systems with microgels may offer new and interesting rheological behaviours including control of the structuring and relaxation times of the network.
5.3 Model systems
Non-aqueous microgels are of particular interest in academia as model systems. They enable the study of fundamental colloidal properties because their Hamaker constants can be controlled by their swelling ratios.69 Microgels are highly tunable and therefore have potential applications in the study of glass transition temperature and phase behaviour.12For detailed, real-space studies on colloids, the hard-sphere model is a key reference system. In order to allow such studies, refractive index and density matching are desired. To achieve this with solid polymer particles, a dense (halogenated) solvent may be required, but unexpected charging effects have been found with such systems.70 Non-aqueous microgels offer a different route to generating model systems as they may contain a significant fraction of solvent, their density and refractive index is close to the solvent by construction.
Microgels in non-aqueous solvents have always been important as model systems; Sieglaff was one of the first to demonstrate a depletion induced phase separation on the addition of free polymer using microgels in non-aqueous solvents71,72 and since then numerous studies on these systems have been conducted. For example, Clarke and Vincent observed a reversible flocculation of PS microgels in EB on addition of free linear PS at the critical polymer/solvent volume fraction. Decreasing the molecular weight of the free PS or increasing the average segment concentration in the swollen microgel particles increases the value of this critical volume fraction.18 They compared their experimental data to the VLW (Vincent, Luckham and Waite) theory which allows for inter-penetration of the free polymer coils with the periphery of the microgel particles and found a good agreement.18
Bartsch et al. used microgels to deliberately introduce a “squishy” particle into their systems. They looked at the interactions of PS and BA microgels cross-linked with DIB and EGDMA in various solvents. They concluded that the interactions were influenced by the cross-link density, particle size and swelling ratio of the microgels. The swelling ratio determines how compressible the microgel particles are at high volume fractions, with higher cross-link densities needed to produce hard sphere behaviours at high volume fractions.65
Additionally a number of studies on PS and DIB microgels in EN have been used to further understand the glass transition dynamics of this system. Bartsch et al. looked at the phase behaviour of two different PS and DIB microgel particles in a good solvent (EN). They showed that particles with a cross-link density of 50 mol% acted as hard spheres whereas particles with a cross-link density of 10 mol% were much softer. Nonetheless, the glass transition dynamics of both sets of particles could be described by mode coupling theory.73
Eckert and Bartsch studied the effect of adding free polymer to a solution of PS and DIB microgels in EN. They showed that the free polymer caused a depletion interaction via short ranged attraction, and this attraction increased the strength of density fluctuations and melted the colloidal glass. However, as they increased the strength of these short ranged attractions the system re-froze; they called this the re-entrant glass transition.74
Microgels can also be used as model systems for macrogels.20 Macroscopic gels have complicated topological structures that can affect their structural and dynamical properties. However, very little is known about how the topology contributes to the network behaviour of gels and therefore, quantitative data on gels is very difficult. Microgels are much easier to handle and purify than macroscopic gels and much more is known about their topology. Consequently, their dynamic and structural properties can be understood better than for macroscopic gels.20 Zillessen and Bartsch used PS microgels which were functionalised with benzophenone groups in organic solvents to produce clusters up to 2 μm in size which can be permanently fixed using photo-cross-linking. These structures can then be analysed in detail, providing information on aggregation and crystal nucleation.75
5.4 Photonic crystals
A further application of microgels is in photonic crystals, which are periodic dielectric materials on a nano scale. They affect the movement of photons in the same way that semi-conductors affect electrons; only letting certain wavelengths of light propagate through their structure. Disallowed bands of light are often referred to as photonic band gaps.76 Photonic crystals have applications in many photonic devices due to their ability to control the flow of light. Examples include in chemical sensors and in micro-lasers.76One way to produce photonic crystals is through self assembly of colloidal particles, such as microgels.76 The resulting colloidal crystals have the advantage for this application that they are simple to produce and cost effective. Furthermore, the colour of the colloidal crystal can be changed through the microgel swelling by infiltrating the particles with different organic solvents, e.g. methanol, ethanol and silicone.77
5.5 Molecular imprinting
Another use of non-aqueous microgels is molecular imprinting. Molecular imprinting is a way of creating a cavity within a polymer matrix that will selectively bind to certain molecules. Fig. 10 shows a scheme for the synthesis of molecular imprinted polymers. Firstly a template molecule is bound to functional molecules via non-covalent bonds, these functional molecules are then covalently linked to the polymer. The template is then removed and a cavity is left which has a specific shape and functionality and will therefore bind selectively to certain molecules.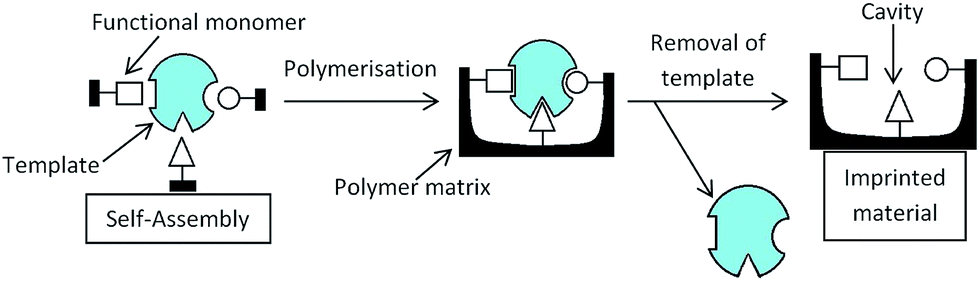 | ||
| Fig. 10 Mechanism of molecular imprinting for cross-linked polymers. This can also be applied to microgel particles. | ||
The advantage of using microgel particles rather than polymers for molecular imprinting is that artificial enzymes can be produced, as microgels are on the same size scale as enzymes.77 Other advantages include their low solution viscosities and their easy characterisation using standard techniques.
Biffis et al. used MMA and EGDMA as monomers to produce molecular imprinting microgels in a number of solvents including cyclohexanone, cyclopentanone and N,N-dimethylformamide. Once the microgels were imprinted, they were tested for their ability to selectively bind to one sugar enantiomer over another. Molecular imprinting is commonly used with non-aqueous microgels as many template molecules used in molecular imprinting are water sensitive.77
5.6 Other applications of non-aqueous microgels
There are a number of other applications of non-aqueous microgel particles that have not yet been mentioned. Firstly, Dyab and Atta used functionalised NIPAM-co-2-acrylamido-2-methylpropane sulfonic acid microgels to stabilise non-aqueous emulsions. At the interface, microgels behave more like linear polymers or proteins than normal solid particles as they can be deformed. They adopt a flattened morphology that prevents the drops from coalescing, even at low surface coverages.79 This has a great impact on the emulsion as the macroscopic properties of an emulsion are determined by the way the microgels adsorb at the interface. These emulsions have a high storage stability and a number of possible applications including pharmaceutical and cosmetic applications as well as nano-reactors of polymerisation of water-sensitive monomers or catalysts.78Microgels are also widely used in oil recovery; where oil and gases are recovered from the earth's crust. Large drills are used to reach the areas where the fuels are stored and fluids are used to push the oil and gas out of the rocks. It is important that during this process there is no uncontrolled release or unwanted loss of oil or gas and therefore the fluids used in this process are carefully engineered and designed to function at the high pressures and temperatures encountered underground.
A large proportion of the oil and gas is stored in tiny pores inside the rock and large pressures are applied to the rock using fluids to create passages in the rocks and reach the stores of oil. Microgels are used because they can enter these pores and then swell in the rock and minimise the loss of fluid. The microgels can then de-swell over time and be released from the rock when the hydrocarbons are recovered.14 Oil recovery often uses aqueous fluids, however, sometimes non-aqueous fluids are also needed, and therefore so are non-aqueous microgels. The microgels needed for this application need to be thermodynamically stable under conditions of high temperature and pressure and often monomers with carbonyl, ether or ester groups are used.14
6 Conclusions
Aqueous microgels can easily be prepared, in a benign solvent, and above all are useful and interesting because of convenient triggers for swelling/deswelling, such as temperature and pH. Non-aqueous microgels are less straightforward to work with in all three respects.There are 5 main mechanisms used to produce microgel particles in non-aqueous solvents, all of which have drawbacks. Making particles in water, and transferring them to non-aqueous solvents introduces a number of additional challenges regarding re-dispersing them without particle aggregation. Making the particles directly in non-aqueous solvents requires very careful consideration of the monomers and solvents used. Arguably one of the major challenges in microgel particle synthesis is deciding which method and materials to use as these can all play a vital role in controlling the properties of the resulting particles, in particular the size and swelling.
Despite its shortcomings, Flory–Rehner theory for the thermodynamics of swollen polymer gels provides a basis for understanding the swelling behaviour of microgel particles, and arguably should be best suited to non-aqueous suspensions. The theory highlights the role of cross-link density, solvent molar volume and polymer solvency (χ parameter) in controlling the extent of swelling. The role of cross-link density was shown to be described well by FRT. In order to achieve swollen microgels, a good solvent and a small molecular volume are required. This rather restricts what polymer/solvent combinations can be used, and known combinations have been reviewed here.
Microgels in non-aqueous solvents have only been shown to swell in response to solvency and osmotic pressure from polymers in solution. Large Bjerrum lengths in non-aqueous solvents prevent charges playing a role, at least inside the microgels. There are ways to promote charges in these solvents and this is one way to potentially introduce more swelling triggers in non-aqueous solvents. Any swelling or de-swelling that is seen in non-aqueous solvents is often much more gradual than swelling in aqueous solvents as the χ parameters have a much smaller variation. This is unlike aqueous systems where a volume phase transition temperature can result in a sharp transition from the particles being swollen to unswollen. Polymer solvent combinations allowing a significant variation of solvency would be especially promising for further study and work in this direction is currently underway on our laboratory.
Despite all this, there are a number of applications already of non-aqueous microgels, mainly in coatings technology, where they are often incorporated into formulations to help control the rheology. Non-aqueous microgels have also been used for photonic crystals, molecular imprinting and in model systems. However, if convenient model systems could be developed showing a (de)swelling transition in response to a convenient trigger, such as temperature, or light perhaps, this would open up opportunities for new applications. Examples would be controlled release/drug delivery applications (of water soluble or sensitive compounds), control of suspension structure and flow behaviour, and responsive photonic materials.
Acknowledgements
We thank Prof. B. Vincent for a critical reading of the manuscript. We would also like to thank Bayer CropScience AG and the EPSRC for the provision of a research studentship.References
- H. Staudinger and E. Husemann, Ber. Dtsch. Chem. Ges., 1935, 68, 1618–1634 CrossRef.
- W. Baker, Ind. Eng. Chem., 1949, 41, 511–520 CrossRef CAS.
- H. Cui, H. Chen, R. Qu, C. Wang, C. Sun, W. Zhou, M. Yu and H. Jiang, J. Appl. Polym. Sci., 2009, 111, 3144–3149 CrossRef CAS.
- Z. Y. Ding, S. M. Ma, D. Kriz, J. J. Aklonis and R. Salovey, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 1189–1194 CrossRef CAS.
- P. B. Zetterlund, N. Alam and M. Okubo, Polymer, 2009, 50, 5661–5667 CrossRef CAS PubMed.
- P. Dowding, B. Vincent and E. Williams, J. Colloid Interface Sci., 2000, 221, 268–272 CrossRef CAS PubMed.
- K. Bayliss, J. S. van Duijneveldt, M. A. Faers and A. W. P. Vermeer, Soft Matter, 2011, 7, 10345–10352 RSC.
- J. Yin, D. Dupin, J. F. Li, S. P. Armes and S. Y. Liu, Langmuir, 2008, 24, 9334–9340 CrossRef CAS PubMed.
- V. Butun, A. Atay, C. Tuncer and Y. Bas, Langmuir, 2011, 27, 12657–12665 CrossRef CAS PubMed.
- N. Sasa and T. Yamaoka, Adv. Mater., 1994, 6, 417–421 CrossRef CAS.
- C. Supasuteekul, A. H. Milani, J. M. Saunders, S. Lally, T. Freemont and B. R. Saunders, Soft Matter, 2012, 8, 7234–7242 RSC.
- F. Scheffold, P. Diaz-Leyva, M. Reufer, N. Ben Braham, I. Lynch and J. L. Harden, Phys. Rev. Lett., 2010, 104, 128304 CrossRef.
- M. G. Gichinga, S. Striegler, N. A. Dunaway and J. D. Barnett, Polymer, 2010, 51, 606–615 CrossRef CAS PubMed.
- Microgel Suspensions: Fundamentals and Applications, ed. A. Fernandez-Nieves, H. M. Wyss, J. Mattsson and D. A. Weitz, Wiley-VCH, Weinheim, 2011, pp. 117–132 Search PubMed.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- C. P. Royall, W. C. K. Poon and E. R. Weeks, Soft Matter, 2013, 9, 17–27 RSC.
- D. Zou, V. Derlich, K. Gandhi, M. Park, L. Sun, D. Kriz, Y. D. Lee, G. Kim, J. J. Aklonis and R. Salovey, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1909–1921 CrossRef CAS.
- J. Clarke and B. Vincent, J. Chem. Soc., Faraday Trans., 1981, 77, 1831–1843 RSC.
- H. Nur, M. J. Snowden, V. J. Cornelius, J. C. Mitchell, P. J. Harvey and L. S. Benee, Colloids Surf., A, 2009, 335, 133–137 CrossRef CAS PubMed.
- M. Antonietti, W. Bremser and M. Schmidt, Macromolecules, 1990, 23, 3796–3805 CrossRef CAS.
- W. Funke, O. Okay and B. Joos-Muller, Adv. Polym. Sci., 1998, 136, 139–234 CrossRef.
- S. T. Camli, F. Buyukserin, M. S. Yavuz and G. G. Budak, Colloids Surf., A, 2010, 366, 141–146 CrossRef CAS PubMed.
- L. H. Cragg and J. A. Manson, J. Polym. Sci., 1952, 9, 265–278 CrossRef CAS.
- W. D. Harkins, J. Am. Chem. Soc., 1947, 69, 1428–1444 CrossRef CAS.
- J. Goodwin, J. Hearn, C. Ho and R. Ottewill, Colloid Polym. Sci., 1974, 252, 464–471 CAS.
- J. S. Song and M. A. Winnik, Macromolecules, 2005, 38, 8300–8307 CrossRef CAS.
- K. Barrett, Dispersion Polymerisation in Organic Media, John Wiley and Sons, New York, 1975 Search PubMed.
- M. Antonietti and C. Rosenauer, Macromolecules, 1991, 24, 3434–3442 CrossRef CAS.
- L. X. Nie, W. Jiang, W. L. Yang, C. C. Wang and S. K. Fu, J. Macromol. Sci., Part A: Pure Appl.Chem., 2005, 42, 623–631 CrossRef PubMed.
- R. Frank, J. Downey, K. Yu and H. Stover, Macromolecules, 2002, 35, 2728–2735 CrossRef CAS.
- H. Y. Jiang, H. Chen, Y. Liang and X. Liu, Polym. Adv. Technol., 2011, 22, 1555–1562 CrossRef CAS.
- K. Li and H. Stover, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3257–3263 CrossRef CAS.
- X. Lu, D. Huang, X. Yang and W. Huang, Polym. Bull., 2006, 56, 171–178 CrossRef CAS.
- J. Downey, G. McIsaac, R. Frank and H. Stover, Macromolecules, 2001, 34, 4534–4541 CrossRef CAS.
- E. Goh and H. Stover, Macromolecules, 2002, 35, 9983–9989 CrossRef CAS.
- M. Antonietti, W. Bremser, D. Muschenborn, C. Rosenauer, B. Schupp and M. Schmidt, Macromolecules, 1991, 24, 6636–6643 CrossRef CAS.
- J. Machotova, J. Snuparek, Z. Cernosek and L. Svoboda, Prog. Org. Coat., 2008, 62, 71–78 CrossRef CAS PubMed.
- E. Bartsch, T. Eckert, C. Pies and H. Sillescu, J. Non-Cryst. Solids, 2002, 307, 802–811 CrossRef.
- A. L. Lovell and M. S. El-Aasser, Emulsion Polymerisation and Emulsion Polymers, John Wiley and Sons, Chichester, 1997 Search PubMed.
- M. Antonietti, R. Basten and S. Lohmann, Macromol. Chem. Phys., 1995, 196, 441–466 CrossRef CAS.
- B. R. Saunders and B. Vincent, Colloid Polym. Sci., 1997, 275, 9–17 CAS.
- K. Kobayashi and M. Senna, J. Appl. Polym. Sci., 1992, 46, 27–40 CrossRef CAS.
- J. Goodwin, R. Ottewill and R. Pelton, Colloid Polym. Sci., 1979, 257, 61–69 CAS.
- M. Antonietti, S. Lohmann and C. Van Niel, Macromolecules, 1992, 25, 1139–1143 CrossRef CAS.
- Y. B. Kim, J. K. Choi, J. A. Yu and J. W. Hong, Synth. Met., 2002, 131, 79–85 CrossRef CAS.
- M. Nair, Prog. Org. Coat., 1992, 20, 53–61 CrossRef CAS.
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- J. Mewis and N. Wagner, Colloidal Suspension Rheology, Cambridge University Press, New York, 2012 Search PubMed.
- R. Kemp, R. Sanchez, K. J. Mutch and P. Bartlett, Langmuir, 2010, 26, 6967–6976 CrossRef CAS PubMed.
- J. B. Thorne, G. J. Vine and M. J. Snowden, Colloid Polym. Sci., 2011, 289, 625–646 CAS.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC Press, New York, 2007 Search PubMed.
- H. N. W. Lekkerkerker and R. Tuinier, Lecture Notes in Physics 833. Colloids and the Depletion Interaction, Springer, 2011 Search PubMed.
- A. Fernandez-Nieves, A. Fernandez-Barbero, B. Vincent and F. J. de las Nieves, Prog. Colloid Polym. Sci., 2000, 115, 134–136 CAS.
- D. W. Holley, M. Ruppel, J. W. Mays, V. S. Urban and D. Baskaran, Polymer, 2014, 55, 58–65 CrossRef CAS PubMed.
- M. Bradley, N. Bruno and B. Vincent, Langmuir, 2005, 21, 2750–2753 CrossRef CAS PubMed.
- P. J. Flory and J. Rehner, J. Chem. Phys., 1943, 2, 512–520 CrossRef PubMed.
- M. Quesada-Perez, J. Alberto Maroto-Centeno, J. Forcada and R. Hidalgo-Alvarez, Soft Matter, 2011, 7, 10536–10547 RSC.
- S. G. Croll, J. Coat. Technol. Res., 2010, 7, 49–55 CrossRef CAS.
- A. Khokhlov, Polymer, 1980, 21, 376–380 CrossRef CAS.
- B. Sierra-Martin, J. Lietor-Santos, A. Ferandez-Barbero, T. Nguyen and A. Ferandez-Nieves, in Microgel Suspensions: Fundamentals and Applications, ed. J. M. A. Fernandez-Nieves, H. M. Wyss and D. A. Weitz, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, ch. Swelling Thermodynamics of Microgel Particles, pp. 73–117 Search PubMed.
- S. Patel, C. Malone, S. amd Cohen, J. Gillmor and R. Colby, Macromolecules, 1992, 25, 5241–5251 CrossRef CAS.
- Polymer Handbook, ed. J. Brandrup and E. H. Immergut, John Wiley and Sons, 3rd edn, 1989 Search PubMed.
- D. Saatweber and B. Vogt-Birnbrich, Prog. Org. Coat., 1996, 28, 33–41 CrossRef CAS.
- S. Ishikura, K. Ishii and R. Midzuguchi, Prog. Org. Coat., 1988, 15, 373–387 CrossRef CAS.
- E. Bartsch, S. Kirsch, P. Lindner, T. Scherer and S. Stolken, Bunsen-Ges. Phys. Chem., Ber., 1998, 102, 1597–1602 CrossRef CAS.
- A. Fessenbecker, P. Galda, T. Ziser, T. Fruh and W. Obrecht, US 8119581, 2012.
- M. Wolfe, Prog. Org. Coat., 1992, 20, 487–500 CrossRef CAS.
- H. Xie, J. Guan and J. Guo, J. Appl. Polym. Sci., 1995, 58, 951–956 CrossRef CAS.
- G. Kaggwa, M. Carey, C. Such and B. Saunders, J. Colloid Interface Sci., 2003, 257, 392–397 CrossRef CAS.
- A. I. Campbell, V. J. Anderson, J. S. van Duijneveldt and P. Bartlett, Phys. Rev. Lett., 2005, 94, 208301 CrossRef.
- C. Sieglaff, J. Polym. Sci., 1959, 41, 319–326 CrossRef CAS.
- H. N. W. Lekkerkerker and R. Tuinier, in Lecture Notes in Physics 833. Colloids and the Depletion Interaction, ed. W. Beihlbock, J. Ehlers, K. Hepp and H. Weidenmuller, Springer, 2011, ch. Historical Overview on Depletion, pp. 15–38 Search PubMed.
- E. Bartsch, T. Eckert, C. Pies and H. Sillescu, J. Non-Cryst. Solids, 2002, 307–310, 802–811 CrossRef CAS.
- T. Eckert and E. Bartsch, Phys. Rev. Lett., 2002, 89, 125701 CrossRef CAS.
- A. Zillessen and E. Bartsch, Langmuir, 2010, 26, 89–96 CrossRef CAS PubMed.
- S. Kim, Y. Gon Seo, Y. Cho, J. Shin, S. Chul Gil and W. Lee, Bull. Korean Chem. Soc., 2010, 31, 1891 CrossRef CAS.
- A. Biffis, N. Graham, G. Siedlaczek, S. Stalberg and G. Wulff, Macromol. Chem. Phys., 2001, 202, 163–171 CrossRef CAS.
- A. K. F. Dyab and A. M. Atta, RSC Adv., 2013, 3, 25662–25665 RSC.
- F. Pinaud, K. Geisel, P. Masse, B. Catargi, L. Isa, W. Richtering, V. Ravaine and V. Schmitt, Soft Matter, 2014, 10, 6963–6974 RSC.
| This journal is © The Royal Society of Chemistry 2014 |