 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surfactant or block copolymer micelles? Structural properties of a series of well-defined n-alkyl–PEO micelles in water studied by SANS
Thomas
Zinn†
a,
Lutz
Willner
a,
Reidar
Lund
*b,
Vitaliy
Pipich
c,
Marie-Sousai
Appavou
c and
Dieter
Richter
a
aJülich Centre for Neutron Science JCNS and Institute for Complex Systems ICS, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: l.willner@fz-juelich.de
bDepartment of Chemistry, University of Oslo, Postboks 1033 Blindern, 0315 Oslo, Norway. E-mail: reidar.lund@kjemi.uio.no
cJülich Centre for Neutron Science JCNS, Forschungszentrum Jülich GmbH, Outstation at MLZ, Lichtenbergstraße 1, 85747 Garching, Germany
First published on 17th April 2014
Abstract
Here we present an extensive small-angle neutron scattering (SANS) structural characterization of micelles formed by poly(ethylene oxide)-mono-n-alkyl ethers (Cn–PEOx) in dilute aqueous solution. Chemically, Cn–PEOx can be considered as a hybrid between a low-molecular weight surfactant and an amphiphilic block copolymer. The present system, prepared through anionic polymerization techniques, is better defined than other commercially available polymers and allows a very precise and systematic testing of the theoretical predictions from thermodynamical models. The equilibrium micellar properties were elaborated by systematically varying the n-alkyl chain length (n) at constant PEO molecular weight or increasing the soluble block size (x), respectively. The structure was reminiscent of typical spherical star-like micelles i.e. a constant core density profile, ∼r0, and a diffuse corona density profile, ∼r−4/3. Through a careful quantitative analysis of the scattering data, it is found that the aggregation number, Nagg initially rapidly decreases with increasing PEO length until it becomes independent at higher PEO molecular weight as expected for star-like micelles. On the other hand, the dependency on the n-alkyl length is significantly stronger than that expected from the theories for star-like block copolymer micelles, Nagg ∼ n2 similar to what is expected for surfactant micelles. Hence the observed aggregation behavior suggests that the Cn–PEOx micelles exhibit a behavior that can be considered as a hybrid between low-molecular weight surfactant micelles and diblock copolymer micelles.
1 Introduction
The self-assembly of block copolymers in micellar structures has been widely studied in theoretical studies,1–9,26 experiments10–17 and computer simulations.18–20 A general overview on this topic can be found in several review articles and books.21–25Diblock copolymers and small surfactant molecules basically show the same spontaneous self-association of single molecules (unimers) into micellar aggregates. Depending on the conditions and molecular parameters, spherical, cylindrical or vesicular micelles are usually formed.22 Nagarajan and Ganesh5 developed a thermodynamic treatment of block copolymers in a selective solvent by deriving the total Gibbs free energy for a micellar solution. Analytical and self-consistent calculations are inherently difficult for such multicomponent systems. However, for block copolymer micelles it has been shown that the pseudo-phase approximation5i.e. that the micelles can be viewed thermodynamically as a distinct “phase”, is a reasonable assumption. This is valid for a very low cmc (critical micelle concentration), i.e. when the fraction of free chains is comparatively small and the aggregation number is large. This assumption is usually fulfilled for amphiphilic block copolymers in water where the interfacial tension assumes large values. Moreover by utilizing the self-similar properties of polymers, scaling theories have been applied with great success.3,4,8,9 These theories provide rather simple predictions of the general dependency of molecular parameters that can be systematically tested by experiments. For low-molecular weight surfactants, however, both the pseudo-phase approximation and scaling theories cannot generally be used and demands much more detailed analysis. It is therefore interesting to study systems which are hybrids between the two, i.e. amphiphilic molecules where one part is polymer-like and the other is of low molecular weight. In this way the limitations of the theories can be elaborated.
Small-angle X-ray/neutron scattering (SAXS/SANS) techniques have proven to be a powerful method in order to examine the structures of micellar aggregates on a nanometer length scale and many comparisons between experimental data and theories have been reported in the literature.13,27–29 Contrary to many other techniques, SAXS/SANS provide quantitative information of the detailed shape and size of nanostructures in solution. In addition, SANS provides additional advantages in terms of contrast variation through relatively simple hydrogen/deuterium substitution that allows the different parts of the micelles (core, shell) to be selectively highlighted. However, there are only very few systematic studies testing the existing thermodynamical theories for micelles by varying molecular parameters and investigating the resulting structures using SANS/SAXS. Amphiphilic block copolymers of the type poly(diene)–PEO, with polyisoprene or polybutadiene, or the saturated analogues PE or PEP, as insoluble blocks and n-alkyl PEOs have been studied more intensively in the past because of their chemical similarities to low-molecular weight non-ionic surfactants, CnEm. For instance, commercially available “Brij” surfactants, Cn–PEO,30,31 poly(ethylene-co-propylene)-b-poly(ethylene oxide), PEP-b-PEO,32–35 poly(butadiene)-b-poly(ethylene oxide) PB-b-PEO36–38 or poly(styrene)-b-poly(ethylene oxide), PS-b-PEO39,40 have been used to study the structure of block copolymer micelles by scattering techniques applying X-rays or neutrons. Cn–PEO surfactants are interesting because they can be purchased in various PEO lengths and thereby bridge the gap between low-molecular weight surfactants and polymeric surfactants. However, as Brij surfactants are industrial products, impurities might exist which requires care when comparing theory and experiments.
In this paper, we present a structural investigation of poly(ethylene oxide)-mono-n-alkyl ether block copolymer micelles by SANS. These Cn–PEO polymers were synthesized using state-of-the-art anionic polymerization leading to very well defined materials in terms of very low degree of impurities and near monodisperse PEO blocks (Mw/Mn ≤ 1.05). Moreover, the control of the synthesis allowed us to accurately vary both the length of the n-alkyl group and the PEO block beyond that for Brij surfactants which are generally only available with C12 or C18 hydrophobic blocks and rather short PEO segments. By a careful quantitative analysis of the SANS data with a core–shell model, we analysed the micellar structure for a series of Cn–PEO5 polymers with n ranging from 18 to 30 and for C27 with three further PEO molecular weights of 10 kg mol−1, 20 kg mol−1 and 40 kg mol−1. In addition from previous work we know that the system is able to attain equilibrium since molecular exchange is active for all n-alkyl–PEO micelles.41 This allows an accurate and sensible comparison with existing thermodynamic predictions which often is complicated for regular amphiphilic block copolymers due to slow equilibration kinetics and non-ergodic behavior.42
2 Theoretical background
In analogy to the micellization of low-molecular weight surfactants A–B block copolymers spontaneously self-assemble when dispersed in a selective solvent. If the solvent is selective for the A block, microdomains of a collapsed B block surrounded by a swollen A block are formed. The individual polymer blocks are characterized by the degree of polymerization NA(B), a statistical segment length lA(B) and monomer volume vA(B) ∼ lA(B)3.The structure of the microdomains primarily depends on the degree of polymerization N = NA + NB, the composition and the interactions between the constituents that are thermodynamically described by the Flory–Huggins interaction parameter χ. With the assumption of a highly asymmetric linear diblock (NA ≫ NB) and the dilute solution limit micellar aggregates typically reveal a spherical shape. The topology of these micelles is divided into two distinct regions: the micellar core with a radius Rc and the micellar corona with a thickness D = Rm − Rc where Rm is the overall micellar radius. The structure of the micellar entity on a thermodynamic level is given by the free energy per aggregated chain in the assembly ![[scr F, script letter F]](https://www.rsc.org/images/entities/char_e141.gif) mic which can be expressed in terms of mentioned parameters of the polymer. mic can then be summarized by three main essential contributions:
mic which can be expressed in terms of mentioned parameters of the polymer. mic can then be summarized by three main essential contributions:
| mic = core + corona + int | (1) |
core and corona contain entropic terms that describe the stretching of the core blocks and osmotic crowding of the corona blocks, respectively. The last term int is the interfacial free energy associated with the creation of an interface separating the core region from the corona. This enthalpic contribution favors micellization and is given by the interfacial area Aint and the interfacial tension γ. The Flory–Huggins theory relates χ via the Helfand-equation to γ: γ ∼ χ1/2. Within a self-consistent mean-field theory the microdomain formation is distinguished to three distinct regimes: the weak (WSL),43 the strong (SSL)44 and the super strong segregation limit (SSSL).8,45 In the WSL the polymer is only weakly perturbed from a Gaussian coil behavior (χN ≥ 10) whereas for sufficiently large polymers and a high enough interfacial tension a sharp interface separates the A and B domains from each other (χN ≫ 10). Furthermore, the microdomain size (e.g. the core radius Rc) is controlled by the number of diblock copolymers (Nagg) in the structure. Applying a simple geometric space filling argument the scaling behavior is given by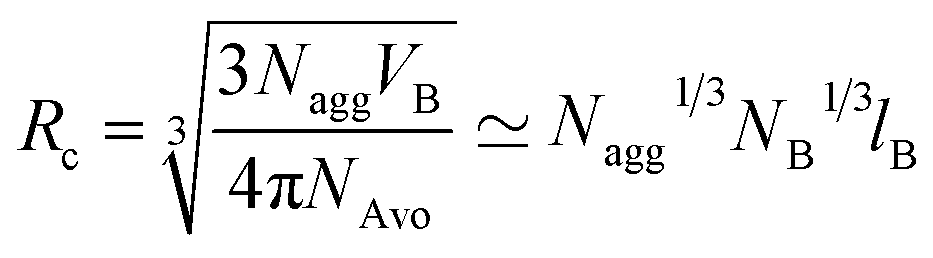 | (2) |
corona. As a consequence in this case a nearly fully elongated conformation of the B-block is assumed.
The micelle free energy per aggregated chain in units of kBT is given by eqn (3).
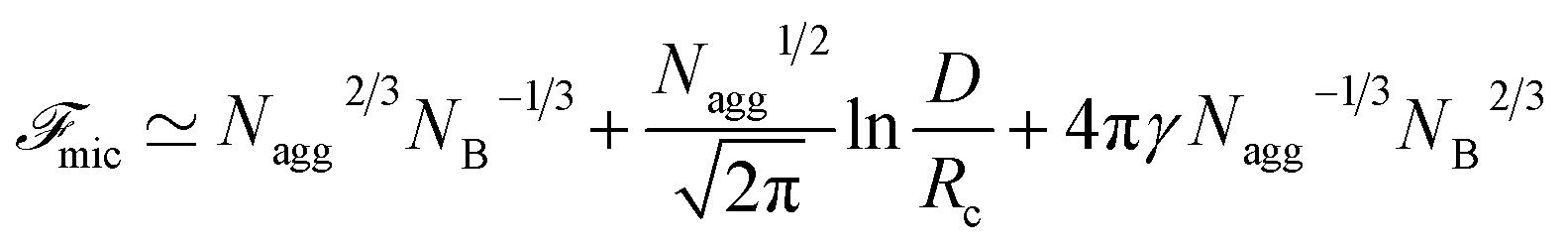 | (3) |
The equilibrium aggregation number of the micellar entity is given by minimization of the free energy with respect to Nagg. This leads to the following scaling expressions
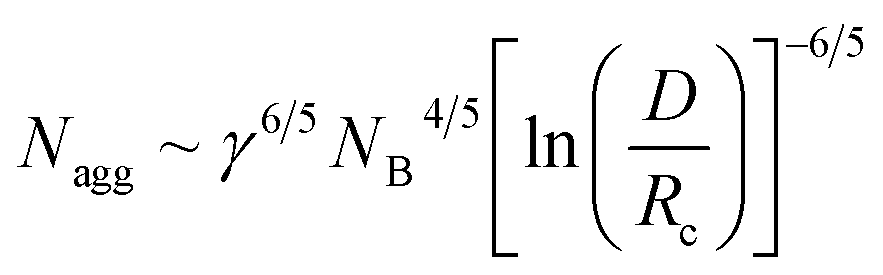 | (4) |
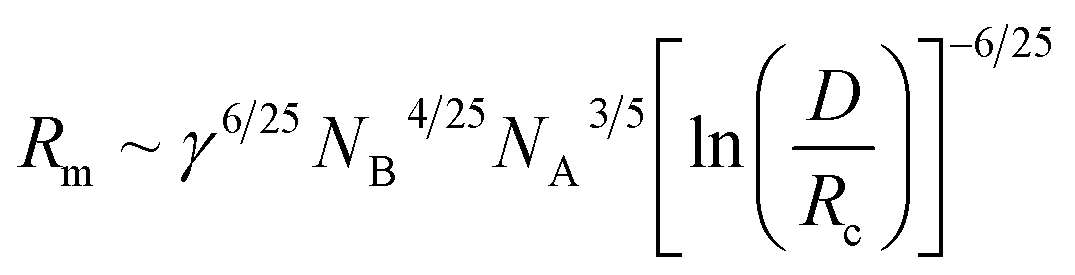 | (5) |
The minimization of eqn (3) leads to a term (NB−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Nagg)1/6 which can be approximated in the limit of NA ≫ NB by 1.3 The core radius is obtained by inserting eqn (4) into (2) which leads to the following expression:
Nagg)1/6 which can be approximated in the limit of NA ≫ NB by 1.3 The core radius is obtained by inserting eqn (4) into (2) which leads to the following expression:
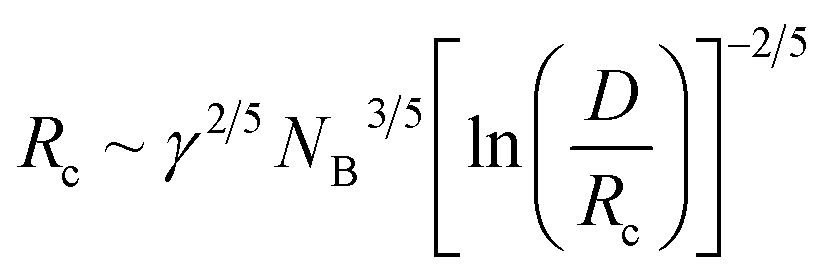 | (6) |
Eqn (4)–(6) are obtained for the SSL but as the γ increases the transition to the SSSL occurs at a critical value γ★ where the core block chains exhibit an almost fully elongated conformation:
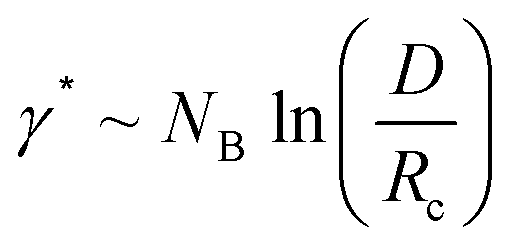 | (7) |
The latter eqn (7) is obtained from eqn (6) by assuming Rc ∼ NB. Inserting eqn (7) into eqn (4) yields a characteristic NB2-dependence on the aggregation number:
| Nagg ∼ NB2 | (8) |
This type of behavior is typically observed for low-molecular weight surfactant micelles. Contrary to amphiphilic block copolymer micelles the NB2-dependence is satisfied inherently. In this case the micellar size and shape are given by geometrical constrains due to the chain packing inside the core and was introduced by Israelachvili and coworkers more than 30 years ago.46 In order to emphasize the difference to the surfactant approach the interfacial tension controls the morphology of the micelle rather than a balance between the two opposing forces of the chain deformation and minimization of the interfacial area.
3 Experimental section
3.1 Materials and solutions
:20 mixture of an 1-alcohol (CnH2n+1OH with n = 18, 21, 24, 27, 28, 30) (Aldrich) with its corresponding potassium-1-alkoxide (CnH2n+1O−K+) as the initiating system. Deuterated polymers were prepared accordingly by polymerizing perdeuterated ethylene oxide, EO-d4 (Eurisotop; 98%D). For polymer C18–PEO5 additionally deuterated 1-octadecanol-d37 (Eurisotop, 98%D) was used whereas for all other polymers the 1-alkohols were always proteated. The polymerizations were carried out under high vacuum in toluene at 95 °C. At this temperature the potassium-1-alkoxides are soluble and the H+/K+ exchange is fast providing homogeneous initiation/propagation conditions. Generally, working with the ethylene oxide monomer requires strong safety precautions because of the high toxicity and inflammability of this material. Moreover, EO is a liquid gas (bp = 10 °C) evolving overpressure in particular in the beginning of the polymerization reaction where not yet much of the EO monomer has been polymerized. Therefore, pressure tested (12 bars) heavy walled Schlenk bombs fitted with Teflon plug valves were taken as polymerization reactors. After 24 h the overpressure had entirely disappeared indicating complete conversion of the monomer. The living polymers were terminated by adding acetic acid leading to a hydroxy group at the terminal position. The polymers were precipitated twice in cold acetone at −20 °C and isolated by centrifugation. Finally, they were dissolved in benzene, filtered and freeze-dried in high vacuum.
The resulting polymers were characterized by a combination of size exclusion chromatography (SEC) and 1H-NMR (proteated polymers). NMR-spectra were recorded in CDCl3. The number average molecular weight of the PEO component was then calculated using the integral intensity of the n-alkyl block as the internal reference. SEC measurements were done with tetrahydrofuran/N,N-dimethylacetamide (85/15) as the eluent at 50 °C using a set of three Agilent PlusPore GPC columns with a continuous distribution of pore sizes and PEO standards for calibration. Determined polydispersity indices were typically small in the order of Mw/Mn ≤ 1.04. The chromatograms, however, revealed small contents (1%) of an impurity at elution volumes corresponding to approximately twice the alkyl–PEO molecular weight. Most likely this is due to the presence of spurious amounts of water which cannot be removed even with rigorous drying procedures for the monomer and solvent. Water can participate in the H+/K+ exchange described above and thus may act as a difunctional initiator for the EO polymerization. Deuterated polymers were only analyzed by SEC. Their number average molecular weights were calculated by relating the SEC data to those of the proteated counterparts and by taking into account the deuteration. The important polymer characteristics are summarized in Table 1.
| Polymer | Labela | M Cn | M n (PEO) |
|---|---|---|---|
| a Type of isotope labelling: (h) hydrogen/(d) deuterium. b Molecular weight of respective n-alkane [g mol−1]. c M n (PEO) in [kg mol−1]. | |||
| C18–PEO5 | hh | 254.5 | 4.0 |
| dd | 292.2 | 4.0 | |
| C21–PEO5 | hh | 296.6 | 4.1 |
| hd | 4.4 | ||
| C24–PEO5 | hh | 338.7 | 4.2 |
| hd | 4.2 | ||
| C27–PEO5 | hh | 380.7 | 4.2 |
| hd | 4.4 | ||
| C27–PEO10 | hh | 10.4 | |
| hd | 10.9 | ||
| C27–PEO20 | hh | 21.2 | |
| hd | 20.4 | ||
| C27–PEO40 | hh | 36.0 | |
| hd | 38.5 | ||
| C28–PEO5 | hh | 394.8 | 4.6 |
| hd | 4.8 | ||
| C30–PEO5 | hh | 422.8 | 4.3 |
| hd | 4.1 | ||
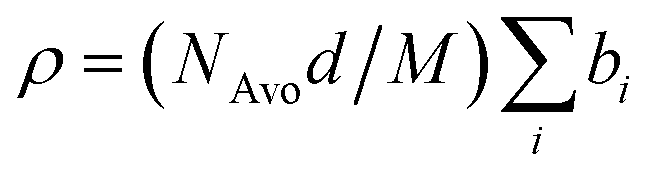 here bi denotes the coherent scattering length of an individual atom in the n-alkyl block, the EO repeat unit or the water molecules. M is the respective molecular weight and NAvo = 6.022 × 1023 Avogadro's number. For the calculation of ρ of the hydrophobic block the chemical composition of the n-alkyl group (CnH2n+1) was taken because this part was considered to be fully segregated forming the core of the micelle. Mass densities were those of the n-alkanes as tabulated in Landolt–Börnstein.47 The calculated scattering length densities for the different n-alkyls are summarized in Table 2. In the case of PEO the solution densities were taken as determined by Sommer et al.:31d(h–PEO) = 1.196 g cm−3 and d(d–PEO) = 1.305 g cm−3. The corresponding scattering length densities were then calculated to be ρh–EO = 0.676 × 1010 cm−2 and ρd–EO = 7.421 × 1010 cm−2, respectively. Scattering length densities of the pure solvents H2O and D2O are ρH2O = −5.599 × 109 cm−2 and ρD2O = 6.357 × 1010 cm−2. For polymers C27–PEOx we have used a H2O–D2O water mixture (ϕD2O = 0.56) as the solvent having an intermediate contrast of ρH2O/D2O = 3.360 × 1010 cm−2. Densities at 20 °C were taken for all calculations. Temperature effects were proven to be negligible in the studied temperature range.
here bi denotes the coherent scattering length of an individual atom in the n-alkyl block, the EO repeat unit or the water molecules. M is the respective molecular weight and NAvo = 6.022 × 1023 Avogadro's number. For the calculation of ρ of the hydrophobic block the chemical composition of the n-alkyl group (CnH2n+1) was taken because this part was considered to be fully segregated forming the core of the micelle. Mass densities were those of the n-alkanes as tabulated in Landolt–Börnstein.47 The calculated scattering length densities for the different n-alkyls are summarized in Table 2. In the case of PEO the solution densities were taken as determined by Sommer et al.:31d(h–PEO) = 1.196 g cm−3 and d(d–PEO) = 1.305 g cm−3. The corresponding scattering length densities were then calculated to be ρh–EO = 0.676 × 1010 cm−2 and ρd–EO = 7.421 × 1010 cm−2, respectively. Scattering length densities of the pure solvents H2O and D2O are ρH2O = −5.599 × 109 cm−2 and ρD2O = 6.357 × 1010 cm−2. For polymers C27–PEOx we have used a H2O–D2O water mixture (ϕD2O = 0.56) as the solvent having an intermediate contrast of ρH2O/D2O = 3.360 × 1010 cm−2. Densities at 20 °C were taken for all calculations. Temperature effects were proven to be negligible in the studied temperature range.
| Label | C18 | C21 | C24 | C27 | C28 | C20 |
|---|---|---|---|---|---|---|
| h | −0.349 | −0.343 | −0.339 | −0.335 | −0.334 | −0.333 |
| d | 6.520 |
3.2 SANS and data evaluation
The small-angle neutron scattering (SANS) experiments were conducted at KWS-1 and KWS-2 instruments located at Heinz-Meier-Leibnitz Zentrum (MLZ) in Garching, Germany. Measurements of Cn–PEO5 were carried out under full contrast, i.e. hh in D2O and hd/dd in H2O. The C27–PEOx samples were measured in the isotopic water mixture (ϕD2O = 0.56) providing the intermediate contrast. The latter contrast condition was chosen in order to study micellar exchange kinetics in these systems. However, a discussion of the kinetics goes beyond the scope of this work. Sample-to-detector distances were 2 and 8 m and 20 m additionally for C27–PEOx with x > 10. Collimation lengths were set identical to sample-to-detector distances, except for 2 m where an 8 m collimation length was chosen in order to avoid detector saturation. KWS measurements were carried out with a neutron wavelength of λ = 7 Å and a wavelength spread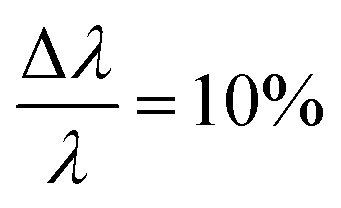 at KWS-1 and
at KWS-1 and 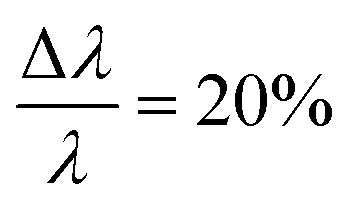 at KWS-2, respectively. The 2 and 8 m settings yield a Q-range of 0.006 ≤ Q [Å−1] ≤ 0.2 and for the 20 m setup we could extend the range down to Q = 0.003 Å−1, where Q = 4π/λsinθ is the momentum transfer and 2θ is the scattering angle. Scattered intensities were corrected for detector sensitivity, empty cell scattering, electronic noise and dead time effects due to detector electronics. The scattering data were set on absolute scale using plexiglass as a calibrated secondary standard. The thus obtained absolute normalized differential scattering cross-section dΣ/dΩ (Q) in cm−1 was further corrected for incoherent scattering contributions. Incoherent scattering of the solvent was measured separately, whereas the incoherent scattering of the polymer was calculated. All data reductions were done by using the computer software QtiKWS available at MLZ in Garching.48
at KWS-2, respectively. The 2 and 8 m settings yield a Q-range of 0.006 ≤ Q [Å−1] ≤ 0.2 and for the 20 m setup we could extend the range down to Q = 0.003 Å−1, where Q = 4π/λsinθ is the momentum transfer and 2θ is the scattering angle. Scattered intensities were corrected for detector sensitivity, empty cell scattering, electronic noise and dead time effects due to detector electronics. The scattering data were set on absolute scale using plexiglass as a calibrated secondary standard. The thus obtained absolute normalized differential scattering cross-section dΣ/dΩ (Q) in cm−1 was further corrected for incoherent scattering contributions. Incoherent scattering of the solvent was measured separately, whereas the incoherent scattering of the polymer was calculated. All data reductions were done by using the computer software QtiKWS available at MLZ in Garching.48
The coherent macroscopic scattering cross-section of the micellar solutions dΣ/dΩ (Q) in the dilute limit i.e. S(Q) ≈ 1 was analyzed according to
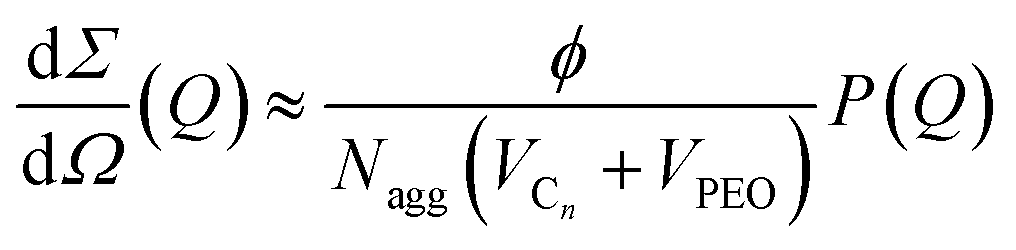 | (9) |
The micellar form factor P(Q) is given by
| P(Q) = (ρCn − ρ0)2Nagg2VCn2Ac2(Q) + (ρPEO − ρ0)2Nagg(Nagg − B(0))VPEO2Ash2(Q) + 2(ρCn − ρ0)(ρPEO − ρ0)Nagg2VCnVPEOAc(Q)Ash(Q) + VPEO2Δρsh2B(Q) | (10) |
 | (11) |
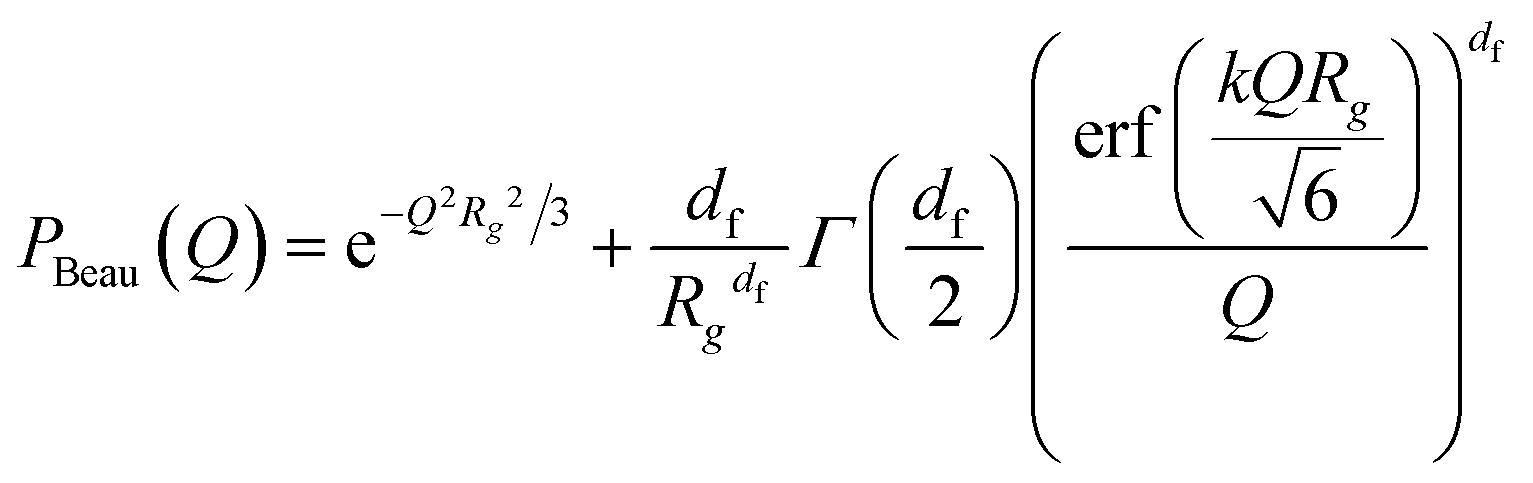 | (12) |
The individual Ac(Q) for the core and the shell Ash(Q) are calculated based on assuming a compact homogeneous core density profile, ncore = const. and the star-like shell density profile, nshell ∼ r−4/3, respectively.52 Thus, the scattering amplitudes can be written as:
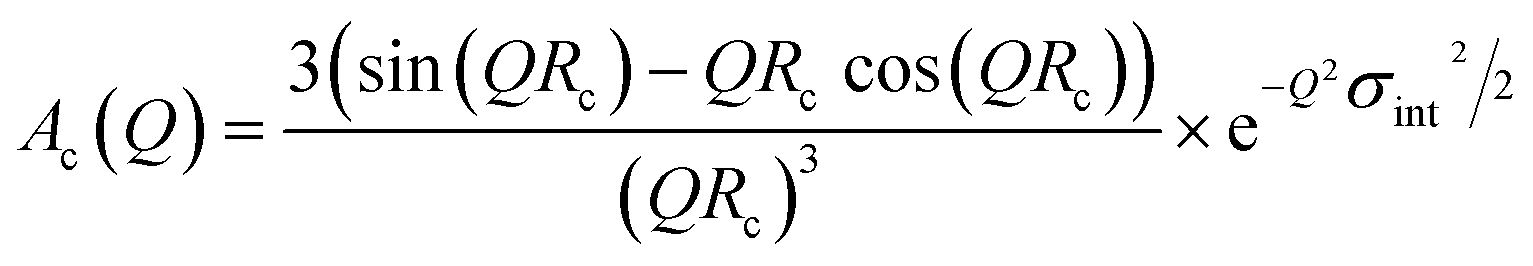 | (13) |
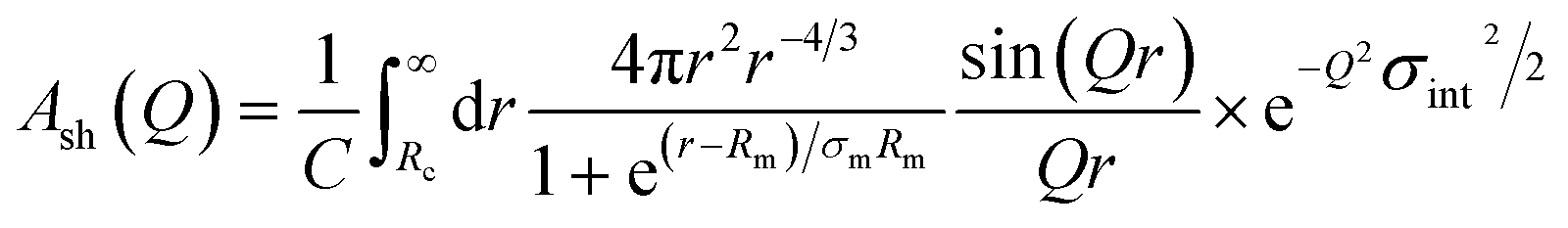 | (14) |
where C is a normalization constant 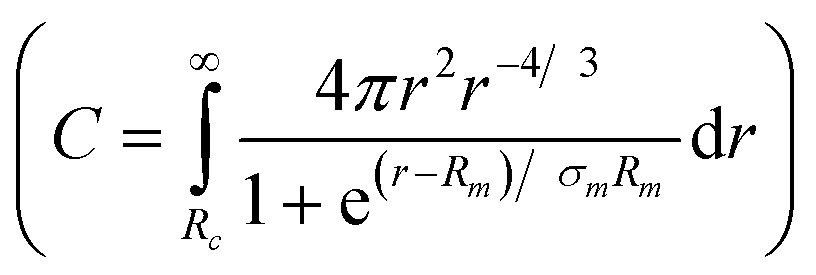 . In eqn (13) and (14) the Gaussian factor gives a smooth core–corona interface where σint is a measure of the surface roughness. The Fermi cut-off function in eqn (14) is used to terminate the corona region to finite size. For the analysis σm was set to 10% of Rm. Finally, data analysis incorporates instrumental resolution effects according to a wavelength spread, finite collimation and detector resolution.53,54
. In eqn (13) and (14) the Gaussian factor gives a smooth core–corona interface where σint is a measure of the surface roughness. The Fermi cut-off function in eqn (14) is used to terminate the corona region to finite size. For the analysis σm was set to 10% of Rm. Finally, data analysis incorporates instrumental resolution effects according to a wavelength spread, finite collimation and detector resolution.53,54
4 Results and discussion
The macroscopic scattering cross-sections, dΣ/dΩ(Q), for Cn−PEOx micelles in dilute solution (ϕ = 0.25) are shown in Fig. 1(a) and (b) in a double logarithmic representation. The scattered intensity is normalized by C = NAvo(VCn + VPEO)/(ϕ(ΔρcVCn + ΔρshVPEO)2) considering concentration, contrast and volumes thus directly yielding Nagg at low Q. For better visibility the scattering curves in Fig. 1(b) have been shifted by different factors. The scattering pattern of the micellar solutions exhibit characteristic features like a low Q Guinier-plateau and at high Q a power law dependence of Q−1.7 reflecting the semi-dilute nature of the polymer shell domain.33 In between the scattered intensity shows a steep decay in a rather narrow Q window. We note that due to the low intensity very close to the incoherent background the apparent slope at high Q sensitively depends on the data reduction. In particular, at low polymer concentration the data statistics is generally poor and the exponent cannot be determined with high precision. However, the necessary structural micellar properties Nagg and Rm were rather unaffected by these variations in the high Q region. Detailed structural parameters were obtained by fitting the data with the spherical core–shell model applying least-square fit routines. Best-fit curves are shown as solid lines in Fig. 1. In order to reduce the number of free parameters the block molecular volumes Vn and VPEO, the polymer volume fractions ϕ and the scattering length densities ρ were taken as obtained from the polymer characterization or calculated as described in the experimental section. These values were hold fixed throughout the whole analysis. ν was found to best describe the data by a value close to 0.2 and was kept constant at this value. Any possible structure factor contributions were considered to be negligible at 0.25% polymer volume fraction. In order to confirm this we have accounted for deviations from the dilute regime by means of a second virial coefficient, A2. As an example the second virial coefficient for C24–PEO5 micelles, A2 ≈ 1.0 × 10−4 cm3 mol g−2, was deduced from three different concentrations, ϕ = 0.25%, 0.5% and 1% (data not shown). From trial fits we could confirm that for the studied low concentrations such a small value of A2 has basically no effect on the results. The important fit parameters are summarized in Table 3 including Rc and D calculated by Rm − Rc. The fits further yield characteristic length scales (“blob”) of the corona between 40 Å and 80 Å. These values depend strongly on the data corrections at high Q where the internal shell structure is essentially determined and therefore, vary rather unsystematically with n. Since the “blob-scattering” has only a minor effect on the global micellar parameters this is not further discussed here. Best fits were obtained by using σint = 5 Å as a value for the core–shell smearing. For data analysis we have neglected any polydispersity effects since micelles were considered to be monodisperse. This assumption can be made for large interfacial tensions in such systems (≈50 m Nm−1) and low cmc (≤2 × 10−5 mol 1−1).55 Additionally, as has already been shown in a previous work41 the aggregation behavior is not significantly affected by H/D isotope labeling (see Table 3). Hence, we can exclude any pronounced isotope effect.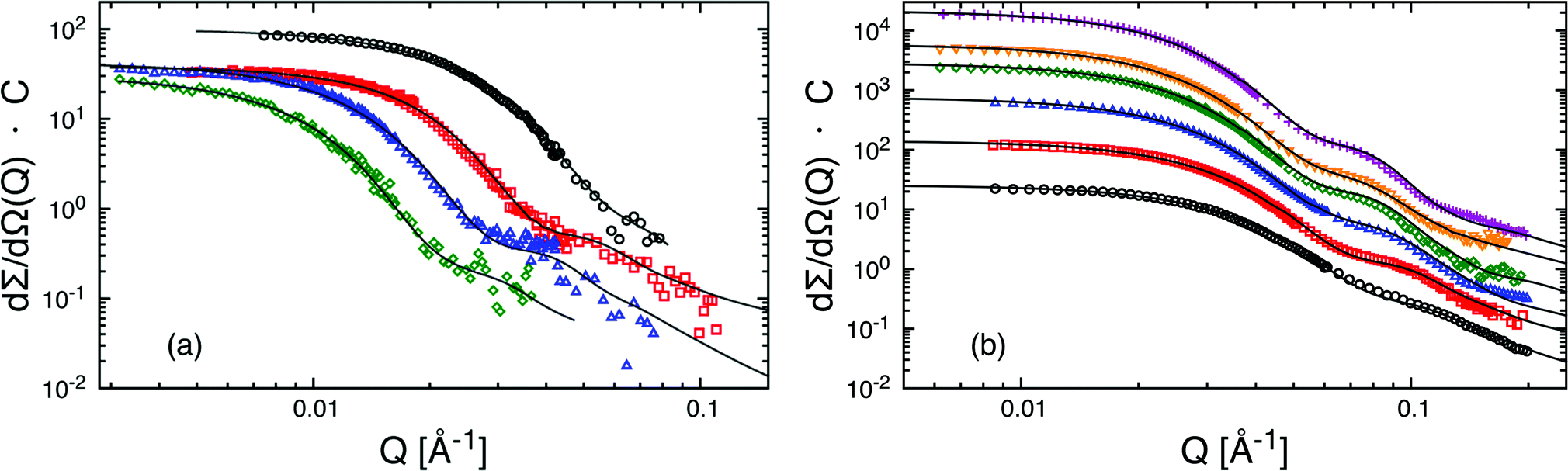 | ||
| Fig. 1 Form factors of fully proteated Cn–PEOx (hh) spherical micelles in dilute aqueous solution at 25 °C: (a) C27–PEOx, x = 5, 10, 20 and 40 kg mol−1 (from right to left) in intermediate contrast and (b) Cn–PEO5, n = 18, 21, 24, 27, 28, 30 (curves from bottom to top), data have been multiplied by constant factors: C21 = 3, C24 = 10, C27 = 30, C28 = 60, C30 = 150. Data have been normalized to a constant C. Core–shell model fits are shown as solid lines. | ||
| Polymer | Label | N agg | R m [Å] | R c [Å] | D [Å] |
|---|---|---|---|---|---|
| C18–PEO5 | hh | 28 | 78 | 15 | 63 |
| dd | 30 | 79 | 16 | 63 | |
| C21–PEO5 | hh | 45 | 87 | 19 | 68 |
| hd | 53 | 89 | 20 | 69 | |
| C24–PEO5 | hh | 75 | 103 | 23 | 80 |
| hd | 80 | 100 | 24 | 76 | |
| C27–PEO5 | hh | 96 | 109 | 26 | 83 |
| hd | 122 | 108 | 27 | 81 | |
| C27–PEO10 | hh | 39 | 142 | 18 | 124 |
| hd | 41 | 147 | 18 | 129 | |
| C27–PEO20 | hh | 41 | 208 | 18 | 190 |
| hd | 38 | 204 | 18 | 186 | |
| C27–PEO40 | hh | 30 | 282 | 16 | 266 |
| hd | 23 | 268 | 15 | 253 | |
| C28–PEO5 | hh | 95 | 110 | 26 | 84 |
| hd | 107 | 112 | 27 | 85 | |
| C30–PEO5 | hh | 125 | 111 | 30 | 81 |
| hd | 120 | 101 | 29 | 72 |
4.1 Effect of PEO molecular weight on micellar structure
The SANS scattering curves shown in Fig. 1(a) demonstrate the influence of PEO molecular weight on the micellar structure for four different PEOx molecular weights x = 5, 10, 20 and 40 kg mol−1 at constant hydrocarbon chain length (n = 27). As can be seen the aggregation number clearly drops when the PEO molecular weight is increased from 5 kg mol−1 to 10 kg mol−1 whereas a further increase of MPEO to 20 kg mol−1 and 40 kg mol−1 does not lead to a significant change in Nagg anymore. The SANS curves additionally show a consistent shift of the Guinier plateau to smaller Q as a natural consequence of increasing micellar size with the PEO molecular weight. The data were analyzed by the core–shell model following the same fit procedure as described in the previous section. Best fits are shown as solid lines in Fig. 1. The important fit parameters are included in Table 3.The found aggregation numbers Nagg are then plotted as a function of the number of EO repeat units, NPEO, in Fig. 2(a). As already qualitatively discussed above there is a steep decay in Nagg from C27–PEO5 to C27–PEO10 while for the higher PEO molecular weights Nagg depends only weakly on the PEO chain length. In the scaling theory for starlike micelles of Halperin3 the dependence of Nagg on the corona chain length is not explicitly considered. There, the aggregation number essentially depends only on the size of the core block. Zhulina and coworkers9 on the other hand have shown that in the limit of long chains Nagg shows a weak logarithmic dependence, Nagg ∼ (lnN)−6/5. This dependence is depicted by the solid line in Fig. 2(a). Apparently, the data points are well represented for large NPEO but do not agree for the initial strong drop between C27–PEO5 and C27–PEO10. On the basis of a pseudo mean-field phase approximation Nagarajan and Ganesh5 proposed that the corona block size has a stronger influence on the aggregation behavior, especially when the solvent is a very good solvent. For PEO–PPO micelles in water they numerically calculated the following empirical scaling relationship: Nagg ∼ N−0.51. This dependence is shown as a dashed line in Fig. 2(a). It can be seen that the experimental trend is well reproduced by this relationship including the data point for C27–PEO5 micelles in intermediate contrast and under full contrast measured in D2O (see triangle Fig. 2(a)). The observed decrease of Nagg can be qualitatively explained by a change of the balance between entropic and enthalpic contributions with the growing PEO block.56 This should lead to a larger steric hindrance of the head-groups on the hydrophobic core surface such that the system is shifted to a new equilibrium structure with smaller aggregation numbers. Furthermore, Fig. 2(b) depicts the micellar shell thickness D as a function of NPEO. We see that the experimental data are in excellent agreement with the theoretical prediction D ∼ N3/5 for star-like micelles3 which is shown by the straight line. Thus, the aggregation behavior at constant n-alkyl chain length is determined by the polymer character of the hydrophilic PEO block.
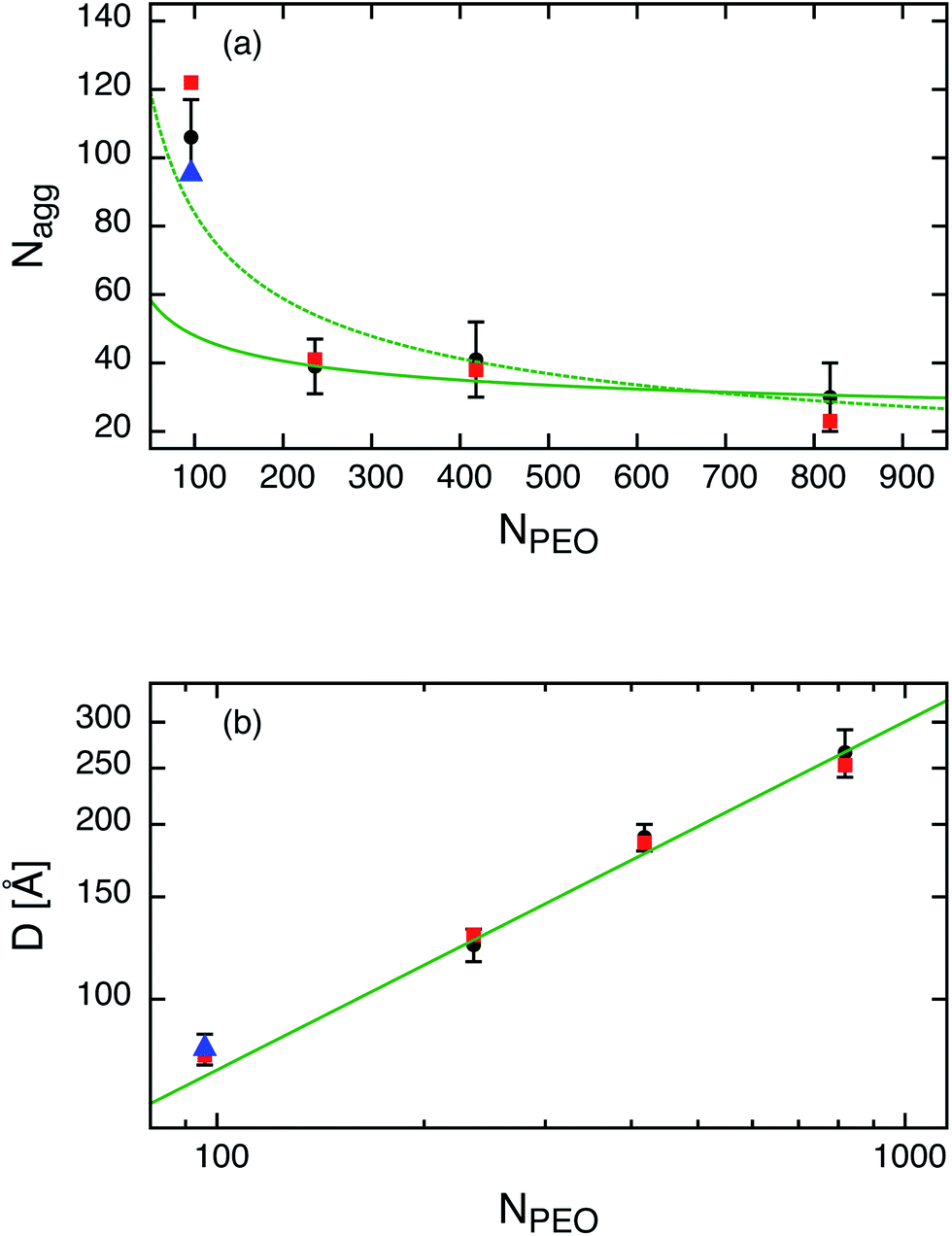 | ||
Fig. 2 (a) Aggregation number Naggvs. number of EO repeat units, NPEO: ( ) hh labeled polymer, ( ) hh labeled polymer, ( ) hd labeled polymer and ( ) hd labeled polymer and ( ) obtained under full contrast in D2O. Solid line represents the scaling law of the micellar star-model,9 dashed line a semi-empirical dependence deduced by Nagarajan and Ganesh for PPO–PEO micelles.5 (b) Shell thickness D vs. NPEO in log–log representation. Solid line represents a slope of 3/5. ) obtained under full contrast in D2O. Solid line represents the scaling law of the micellar star-model,9 dashed line a semi-empirical dependence deduced by Nagarajan and Ganesh for PPO–PEO micelles.5 (b) Shell thickness D vs. NPEO in log–log representation. Solid line represents a slope of 3/5. | ||
4.2 Effect of hydrocarbon chain length
The C18–PEO5 micelles were compared to micelles formed by a commercial material Brij700 of similar chemical composition. Sommer et al.31 have extensively studied the structure of Brij700 by using SANS and SAXS. They found a core radius of Rc = 15–17 Å and an aggregation number of Nagg = 30 in very good agreement with the present results. We take this now as a starting point to systematically discuss the effect of the n-alkyl chain length at a constant PEO molecular weight of 5 kg mol−1.The scattering data presented in Fig. 1(b) for the different hh Cn–PEO5 micellar solutions in D2O show that the intensity increases with n, directly demonstrating a growth of the micelles in terms of an increasing Nagg accompanied by an increasing core size. Although the PEO molecular weight stays constant the shell thickness D increases slightly (see Table 3). This might be due to a slightly higher PEO density near the core surface leading to a stronger chain stretching of the PEO block. It should be stressed again that the interfacial tension, γ, of the water–n-alkane interface is almost constant (≈50 m Nm−1) within the higher members of the homologous series of n-alkanes.55 Thus, any effect on the micellar properties is mainly due to the increase of the hydrocarbon chain length. The aggregation number Nagg as a function of the n-alkyl chain length is depicted in Fig. 3. We included recent experimental data found for poly(ethylene oxide)-mono-n-alkyl ethers Cn–PEO.30,31,57,58 In order to exclude any effects of the hydrophilic block on the aggregation number Nagg was scaled by NPEO0.51 which was found above to be a reasonable description of the PEO length on Nagg. We observe that Nagg systematically increases with increasing n. The system almost reveals the characteristic n2 dependence for the aggregation number that is denoted by the solid line in Fig. 3. The deviation from this behavior is larger for the C18–PEO5 and C21–PEO5 but still in good agreement with the n2 power law. The n2 scaling can then be associated by either assuming a simple geometrical model as for low-molecular weight surfactants that inherently give the n2 scaling law26 or the super strong segregation regime as outlined in the theoretical section. Given the hybrid nature of our system, it is tempting to speculate that this behavior reflects the surfactant properties of the hydrocarbon core. This is furthermore supported by the fact that the area a per molecule on the core surface does not vary substantially with n. Here we find a mean value of about (93 ± 6) Å2 which suggest that Nagga = 4πRc2 is similar to what is found for surfactant micelles.46 Thus, the n-alkyl chain linearly increases with n and if we allow a homogeneous compact core,59 it follows that Nagg scales with n2. A comparison of Rc with the maximal possible length, lmax, of an alkane chain 1.53 + 1.265 (n − 1) [Å]59 shows that the experimental values found for the micelles in the core are systematically smaller. For example for C24 we find Rc = 23 Å whereas lmax = 31 Å. Apparently, the alkyl chains do not assume a fully extended all-trans configuration. This was also observed by Sommer et al.31 on Brij700 where Rc is reduced by a factor of about 0.75 for C18. This corresponds to a more flexible conformation in the bulk state i.e. the all-trans configuration is perturbed due to kinks along the backbone. According to Tanford the average chain length of a more flexible n-alkyl chain is given by lflex = 1.53 + 0.925(n − 1) [Å].59 The latter analytical expression nicely agrees with the experimental data e.g. lflex[C24] = 23 Å. Thus, the conformation of the alkyl chains can be considered as more flexible which coincides with the assumption that the spherical core has a homogeneous density profile. It should be mentioned that long alkyl chains might crystallize60,61 partly resulting in a non-spherical core domain. Crystalline micellar cores together with the high interfacial tension and a temperature insensitive aggregation number would support micelles in the super-strong segregation limit (SSSL) where the micellar coronas are still spherical. Since the scattering data are an average over an ensemble of micelles, any deviation of the micellar core from spherical geometry is not easy to deduce directly. The rather broad core–corona interface (σint ≈ 5 Å) might be an indication for a more elliptical shape of the core. Since, our scattering data cannot capture this possibility, it has not been considered here. But there is evidence of a phase transition, differential scanning calorimetry, density measurements and SAXS data suggest that n-alkyl micellar cores are partly crystalline at low enough temperatures. The discussion of these results goes beyond the scope of the present work and will be shown in an upcoming publication.
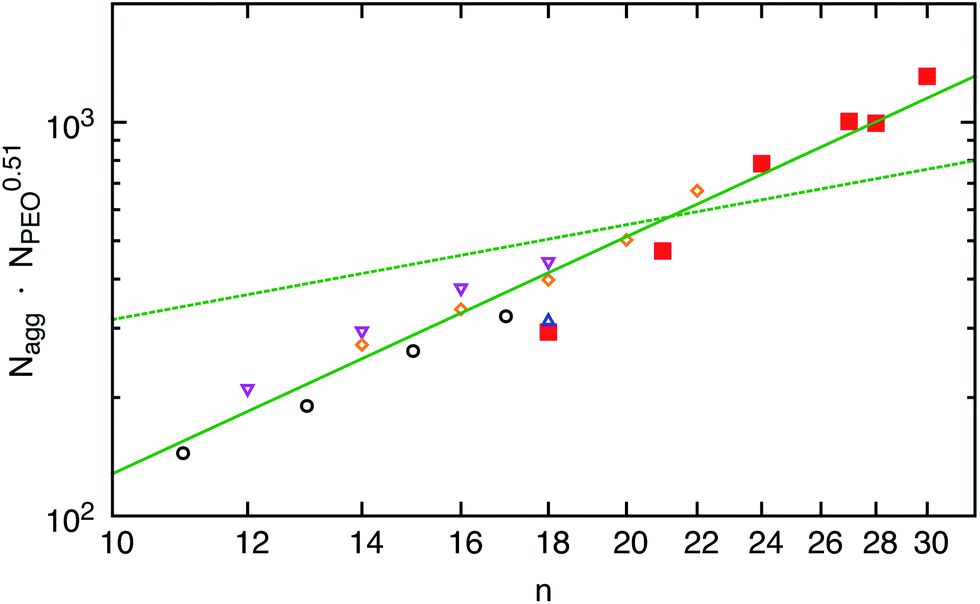 | ||
Fig. 3 Aggregation number Nagg as a function of n: ( ) Cn–PEO5, ( ) Cn–PEO5, ( ) CnE40,57 ( ) CnE40,57 ( ) Brij700,31 ( ) Brij700,31 ( ) Cn–PEO5,58 and ( ) Cn–PEO5,58 and ( ) Cn–PEO5.30 The solid line has a slope of 2 and the dashed line of 4/5 expected form the star-model. ) Cn–PEO5.30 The solid line has a slope of 2 and the dashed line of 4/5 expected form the star-model. | ||
We note that for amphiphilic micelles the same scaling law is theoretically predicted by the SSSL8 and experimentally found by Förster et al.13 A comparison of Nagg to the theoretically predicted scaling laws for star-like block copolymer micelles3 reveals no agreement with our data. The expected power laws for star-like micelles Nagg ∼ N4/5 are too weak and clearly not represented by our data (see dashed line in Fig. 3). In addition, the effect of temperature on the micelles in thermal equilibrium was studied for C24–PEO5 and C30–PEO5 micelles in a temperature range between 20 °C and 60 °C. This is depicted in Fig. 4 which shows the effect of temperature on the aggregation number Nagg and the corona thickness D for C24–PEO5 micelles in water. We find that the aggregation number is within the experimental uncertainty independent of temperature which might be explained by the fact that the interfacial tension is almost temperature insensitive.33 This result coincides with recent findings by Sommer et al.62 for Brij700 in water. Moreover, there is the trend of decreasing micellar size. Since Nagg does not change with temperature this shrinkage is associated with variations in the PEO interactions in water. It is a well-established fact that PEO exhibits a large number of conformations strongly depending on temperature.63,64 As the temperature increases the hydration becomes less effective which leads to a conformational change i.e. a more coiled conformation of PEO. The discussion of the temperature effect follows a model that explains the existence of a lower critical solution temperature (LCST) in PEO–water systems. Thus, the conformation change is not the only reason for the PEO shrinkage but rather the induced decrease of solvent quality. The experimental results however are in qualitive agreement with recent computer simulations.20
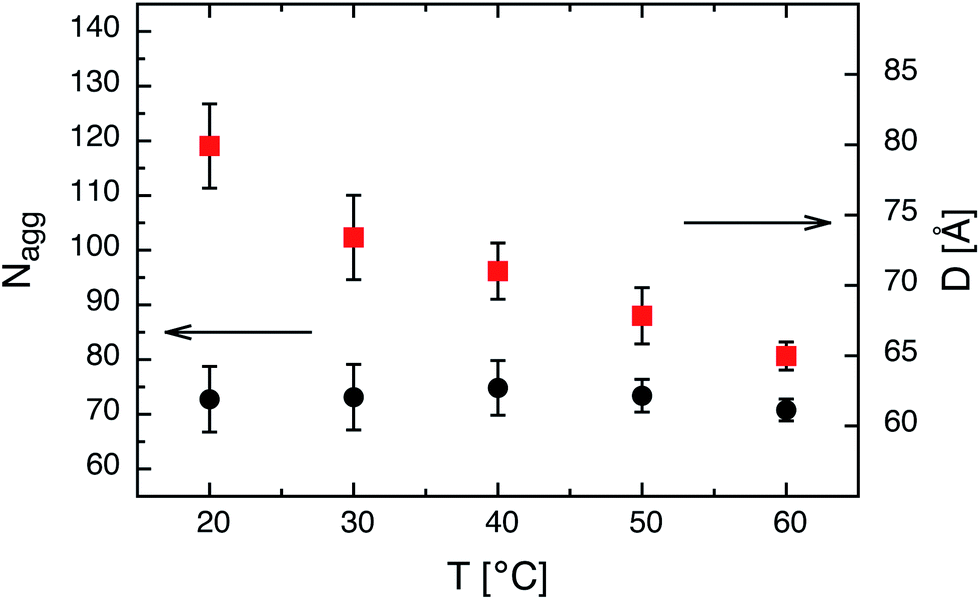 | ||
Fig. 4 Aggregation number Nagg and corona thickness D as a function of temperature T for C24–PEO5 micelles: ( ) Nagg and ( ) Nagg and ( ) D. ) D. | ||
5 Concluding remarks
Poly(ethylene oxide) mono n-alkyl ether diblock copolymers, Cn−PEOx, with n = 18, 21, 24 27, 28, 30 and x = 5, 10, 20, 40 kg mol−1 in aqueous solution form spherical aggregates as determined by SANS. Details of the micellar structure were extracted by applying a sophisticated core–shell model including as main ingredients a diffuse star density profile for the corona, ϕ(r) ∼ r−4/3, and a constant density profile for the core region. By this, excellent description of all SANS data was feasible providing accurate structural parameters of the micelles as a function of n and x. This allows a systematic discussion with respect to current thermodynamic models.In particular, we experimentally verified the predicted effects of the hydrophobic and hydrophilic chain length on the aggregation behavior. As demonstrated the aggregation number of the formed micelles increases quadratically with the length of the n-alkyl chain. By changing the molecular weight of the PEO block the aggregation number consistently changes as predicted by the empiric power-law numerically obtained by Nagarajan and Ganesh for PPO–PEO. Moreover, the core size Rc is in accordance with an expression for the chain length of a flexible n-alkyl chain which coincides with the Tanfords model for a liquid-like hydrocarbon chain i.e. hydrocarbon chain having kinks along the chain. This interpretation is also related to simple geometrical constraints on the chain packing as known from low-molecular weight surfactant micelles. However, both the SSSL theory for amphiphilic block copolymers and the surfactant theory predict that the aggregation number follows a quadratic dependence on the hydrophobic block. Considering the hydrophobic n-alkyl block the system is still a surfactant but with the rather long PEO block a polymer character is imposed on the system. The conclusion however is that the chemical hybrid character of Cn–PEOx between a surfactant molecule and a block copolymer is also reflected in the equilibrium micellar properties. Thus, we consider Cn–PEOx polymers as a hybrid material which might close the missing gap to micelles prepared from low-molecular weight non-ionic CnEm surfactants carrying only short n-alkyl chains and EO head-groups, respectively.
Finally, we point out that the detailed characterization of the size and shape of Cn–PEOx micelles serves as an important prerequisite to study the equilibrium chain exchange kinetics of these micellar entities. Details of this study will be presented in a forthcoming paper.
Acknowledgements
R.L. greatly acknowledges grants from the Norwegian Research Council, under the SYNKNOYT program (218411 and 228573).References
- J. Noolandi and K. M. Hong, Macromolecules, 1983, 16, 1443–1448 CrossRef CAS.
- L. Leibler, H. Orland and J. C. Wheeler, J. Chem. Phys., 1983, 79, 3550–3557 CrossRef CAS PubMed.
- A. Halperin, Macromolecules, 1987, 20, 2943–2946 CrossRef CAS.
- T. Birshtein and E. Zhulina, Polymer, 1989, 30, 170–177 CrossRef CAS.
- R. Nagarajan and K. Ganesh, J. Chem. Phys., 1989, 90, 5843 CrossRef CAS PubMed.
- D. Izzo and C. M. Marques, Macromolecules, 1993, 26, 7189–7194 CrossRef CAS.
- Z. Gao and A. Eisenberg, Macromolecules, 1993, 26, 7353–7360 CrossRef CAS.
- A. N. Semenov, I. A. Nyrkova and A. R. Khokhlov, Macromolecules, 1995, 28, 7491–7500 CrossRef CAS.
- E. B. Zhulina, M. Adam, I. LaRue, S. S. Sheiko and M. Rubinstein, Macromolecules, 2005, 38, 5330–5351 CrossRef CAS.
- M. Wilhelm, C. L. Zhao, Y. Wang, R. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- J. R. Quintana, M. Villacampa, M. Munoz, A. Andrio and I. A. Katime, Macromolecules, 1992, 25, 3125–3128 CrossRef CAS.
- J. R. Quintana, M. Villacampa, A. Andrio, M. Munoz and I. A. Katime, Macromolecules, 1992, 25, 3129–3136 CrossRef CAS.
- S. Förster, M. Zisenis, E. Wenz and M. Antonietti, J. Chem. Phys., 1996, 104, 9956–9970 CrossRef PubMed.
- A. Jada, G. Hurtrez, B. Siffert and G. Riess, Macromol. Chem. Phys., 1996, 197, 3697–3710 CrossRef CAS.
- H. Iatrou, L. Willner, N. Hadjichristidis, A. Halperin and D. Richter, Macromolecules, 1996, 29, 581–591 CrossRef CAS.
- P. Hickl, M. Ballauff and A. Jada, Macromolecules, 1996, 29, 4006–4014 CrossRef CAS.
- J. Bang, S. Jain, Z. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Macromolecules, 2006, 39, 5583 CrossRef CAS.
- K. Binder and M. Müller, Curr. Opin. Colloid Interface Sci., 2000, 5, 314–322 CrossRef.
- A. Milchev, A. Bhattacharya and K. Binder, Macromolecules, 2001, 34, 1881–1893 CrossRef CAS.
- S. Hezaveh, S. Samanta, G. Milano and D. Roccatano, J. Chem. Phys., 2012, 136, 124901 CrossRef PubMed.
- A. Halperin, M. Tirrell and T. P. Lodge, Adv. Polym. Sci., 1992, 100, 31–71 CrossRef CAS.
- P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers: Self-Assembly and Applications, Elsevier Science, 2000 Search PubMed.
- C. Svaneborg and J. S. Pedersen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 64, 010802 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- I. Hamley, Block Copolymers in Solution: Fundamentals and Applications, John Wiley & Sons, 2005 Search PubMed.
- R. Nagarajan and E. Ruckenstein, Langmuir, 1991, 7, 2934–2969 CrossRef CAS.
- A. Poppe, L. Willner, J. Allgaier, J. Stellbrink and D. Richter, Macromolecules, 1997, 30, 7462–7471 CrossRef CAS.
- J. S. Pedersen, C. Svaneborg, K. Almdal, I. W. Hamley and R. N. Young, Macromolecules, 2003, 36, 416–433 CrossRef CAS.
- S. Manet, A. Lecchi, M. Impéror-Clerc, V. Zholobenko, D. Durand, C. L. P. Oliveira, J. S. Pedersen, I. Grillo, F. Meneau and C. Rochas, J. Phys. Chem. B, 2011, 115, 11318–11329 CrossRef CAS PubMed.
- F. Renou, T. Nicolai, E. Nicol and L. Benyahia, Langmuir, 2009, 25, 515–521 CrossRef CAS PubMed.
- C. Sommer, J. S. Pedersen and V. M. Garamus, Langmuir, 2005, 21, 2137–2149 CrossRef CAS PubMed.
- L. Willner, A. Poppe, J. Allgaier, M. Monkenbusch, P. Lindner and D. Richter, EPL, 2000, 51, 628 CrossRef CAS.
- R. Lund, L. Willner, J. Stellbrink, A. Radulescu and D. Richter, Macromolecules, 2004, 37, 9984–9993 CrossRef CAS.
- H. Kaya, L. Willner, J. Allgaier, J. Stellbrink and D. Richter, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 499–501 CrossRef.
- G. V. Jensen, Q. Shi, G. R. Deen, K. Almdal and J. S. Pedersen, Macromolecules, 2012, 45, 430–440 CrossRef CAS.
- S. Förster, B. Berton, H.-P. Hentze, E. Krämer, M. Antonietti and P. Lindner, Macromolecules, 2001, 34, 4610–4623 CrossRef.
- Y. Deng, R. Young, A. Ryan, J. Fairclough, A. Norman and R. Tack, Polymer, 2002, 43, 7155–7160 CrossRef CAS.
- S. Jain, M. H. E. Dyrdahl, X. Gong, L. E. Scriven and F. S. Bates, Macromolecules, 2008, 41, 3305–3316 CrossRef CAS.
- K. Mortensen, W. Brown, K. Almdal, E. Alami and A. Jada, Langmuir, 1997, 13, 3635–3645 CrossRef CAS.
- H. Yu, A. Natansohn, M. A. Singh and I. Torriani, Macromolecules, 2001, 34, 1258–1266 CrossRef CAS.
- T. Zinn, L. Willner, R. Lund, V. Pipich and D. Richter, Soft Matter, 2012, 8, 623–626 RSC.
- R. Lund, L. Willner and D. Richter, Adv. Polym. Sci., 2013, 51–158 CrossRef.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- A. N. Semenov, JETP Lett., 1985, 88, 733–742 Search PubMed.
- I. A. Nyrkova, A. R. Khokhlov and M. Doi, Macromolecules, 1993, 26, 3601–3610 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- R. C. Wilhoit, K. N. Marsh, X. Hong, N. Gadalla and M. Frenkel, in Landolt–Börnstein – Group IV Physical Chemistry, 1996 Search PubMed.
- A. Radulescu, V. Pipich, H. Frielinghaus and M. S. Appavou, J. Phys.: Conf. Ser., 2012, 351, 012026 CrossRef.
- J. S. Pedersen and C. Svaneborg, Curr. Opin. Colloid Interface Sci., 2002, 7, 158–166 CrossRef CAS.
- R. Lund, V. Pipich, L. Willner, A. Radulescu, J. Colmenero and D. Richter, Soft Matter, 2011, 7, 1491–1500 RSC.
- G. Beaucage, J. Appl. Crystallogr., 1996, 29, 134–146 CrossRef CAS.
- M. Daoud and J. Cotton, J. Phys., 1982, 43, 531–538 CAS.
- J. S. Pedersen, J. Phys. IV, 1993, 03, 491–498 CrossRef CAS.
- J. G. Barker and J. S. Pedersen, J. Appl. Crystallogr., 1995, 28, 105–114 CrossRef CAS.
- M. Ameri, D. Attwood, J. H. Collett and C. Booth, J. Chem. Soc., Faraday Trans., 1997, 93, 2545–2551 RSC.
- M. R. Bohmer and L. K. Koopal, Langmuir, 1990, 6, 1478–1484 CrossRef CAS.
- C. Booth, D. Attwood and C. Price, Phys. Chem. Chem. Phys., 2006, 8, 3612–3622 RSC.
- F. Lafléche, D. Durand and T. Nicolai, Macromolecules, 2003, 36, 1331–1340 CrossRef.
- C. Tanford, The Hydrophobic Effect: Formation of Micelles and Biological Membranes, John Wiley & Sons, 1980 Search PubMed.
- P. R. Knowles, R. J. Barlow, F. Heatley, C. Booth and C. Price, Macromol. Chem. Phys., 1994, 195, 2547–2558 CrossRef CAS.
- R. G. Alamo, L. Mandelkern, G. M. Stack, C. Krohnke and G. Wegner, Macromolecules, 1994, 27, 147–156 CrossRef CAS.
- C. Sommer, J. S. Pedersen and P. Stein, J. Phys. Chem. B, 2004, 108, 6242–6249 CrossRef CAS PubMed.
- R. Kjellander and E. Florin, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2053–2077 RSC.
- M. J. Hey, S. M. Ilett and G. Davidson, J. Chem. Soc., Faraday Trans., 1995, 91, 3897–3900 RSC.
Footnote |
| † Present address: Department of Chemistry, University of Oslo, Postboks 1033 Blindern, 0315 Oslo, Norway. |
| This journal is © The Royal Society of Chemistry 2014 |