 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge and sequence effects on the self-assembly and subsequent hydrogelation of Fmoc-depsipeptides†
Mary M.
Nguyen
,
Kevin M.
Eckes
and
Laura J.
Suggs
*
Department of Biomedical Engineering, The University of Texas at Austin, 107 W Dean Keeton Street, Austin, TX 78712, USA. E-mail: m.nguyen@utexas.edu; suggs@utexas.edu; Fax: +1 512 471 0616; Tel: +1 512 232 8593
First published on 20th February 2014
Abstract
Herein we report on the self-assembly of a family of Fmoc-depsipeptides into nanofibers and hydrogels. We show that fiber formation occurs in depsipeptide structures in which the fluorenyl group is closely associated and that side-chain charge and sequence affect the extent of self-assembly and subsequent gelation. Using fluorescence emission spectroscopy and circular dichroism, we show that self-assembly can be monitored and is observed in these slow-gelling systems prior to hydrogel formation. We also demonstrate that the ionic strength of salt-containing solutions affects the time at which self-assembly results in gelation of the bulk solution. From transmission electron microscopy, we report that morphological changes progress over time and are observed as micelles transitioning to fibers prior to the onset of gelation. Gelled depsipeptides degraded at a slower rate than non-gelled samples in the presence of salt, while hydrolysis in water of both gels and solution samples was minimal even after 14 days. Our work shows that while incorporating ester functionality within a peptide backbone reduces the number of hydrogen bonding sites available for forming and stabilizing supramolecular assemblies, the substitution does not prohibit self-assembly and subsequent gelation.
Introduction
Peptide-based structures have advantages as biomaterials because of the potential for controlled biologic function.1–3 Based on their ability to form higher-ordered structures, they are also able to self-assemble into self-supporting gels, which have application as potential injectable carriers in drug delivery4 and tissue engineering.5,6 The designer self-complementary peptides, EAK,7,8 RAD,7,8 and KFE,9,10 have been well cited in the literature, and their chemical structures alternate hydrophobic and hydrophilic domains which influence the self-assembly into nanofibers, nanotubes, or hydrogels.5,6,11–14 Self-assembly is influenced by the length, sequence, and composition of the chemical backbone within specific environmental conditions, and predicting these processes is not straightforward. For example, KFE8 and KFE16 exhibit similar gelation conditions, while KFE12, with an intermediate length, requires a lower concentration of salt to form a gel. Consistent with the EAK and RAD peptide assembly, each molecule must pack together as a beta-strand with other oligopeptides to form a beta-sheet, suggesting that KFE8 is not long enough to maintain this structure at low salt concentrations. On the other hand, KFE16 peptides have more bonds and therefore more degrees of freedom. Thus, bond rigidity is increased to stabilize the beta-strand and therefore entropy is decreased, causing a decrease in the propensity to self-assemble at lower salt concentrations.Self-assembly has also been studied on short peptide structures with conjugated aromatic groups at the N-terminal, such as the Fmoc moiety.15 Fmoc-peptides are relatively small molecules, with a molecular weight of less than 1000 Da and usually self-assemble in water to form long, fibrous structures. Fmoc-peptides are very robust in their ability to self-assemble and subsequently form gels. While the assembly of Fmoc-peptides is not well-understood, it has been proposed that the hydrophobicity and planar character of the Fmoc group, the ability of the peptide chain to hydrogen bond, and interactions among adjacent peptide residues may all contribute to the formation of nanostructured fibers and ultimately to hydrogels.16–21 For example, a library of seven Fmoc-dipeptides were evaluated for successful gelation, and results indicate that gelation was highly dependent on the interaction of the fluorenyl groups, as circular dichroism spectrum of all gels gave rise to peaks indicative of π–π* transitions in the Fmoc structure. In samples that did not form gels, no contribution from the aromatic region was observed with spectroscopic methods.22 In a separate study, Fmoc-Phe-Gly forms a gel under 2 conditions while a simple change in the sequence to Fmoc-Gly-Phe does not.16 Our group has previously demonstrated both experimentally and computationally that while the stacking of the Fmoc group of Fmoc-Ala-Ala was critical in fiber formation and subsequent gelation, the formation of β-sheet-like hydrogen bonds between molecules was not prevalent.23
The structure and function of short self-complementary peptides can be altered upon modification of the native peptide backbone.24 Altering the peptide backbone with esters to form depsipeptides, can confer properties such as faster degradation while potentially preserving biologic properties,25–27 self-assembly,28,29 and hydrogel formation.25 Incorporating esters into the peptide backbone reduces the potential for hydrogen bonding interactions, however, linear depsipeptides based on amylin sequences have been reported to self-assemble into helical ribbons and peptide nanotubes.29 Amylin(20–29) derivatives were modified with esters at positions 24, 26, and 28; self-assembly of the depsipeptides was unexpected, as a single ester substitution prevented gelation. The depsipeptide did not exhibit β-sheet conformation as is traditionally observed for amylin fibers, suggesting that folding other than β-sheet character is responsible for the self-assembly.
In an effort to capitalize on both the robust assembly and gelation of Fmoc-peptides as well as the hydrolytic sensitivity of depsipeptides, we are interested in developing Fmoc-depsipeptides as a class of biomaterials. Furthermore, we propose to use these materials to investigate the role of inter-chain hydrogen bonding, side-chain charge, and sequence specificity on self-assembly and gelation. The goal of this work is to demonstrate the self-assembly of Fmoc-depsipeptides into nanofibers and hydrogels. We show that the depsipeptide side chain charge and sequence affect the extent to which nanostructured supramolecular fibers form. We also investigated how the ionic strength of salt-containing solutions affects the time at which self-assembly results in gelation of the bulk solution.
Materials and methods
Depsipeptide design
The depsipeptides developed by our group were inspired by both short, self-complementary peptides5,12,14,30–35 and Fmoc-dipeptides.20,21,36,37 The depsipeptide library was synthesized with standard Fmoc-solid phase methods with depsi–dipeptides as the coupling building block (Fig. 1).38 The depsipeptides have an alternating sequence of hydrophilic and hydrophobic residues; they are composed of di–depsipeptide units of lysine (K) or aspartic acid (D) and lactic acid (Lac). The charges are aligned in a block-like manner for structures containing K and D residues, and an alanine (A) or phenylalanine (F) residue was positioned at the C-terminal to reduce undesired side-products.38 The depsipeptides were named according to the N-terminal amino acid and the linear order of various residues followed by the total number of residues. The design strategy of alternating hydrophobic Lac residues with the charged residues K and D was adapted from the sequence of the EAK16 peptide, which has a hydrophobic alanine (A) residue that alternates with either the hydrophilic glutamic acid (E) or lysine (K) amino acids, AEAEAKAKAEAEAKAK. We were also interested in investigating how the Fmoc group influences the self-assembly of our depsipeptide library. Many Fmoc-dipeptides have been found to self-assemble into ordered structures,37 and spectral changes due to the association and orientation of fluorenyl groups within self-assembled systems have been well-cited using fluorescence spectroscopy21,39 and circular dichroism.22,28 | ||
| Fig. 1 Depsipeptide library. The depsipeptides were named according to the N-terminal amino acid and the order of various residues, where K is lysine, Lac is lactic acid, D is aspartic acid and A or F is the residue at the C-terminus, alanine or phenylalanine respectively. The shorthand name is followed by the total number of residues. Fmoc-DAKA-8 is the peptide equivalent of Fmoc-D-Lac-K-A-8. | ||
Depsipeptide synthesis
Abbreviations: Boc, tert-butyloxycarbonyl; OtBu, tert-butyl; D2O, deuterium oxide; DCM, dichloromethane; DMAP, 4-dimethylaminopyridine; DMF, dimethylformamide; DIC, N,N′-diisopropylcarbodiimide; Fmoc, 9-fluorenylmethyloxycarbonyl; TFA, trifluoroacetic acid; TIPS, triisopropylsilane. TFA, DCM, DMF, and diethyl ether were from Fisher Scientific. DIPEA was purchased from Acros Organics. D2O was from Cambridge Isotope Laboratories, Inc. Fmoc-amino acids and DMAP were purchased from EMD Biosciences. Fmoc-Phe-Wang resin, Fmoc-Ala-Wang resin, and DIC were from Anaspec. Piperidine was from Alfa Aesar. TIPS was purchased from Acros.Fmoc-K-Lac-8, Fmoc-K-Lac-D-A-8, and Fmoc-D-Lac-K-A-8 were prepared as reported in earlier work.38 Fmoc-D-Lac-K-F-8 was prepared using the same methods with the exception of first coupling to an Fmoc-Phe-Wang resin (30%). Fmoc-DAKA-8 was synthesized with manual solid phase peptide synthesis methods (37%). Fmoc-Ala-Wang resin was coupled to Fmoc-Lys(Boc)-OH in DMF and DCM with DIC and DMAP. After 2 hours, the resin was washed with DMF and DCM. Upon removing the Fmoc group in 20% piperidine in DMF, Fmoc-Ala-OH in DMF and DCM was coupled to the resin with DIC and DMAP. After 2 hours, the resin was washed with DMF and DCM. The process of removing the Fmoc-group, coupling with Fmoc(Boc)-OH, Fmoc-Ala-OH, or Fmoc-Asp(OtBu)-OH, and washing with DMF and DCM was repeated until the desired sequence was obtained. The peptide was cleaved with TFA/water/TIPS (95:2:3) for 3 hours, precipitated in diethyl ether, and purified with HPLC. The depsipeptides were characterized using liquid chromatography-mass spectrometry (LC-MS) employing electrospray ionization (ESI).
Sample preparation
Samples were dissolved in either pure distilled and deionized water, phosphate buffered saline (PBS), or PBS with an additional 10 mM sodium chloride (PBS + NaCl). PBS without calcium and magnesium was used and had a concentration of NaCl of 137 mM before additional salt was added. Unless otherwise specified, samples were prepared at a concentration of 10 mg ml−1. The pH of the PBS and PBS + NaCl bulk solutions were adjusted with 0.1 N HCl or 0.1 N NaOH. The samples were kept at room temperature and visually observed for hydrogel formation every 18–24 hours. Gelation was defined as the inability of the solution to flow under vial inversion.Stability study
The hydrolysis products of Fmoc-D-Lac-K-A-8 were observed over 14 days at room temperature using HPLC-mass spectrometry. The Fmoc-depsipeptide formed a gel in water, PBS, and PBS + NaCl at a concentration of 10 mg ml−1 over 3 weeks, 6 days, and 2 days respectively. Samples were analyzed post-gelation. After 4, 7, 10, and 14 days, 100 μl was collected and diluted in 900 μl of respective solution. Non-gelled samples were prepared under the critical gel concentration of 1 mg ml−1 and read after 4, 7, 10, and 14 days.Fluorescence experiments
Fluorescence emission spectra were collected on a Molecular Devices plate reader provided by the DNA Sequencing Facility at the Institute of Cellular and Molecular Biology. Samples were read in 96-well black plates with an excitation of 265 nm from 300–500 nm and the respective solution background was subtracted from sample spectrum prior to data analysis.Transition emission microscopy (TEM)
Images were obtained using a FEI Tecnai Transmission electron microscope operating at an acceleration voltage of 80 kV. Samples (5 μl) were placed onto glow-discharged or untreated carbon coated copper grids for 10 seconds, removing the excess with filter paper. Water (5 μl) was placed on grids containing samples prepared in PBS or PBS + NaCl. This process was repeated twice, and the samples were then negatively stained with uranyl acetate (5 μl).Fourier transform infrared spectroscopy (FTIR)
FTIR experiments were performed with dry or wet samples on a Bruker Vertex 70 FTIR spectrometer provided by the Webb group in the Department of Chemistry at the University of Texas at Austin. The spectrometer was equipped with an A518/Q horizontal reflection cell (Bruker) for illuminating the sample at a grazing angle of 80° with respect to the surface. The reflection cell was purged with N2 for 1 hour prior to starting experiments and handled by methods reported in the literature.40 Spectra were recorded using a mercury cadmium telluride (MCT) detector, with 100–400 scans between 400 and 4000 cm−1 at a resolution of 4 cm−1. Samples were also analyzed using an ATR cell equipped with a germanium crystal. The sample substrates consisted of silicon wafers coated with chromium and gold, which were generously prepared and provided by the Webb group. The surfaces were cleaned with either hydrogen flame annealing or with a piranha wash. The latter method was followed by rinses in concentrated HCl, high purity water, and ethanol, then dried with N2. Fmoc-D-Lac-K-A-8 gelled in water, PBS, and PBS + NaCl at a concentration of 10 mg ml−1 over 3 weeks, 6 days and 2 days, respectively, prior analysis. The samples were dried overnight on clean wafers and analyzed by placing the sample side of the wafer face down on the reflection (or ATR) cell crystal. Non-gelling solutions of Fmoc-D-Lac-K-A was dissolved in water, PBS, or PBS + NaCl, immediately placed on the wafer, and dried overnight. Samples prepared in PBS + NaCl caused extensive crystallization of salts upon drying; these samples were thus analyzed wet. To analyze non-self-assembled samples, a freshly prepared sample (20 μl, 10 mg ml−1), mixed less than 2 minutes prior to measurement, was placed on the cell and sandwiched with a clean wafer. Fmoc-D-Lac-K-A-8 gelled in PBS + NaCl at a concentration of 10 mg ml−1 over 2 days prior analysis. Gelled Fmoc-DAKA-8 was prepared at a concentration of 10 mg ml−1 in water and aged at least 2 days. Turbidity was observed instantly upon dissolving Fmoc-DAKA-8 in water, thus it was not possible to investigate a pre-assembled state by taking scans soon after mixing. Adding 1.0 N NaOH to adjust the pH of Fmoc-DAKA-8 in water yielded a non-gelled state, which was used for analysis in solution. Scans of the respective solution served as the background spectrum, which was subtracted from sample spectra.Circular dichroism (CD)
CD was collected on a Jasco J-815 CD Spectrometer. Fmoc-D-Lac-K-A-8 was dissolved in PBS + NaCl, PBS, and water at 10 mg ml−1 and aged for 4 days. Fmoc-DAKA-8 was dissolved in PBS + NaCl, PBS, and water at 10 mg ml−1 initially and aged for 24 hours. The concentrations in PBS + NaCl and PBS were adjusted to bring the mixture to dissolution. The sample (50–100 μl) was pipetted onto a cylindrical cuvette (0.1 mm) and carefully sandwiched as to avoid air bubbles. CD spectra were measured over the range 180–350 nm in with a scanning speed of 200 nm min−1, data pitch of 1 nm, and 3 accumulations. The optical chamber was continually flushed with nitrogen.Rheometry
Depsipeptide solutions were prepared and allowed to gel in hollow cylindrical molds with a diameter of 8 mm. The gelled samples were carefully removed and placed on the stage of the rheometer. Measurements were collected on an Anton Paar Physica MCR 101 rheometer using an 8 mm diameter parallel plate geometry at room temperature and a fixed gap of 0.8–1.30 mm. The storage and loss moduli were measured with a frequency sweep test (0.1–100 Hz) with a fixed strain amplitude (1.0%) and were averaged over 4–5 samples.Results and discussion
Spectroscopic and morphological analysis of depsipeptide self-assembly
We investigated excimer formation of the fluorenyl group as an indicator of close association and self-assembly among the depsipeptides Fmoc-K-Lac-K-A-8, Fmoc-K-Lac-D-A-8, and Fmoc-D-Lac-K-A-8. The fluorescence emission spectrum of Fmoc-K-Lac-K-A-8 in PBS or PBS + NaCl within the pH range of 6.0–9.0 shows a peak at 310 nm (Fig. S1A†), which is comparable to the fluorescence emission spectra collected on non-assembled Fmoc-peptides: Fmoc-Lys(Boc)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Lys(Boc)-Lac-OH, and Fmoc-Asp(OtBu)-Lac-OH (Fig. S1B†). These results suggest the peak at 310 nm is the emission maximum of the fluorenyl group of each Fmoc-depsipeptide in its monomeric state, and does not correlate to a state of ordered assembly.The emission spectra of Fmoc-K-Lac-D-A-8 in ionic solutions 6 days after preparation show a shoulder peak centered around 370 nm that is absent in water (Fig. 2A). TEM images of this depsipeptide show the formation of unordered aggregates in PBS and PBS + NaCl after 11 days (Fig. 2B), while a mixture of globular structures approximately 75 nm in diameter and short fiber-like structures 200–350 nm long are observed in water. Taken with the emission spectra, this result indicates that in water, Fmoc-K-Lac-D-A-8 may undergo self-assembly to some extent. The shoulder peak of the fluorescence spectra at 370 nm has been correlated with extensive aromatic stacking of fluorenyl groups,8 which has been reported to contribute to the self-assembly of certain Fmoc-dipeptides into micelles.21,39 In our system, it is clear some self-assembly has taken place but has not resulted in regular micelle-like structures; it is possible that the increased number of residues or steric hindrance due to large lysine side chains adjacent to the fluorenyl groups prevents formation of true micelles. These results also indicate the association of the fluorenyl groups is influenced by depsipeptide sequence.
 | ||
| Fig. 2 Fluorescence emission spectra (A) and TEM (B) of Fmoc-K-Lac-D-A-8. When prepared in PBS + NaCl and PBS at 10 mg ml−1, a broad shoulder around 370 nm is observed, whereas in water the shoulder peak is not as pronounced, perhaps indicating a small degree of self-assembly. Emission spectra displayed in (A) were collected 6 days after sample preparation. TEM samples were stained with uranyl acetate, and the collected images show globular, unordered networks in ionic solutions. Short, fiber-like morphologies are observed only in aqueous solutions, indicating some self-assembly has taken place. Scale bar = 100 nm. | ||
Excimer formation between multiple aromatic ring structures, such as the Fmoc group, is readily investigated with fluorescence emission spectroscopy. Prior work with covalently-bound fluorine dimers demonstrated different emission peak energies depending on syn- or anti-arrangement.41 Multiple groups have inferred specific orientations for Fmoc groups during the self-assembly process of supramolecular fibrils, such as π-stacking, anti-parallel, or parallel association of the fluorenyl structures.20,42 Recent work has demonstrated that extensive, close aromatic stacking produces the most dramatic red shift while other, less efficient modes of aromatic overlap produce a smaller red shift over that of the monomer.43,44 In comparing the fluorescence emission spectrum of Fmoc-K-Lac-K-A-8 (Fig. S1A†) with Fmoc-K-Lac-D-A-8, our results suggest that the presence of aspartic acid in the latter depsipeptide facilitates closer interaction, and possibly excimer formation, between fluorenyl groups.
In contrast to Fmoc-K-Lac-D-A-8, Fmoc-D-Lac-K-A-8 self-assembled to ultimately form a hydrogel. Gelation was defined as the inability of the solution to flow under vial inversion, and the depsipeptide formed a hydrogel in water, PBS, and PBS + NaCl over 3 weeks, 6 days, and 2 days respectively (Table 1). After 2 days in PBS + NaCl, a red shift from 310 nm to 320 nm was observed with fluorescence spectroscopy (Fig. 3A). Similarly, the peak at 320 nm is observed in the PBS solution after 6 days (Fig. 3B), consistent with the time at which gelation was observed. Fmoc-DAKA-8, the peptide analogue of Fmoc-D-Lac-K-A-8, was also evaluated and demonstrated a similar fluorescence shift in water (Fig. 3C).
| Solvent | Calculated ionic strength in bulk (mM) | Time hydrogel observed | |
|---|---|---|---|
| PBS + NaCl | 175 | 2 days |

|
| PBS | 165 | 6 days | |
| Water | 1.46 × 10−4 | >3 weeks |
 | ||
| Fig. 3 Fluorescence emission spectra of Fmoc-D-Lac-K-A-8 in PBS + NaCl (A) and PBS (B) were collected after 2 hours, 2 days, and 6 days. In PBS + NaCl, both a red shift from 310 to 320 nm and decrease in intensity is observed upon the formation of a gel after 2 days. Similarly, the same trend is observed in PBS after 6 days. The peptide equivalent, Fmoc-DAKA-8 forms a gel in water overnight; emission spectra were collected after 2 hours, 2 days, and 6 days in water (C). | ||
The morphology of Fmoc-D-Lac-K-A-8 pre- and post-gelation was observed with TEM after 18 hours and 4 days. The study was designed to observe intermediate self-assembled structures prior to gelation, as gelation occurred after 2 days in PBS + NaCl, 6 days in PBS, and over 3 weeks in water. After 18 hours, fibers were observed in PBS and PBS + NaCl samples, while spherical aggregates were present in water (Fig. 4A–C). After 4 days, fibers are observed in all three solutions (Fig. 4D–F), similar to fibrous structures seen in Fmoc-DAKA-8 gels at the same time (Fig. S2†). Compared to the co-assembly of short, oppositely charged Fmoc-peptide hydrogels,19 in which the anti-parallel stacking of the fluorenyl groups resulted in the formation of nanofibers and ultimately supramolecular hydrogels, the Fmoc-depsipeptide nanofibers were observed under all conditions after 4 days. However, the less highly stacked positioning of the Fmoc-groups, as evidenced by fluorescence emission, was only evident in PBS + NaCl (Fig. 3A), at which point a gel had already formed. Correlating the association of the fluorenyl groups to the self-assembly of fiber networks observed in Fmoc-peptides,21,39 our results suggest that the efficiency of aromatic overlap in the Fmoc-excimer is solution-dependent.
 | ||
| Fig. 4 TEM images of Fmoc-D-Lac-K-A-8. The depsipeptide was prepared at 10 mg ml−1 in PBS + NaCl (A and D), PBS (B and E), and water (C and F). The images were collected after 18 hours (A–C, scale bar = 100 nm) and after 4 or 5 days (D–F, scale bar = 200 nm). After 18 hours, relatively short fiber networks are observed in PBS + NaCl and PBS, while globular aggregates are shown in water. After 4 or 5 days, fibers are observed for all samples. | ||
Spectral changes occur upon self-assembly of both fibers and hydrogels, although spectral changes over time are more pronounced around the gelation point; therefore, conditions under which self-assembly occurs, and therefore the emission spectra evolve, more quickly over time also cause a more rapid onset of gelation. As a comparison, Fmoc-DAKA-8 was observed to form a viscous, slightly turbid solution within minutes after mixing with water, indicating initial self-assembly. Fmoc-DAKA-8 self-assembled to a gel-forming density after 18 hours. The emission spectrum (Fig. 3C) is consistent with those of the gelled samples of the depsipeptide. The spectral shift to 320 nm is characteristic of less efficient aromatic overlap attributed to anti-parallel alignment of fluorenyl groups for Fmoc-dipeptides.21 Within our system, less efficient overlap of the Fmoc groups is observed in gelled samples, and similar emission trends have been reported in Fmoc-peptide families post-gelation.20,21,45 In water, self-assembly leads to critical gelation density only after 3 weeks. During the initial stages of assembly, it is possible that the fluorenyl group transitions from an orientation with a very high degree of stacking to a much less ordered arrangement. The process may influence the formation of micelle-like structures, which then aggregate to form fibrils in which Fmoc-groups are associated mainly within the core of the fiber. The transition into fibers causes an increase in intensity as the Fmoc groups are no longer quenched, and over time, the decrease of the fluorescence emission at 320 nm indicates the self-assembly into a fiber network.21,39
The chemical structure of our depsipeptides are designed with alternating esters within a native peptide backbone, thus the effects of hydrogen bonding are reduced. Given that self-assembly is modulated by a number of non-covalent interactions, including hydrogen bonding, Fmoc-D-Lac-K-A-8 was investigated at both early and late stages of self-assembly with IR spectroscopy in water (Fig. 5A) and PBS (Fig. 5B). Results show very little difference between the pre-gelled and post-gelled samples in water, which have peak positions at 1743, 1680, and 1544 cm−1, referring to the ester carbonyl stretch, amide I and amide II band, respectively. Similar peak patterns are observed between the pre- and post-gelled samples in PBS, with the exception of a splitting pattern at 1660 and 1643 cm−1 in the hydrogel. Fmoc-D-Lac-K-A-8 in PBS + NaCl post-gelation was compared to Fmoc-DAKA-8 in solution and a hydrogel (Fig. 5C), as described in the methods section. The depsipeptide hydrogel shows peak positions around 1741, 1686, and 1545 cm−1, which is consistent with the data observed in water. The IR spectrum of Fmoc-DAKA-8 in a non-gelled form shows sharp peaks at 1657 and 1543 cm−1 with a broad shoulder around 1579 cm−1. In gelled form, the peptide equivalent has sharp peaks at 1680 and 1628 cm−1, which are similar to peak patterns associated with β-sheet structure.20 This result can be compared to other depsipeptide systems, in which β-sheet folding is observed in the peptide counterpart but absent in the modified structure,29,25 suggesting that β-sheet interactions may be partially disrupted by the inclusion of ester bonds into the peptide backbone. Our results suggest that a higher order structure other than the more commonly reported β-sheet formation between peptide chains in Fmoc-peptide systems contributes to the self-assembly and gelation of Fmoc-D-Lac-K-A-8.
 | ||
| Fig. 5 FTIR spectra of Fmoc-D-Lac-K-A-8. The depsipeptide was prepared in water (A) and PBS (B) in solution and in gelled form. In water, the peaks pre- and post-gelation are relatively similar. In PBS, a splitting pattern in the gelled sample at 1660 and 1643 cm−1 is observed The depsipeptide gel was prepared in PBS + NaCl and compared to the peptide equivalent, Fmoc-DAKA-8 (C) in solution or as a hydrogel. All spectra were normalized to the amide II absorption peak at 1545 cm−1. | ||
While the morphology of Fmoc-D-Lac-K-A-8 nanostructures, as shown by TEM, indicates the formation of micelles and fibers in water after 18 hours and 4 days respectively, the IR data on pre- and post-gelation do not provide any additional information to allow us to distinguish between different stages of self-assembly. Our data indicates, however, that the rate of self-assembly and subsequent gelation is decreased for hydrogen bonding-deficient materials such as the depsipeptides described herein.
Helical stacking of the fluorenyl groups
CD spectra were collected for Fmoc-D-Lac-K-A-8 after 4 days at 10 mg ml−1 in PBS + NaCl, PBS, and water. At the time of the data collection, gelation was only observed in PBS + NaCl. The spectra show positive dichroism from 260–302 nm for samples prepared in the salt containing solutions (Fig. 6A), evidence that self-assembly has proceeded to a greater extent in these solutions. CD data for Fmoc-DAKA-8 was collected after 24 hours in PBS + NaCl, PBS, and water (Fig. 6), consistent with the time at which hydrogel formation is observed. The peptide was less soluble in the salt containing solutions and did not self-assemble to an extent that would form gels in PBS or PBS + NaCl at 5 or 2.5 mg ml−1. In water, gelation of Fmoc-DAKA-8 was observed after 18 hours, and peaks between 260 nm and 310 nm were observed (Fig. 6B). When observed pre-gelation, CD of Fmoc-DAKA-8 shows similar peak patterns observed in the gelled form of the peptide (data not shown), confirming that self-assembly has occurred prior to the point of gelation. A similar spectral feature, the positive peaks from 260–310 nm, is observed for other Fmoc-peptide gel systems in which the fluorenyl groups are thought to be stacked in a helical manner.22 For both the depsipeptide and peptide, helical stacking of the fluorenyl group was observed in gelled forms or in samples very close to gelation. These results suggest that the helical stacking of fluorenyl groups is a critical step in the self-assembly process that precedes the onset of gelation and appears to be a process largely unaffected by ester substitutions in the peptide backbone. | ||
| Fig. 6 CD spectra of Fmoc-D-Lac-K-A-8 in PBS + NaCl, PBS, and water (A) and Fmoc-DAKA-8, the peptide equivalent (B). The depsipeptide was prepared at 10 mg ml−1 and aged for 4 days. In PBS + NaCl and PBS, gels formed after 2 days and 6 days respectively. Strong absorbance peaks from 270–300 nm are observed, which have been reported to be helical stacking of the Fmoc-group. No gels formed in water at the time the CD was collected. Fmoc-DAKA-8 only exhibited gelation in water due to poor solubility in PBS and PBS + NaCl, therefore only the CD spectrum of the peptide in water shows a similar positive dichroism from 270–300 nm indicating helical-like association of Fmoc groups. | ||
Hydrolytic degradation of Fmoc-D-Lac-K-A-8
The stability of non-gelled and gelled samples of Fmoc-D-Lac-K-A-8 was analyzed in PBS + NaCl and water (Fig. 7) using HPLC-mass spectrometry (LC-MS). The degradation profiles in PBS (Fig. S3†) and PBS + NaCl are comparable, thus we chose to compare stability between one ionic and aqueous solution for simplicity. Low concentration samples (1 mg ml−1) were used as the non-gelled components for both solutions, as gelation has only been observed at 10 mg ml−1. In PBS + NaCl, the non-gelled sample is completely degraded after 10 days. In the gelled form, intact depsipeptide remains in the mixture after 14 days. The degradation products are most likely a result of hydrolysis of Fmoc-D-Lac-K-A-8 at the ester bonds, as the mass-to-charge (m/z) values associated with the new peaks in the chromatogram correlate to the predicted hydrolyzed structures (Fig. S4†). In water, the depsipeptide is relatively stable in both the non-gelled and gelled state, though it appears slightly less degraded in the gelled state. Within gelled samples, individual molecules are organized into supramolecular assemblies and therefore the depsipeptide backbone may be less accessible to water molecules and hydronium and hydroxide ions than in the solution state. Tian et al. observed nearly identical behaviour in solutions and gels from depsipeptides containing only one backbone ester substitution.25 It is unclear why degradation occurred faster in salt-containing solutions than in pure water; perhaps the charged ion species shield oppositely charged peptide side chains that would otherwise repulse hydronium and/or hydroxide ions responsible for ester hydrolysis.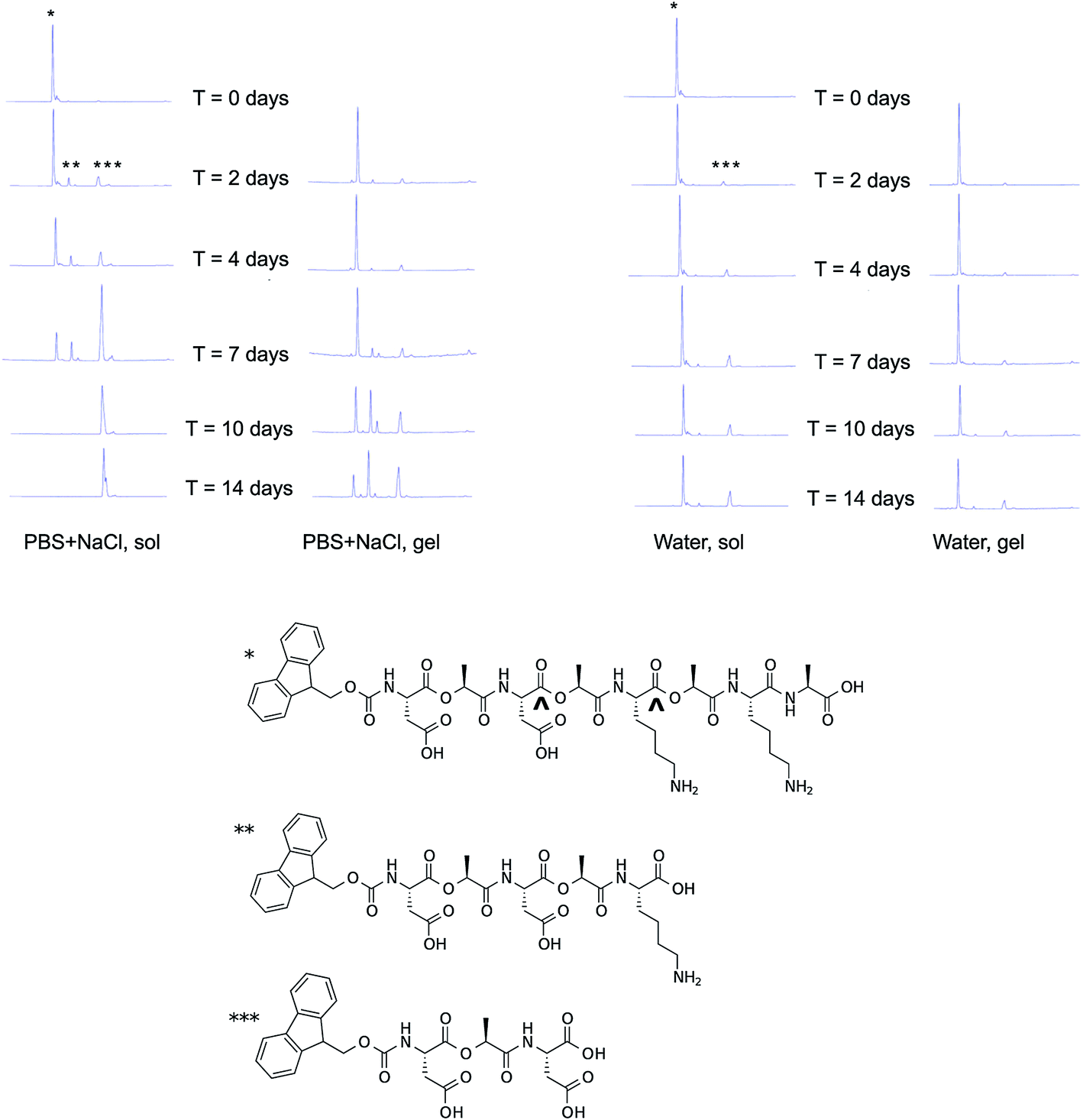 | ||
| Fig. 7 Degradation of Fmoc-D-Lac-K-A-8 over time. Degradation of the depsipeptide was evaluated with HPLC-MS in PBS + NaCl (top left), and water (top right) in a non-gelled and a gelled sample. The chemical structure of Fmoc-D-Lac-K-A-8, represented by the single asterisk (*) is hydrolyzed at two potential ester sites (^), to yield the two predicted degradation products, represented by a double (**) and triple (***) asterisk. As shown by peak intensity changes in the chromatograms over time, the depsipeptide in solution degrades after 2 days and is completely hydrolyzed in the presence of salt after 7 days. In the gelled form, the rate of hydrolysis is slower and is mostly degraded in PBS + NaCl after 14 days. In water, both the depsipeptide in solution and as a gel showed the slowest rate of gelation. The study conducted in PBS and the masses of these structures are provided in the ESI (Fig. S2 and S3†). | ||
Effect of increasing electrostatic interactions or hydrophobicity on gelation
Of all the structures investigated within our depsipeptide library, Fmoc-D-Lac-K-A-8 alone forms a hydrogel after 2 days, 6 days, or 3 weeks in PBS + NaCl, PBS, and water, respectively. Fmoc-D-Lac-K-A-16 (Fig. S5A†) was synthesized in an attempt to investigate the effect of increasing electrostatic interactions on self-assembly. No gelation was observed at 10 mg ml−1 in water, PBS, or PBS + NaCl, and the emission spectra show no unique patterns after 5 days (Fig. S5B†). In another attempt, we increased the overall hydrophobicity of the structure by adding a phenylalanine (F) to synthesize Fmoc-D-Lac-K-F-8. Gelation was observed in water within minutes at 5, 10, and 20 mg ml−1. As a comparison, the mechanical properties of Fmoc-D-Lac-K-A-8 and Fmoc-D-Lac-K-F-8 gels were analyzed using parallel-plate rheometry (Table 2). Results show that increasing the overall hydrophobicity of the depsipeptide increases the gel strength, a trend that has been reported in self-assembling Fmoc-dipeptide systems.16| Depsipeptide | Concentration (mg ml−1) | Solvent | G′ (Pa) | G′′ (Pa) |
|---|---|---|---|---|
| Fmoc-D-Lac-K-A-8 | 10 | PBS + NaCl | 34 ± 19 | 9 ± 3 |
| Fmoc-D-Lac-K-F-8 | 5 | Water | 106 ± 23 | 12 ± 2 |
Fmoc-D-Lac-K-F-8 was less soluble in PBS and PBS + NaCl than in water and did not form a hydrogel in the salt-containing solutions even after 7 days. The observed fluorescence emission peaks at 310 nm in PBS and PBS + NaCl (Fig. 8) reflect the absorbance of the fluorenyl group in solution, and does not correlate to a state of ordered assembly. In water, the association of Fmoc-groups is close but less extensively stacked as observed with the less hydrophobic Fmoc-D-Lac-K-A-8. J-aggregate features have been observed in the emission spectrum of Fmoc-Phe-Phe-OH gels, suggesting that the phenylalanine residue interacts with the fluorenyl group.21 This absorbance was not detected in our system, suggesting that when Fmoc-D-Lac-K-F-8 self-assembles into a fiber-like nanostructure, the phenylalanine ring does not associate closely with the Fmoc-group. The chemical backbone of the depsipeptide differs from Fmoc-Phe-Phe-OH in length, incorporation of esters, and the fact that it is self-complementary. All of these effects have been reported to affect self-assembly in other Fmoc-peptide systems,29,19 thus, we would not expect our results to directly trend with a system limited by two peptide residues.
 | ||
| Fig. 8 Fluorescence emission spectra of Fmoc-D-Lac-K-F-8. In water, a peak at 320 nm is observed and the depsipeptide also forms a gel under those conditions. The observed peaks at 310 nm in PBS and PBS + NaCl reflect the absorbance of the fluorenyl group in solution, as gels did not form under these conditions. | ||
Importance of fluorenyl association for Fmoc-depsipeptide gelation
The self-assembled structures of well-studied peptide families have been reported in the literature and rely heavily on spectroscopic data that correlate to specific secondary folding characteristics in better-characterized protein systems. Observations of secondary structures were absent in the analysis of Fmoc-D-Lac-K-A-8 because such interpretation with CD and IR are based on the chemical features of the native peptide backbone. The incorporation of esters within the depsipeptides disrupts both the flexibility of the structures and their ability to hydrogen bond, thus the comparison of depsipeptides to peptides may be limited with these techniques. Our results suggest that depsipeptides assemble into ordered structures, such as fibers and hydrogels, and the close association of fluorenyl groups is an important indicator of self-assembly and gelation of our materials.The gelation of our family of Fmoc-depsipeptides is influenced by the sequence of charged residues along the chemical backbone, the presence of salt, and the overall hydrophobicity of the structure. Fmoc-D-Lac-K-A-8 formed fibers and hydrogels in water, PBS, and PBS + NaCl. The structural data reveals that the fluorenyl groups are aligned in a closely associated manner in gelled samples, and the data is consistent with Fmoc-peptides that show helical stacking of the fluorenyl groups. Replacing alanine residues with lactic acid contributes to a loss of hydrogen bonding ability due to ester inclusion, which may contribute to slowing the self-assembly process and therefore extend the time to the onset of gelation. The peptide Fmoc-DAKA-8 forms a gel in water overnight, while the depsipeptide Fmoc-D-Lac-K-A-8 in the same solution forms a gel in 3 weeks. Salt-containing solutions most likely act to shield the charged residues, which may help to drive the self-assembly of Fmoc-D-Lac-K-A-8 more rapidly in PBS + NaCl than in PBS or water. Incorporating a phenylalanine residue to the depsipeptide, Fmoc-D-Lac-K-F-8, leads to hydrogel formation in water within minutes, suggesting that hydrophobic interactions may facilitate rapid self-assembly leading to gel formation in the absence of side-chain shielding salts. The hydrogen bonding sites are reduced due to the presence of the ester bond, however this structural modification does not completely disrupt self-assembly and subsequent gelation.
Conclusions
In this work, we synthesized and assayed the self-assembly and gelation capability of a small family of Fmoc-protected depsipeptide oligomers. It is clear that incorporation of regular esters into a native peptide backbone does not prohibit the self-assembly into supramolecular nanostructures that in some cases, form networks dense and entangled enough to cause gelation in water. We have demonstrated that the self-assembly of Fmoc-depsipeptides leading to hydrogel formation is dependent on charge, sequence, and salt concentration. The close association and possibly helical stacking of fluorenyl groups, as determined by fluorescence emission and CD spectroscopies, corresponds with the onset of self-assembly and precedes gelation, and the rate at which the self-assembled fibrillar networks become dense enough to form gels is dependent on the ionic character of the solution. Depsipeptide molecules within gel-forming supramolecular assemblies are protected from degradation relative to those in solution, and the presence of salt increases the hydrolysis rate of the depsipeptide molecules at the ester bond sites. Within our system, we have found that increasing the overall hydrophobicity of the structure increases both gelation rate and gel strength. Our depsipeptide library can therefore be tailored to control the formation rate and strength of the self-assembled gels, allowing them to be applied to a broad range of biomedical applications requiring materials with hydrolytic susceptibility, such as tissue scaffolds and drug delivery systems.Acknowledgements
The authors would like to thank Dr Lauren Webb, Annette Raigoza, Christina Ragain, and Jason Dugger for access and use of their FTIR; Dr Brent Iverson, Dr John Hardy, and Cameron Peebles for insightful discussions; and Ryan Stowers for his thoughtful review of this manuscript. This work was supported by the Welch Foundation and the National Institutes of Health (R21HL102806).References
- M. E. Davis, J. P. M. Motion, D. A. Narmoneva, T. Takahashi, D. Hakuno, R. D. Kamm, S. G. Zhang and R. T. Lee, Circulation, 2005, 111, 442–450 CrossRef CAS PubMed.
- A. L. Sieminski, A. S. Was, G. Kim, H. Gong and R. D. Kamm, Cell Biochem. Biophys., 2007, 49, 73–83 CrossRef CAS PubMed.
- T. C. Holmes, S. de Lacalle, X. Su, G. S. Liu, A. Rich and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- Z. L. Luo, B. Akerman, S. G. Zhang and B. Norden, Soft Matter, 2010, 6, 2260–2270 RSC.
- Y. Loo, S. Zhang and C. A. E. Hauser, Biotechnol. Adv., 2012, 30, 593–603 CrossRef CAS PubMed.
- Y. Yang, U. Khoe, X. Wang, H. Akihiro, Y. Hidenori and S. Zhang, Nano Today, 2009, 4, 193–210 CrossRef CAS PubMed.
- Z. L. Luo, X. J. Zhao and S. G. Zhang, Macromol. Biosci., 2008, 8, 785–791 CrossRef CAS PubMed.
- S. G. Zhang, T. C. Holmes, C. M. Dipersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385–1393 CrossRef CAS.
- A. Saiani, A. Mohammed, H. Frielinghaus, R. Collins, N. Hodson, C. M. Kielty, M. J. Sherratt and A. F. Miller, Soft Matter, 2009, 5, 193–202 RSC.
- M. R. Caplan, E. M. Schwartzfarb, S. G. Zhang, R. D. Kamm and D. A. Lauffenburger, Biomaterials, 2002, 23, 219–227 CrossRef CAS.
- Y. Loo, S. G. Zhang and C. A. E. Hauser, Biotechnol. Adv., 2012, 30, 593–603 CrossRef CAS PubMed.
- F. Gelain, A. Horii and S. G. Zhang, Macromol. Biosci., 2007, 7, 544–551 CrossRef CAS PubMed.
- A. Horii, X. Wang, F. Gelain and S. Zhang, PLoS One, 2007, 2, e190 Search PubMed.
- C. A. E. Hauser and S. Zhang, Chem. Soc. Rev., 2010, 39, 2780–2790 RSC.
- D. J. Adams, Macromol. Biosci., 2011, 11, 160–173 CrossRef CAS PubMed.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- H. Wang, Z. Yang and D. J. Adams, Mater. Today, 2012, 15, 500–507 CrossRef CAS.
- X. D. Xu, C. S. Chen, B. Lu, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, J. Phys. Chem. B, 2010, 114, 2365–2372 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- X. Mu, K. M. Eckes, M. M. Nguyen, L. J. Suggs and P. Ren, Biomacromolecules, 2011, 13, 3562–3571 CrossRef PubMed.
- S. Rotem and A. Mor, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1582–1592 CrossRef CAS PubMed.
- Y. F. Tian, G. A. Hudalla, H. Han and J. H. Collier, Biomater. Sci., 2013, 1, 1037–1045 RSC.
- T. Ohyama, H. Oku, M. Yoshida and R. Katakai, Biopolymers, 2001, 58, 636–642 CrossRef CAS.
- H. Oku, T. Ohyama, A. Hiroki, K. Yamada, K. Fukuyama, H. Kawaguchi and R. Katakai, Biopolymers, 2004, 75, 242–254 CrossRef CAS PubMed.
- X. Y. Yang, M. Wang and M. C. Fitzgerald, Bioorg. Chem., 2004, 32, 438–449 CrossRef CAS PubMed.
- R. C. Elgersma, T. Meijneke, G. Posthuma, D. T. S. Rijkers and R. M. J. Liskamp, Chem.–Eur. J., 2006, 12, 3714–3725 CrossRef CAS PubMed.
- M. Altman, P. Lee, A. Rich and S. G. Zhang, Protein Sci., 2000, 9, 1095–1105 CrossRef CAS PubMed.
- F. Gelain, A. Lomander, A. L. Vescovi and S. G. Zhang, J. Nanosci. Nanotechnol., 2007, 7, 424–434 CrossRef CAS PubMed.
- S. G. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS.
- S. G. Zhang, Biotechnol. Adv., 2002, 20, 321–339 CrossRef CAS.
- S. Zhang, D. M. Marini, W. Hwang and S. Santoso, Curr. Opin. Chem. Biol., 2002, 6, 865–871 CrossRef CAS.
- X. J. Zhao and S. G. Zhang, Chem. Soc. Rev., 2006, 35, 1105–1110 RSC.
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2011, 6, 475–479 RSC.
- J. F. Smith, T. P. J. Knowles, C. M. Dobson, C. E. MacPhee and M. E. Welland, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15806–15811 CrossRef CAS PubMed.
- M. M. Nguyen, N. Ong and L. Suggs, Org. Biomol. Chem., 2013, 11, 1167–1170 CAS.
- J. W. Sadownik, J. Leckie and R. V. Ulijn, Chem. Commun., 2011, 47, 728–730 RSC.
- I. F. Gallardo and L. J. Webb, Langmuir, 2012, 28, 3510–3515 CrossRef CAS PubMed.
- D. Schweitzer, K. H. Hausser and M. W. Haenel, Chem. Phys., 1978, 29, 181–185 CrossRef CAS.
- Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC.
- H. K. Kang, D. E. Kang, B. H. Boo, S. J. Yoo, J. K. Lee and E. C. Lim, J. Phys. Chem. A, 2005, 109, 6799–6804 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587–10589 RSC.
- H. M. Wang, C. H. Yang, M. Tan, L. Wang, D. L. Kong and Z. M. Yang, Soft Matter, 2011, 7, 3897–3905 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of the following: fluorescence emission spectra of Fmoc-amino acids and Fmoc-depsidipeptides, results of a degradation study of Fmoc-D-Lac-K-A-8 in PBS and mass spectra of degradation products, and fluorescence spectra of Fmoc-D-Lac-K-A-16. See DOI: 10.1039/c4sm00009a |
| This journal is © The Royal Society of Chemistry 2014 |