 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of caulonodin lasso peptides revealed unprecedented N-terminal residues and a precursor motif essential for peptide maturation†
Marcel
Zimmermann
,
Julian D.
Hegemann
,
Xiulan
Xie
and
Mohamed A.
Marahiel
*
Department of Chemistry, Biochemistry, Philipps-University Marburg, Hans-Meerwein-Strasse 4 and LOEWE-Center for Synthetic Microbiology, D-35032, Marburg, Germany. E-mail: marahiel@staff.uni-marburg.de
First published on 19th June 2014
Abstract
Lasso peptides, a peculiar family of ribosomally assembled and post-translationally modified peptides (RiPPs), possess a fascinating 3D structure, which can confer rigidity and stability against chemical and thermal denaturation. Their distinctive “lariat knot” structure is accountable for their antibacterial, enzyme inhibitory and receptor antagonist activities. While the biosynthetic machinery was recently characterized, the rules concerning the formation of this unique lasso structure on the basis of their peptide sequences remain elusive. Restrictions such as the length of the peptide, the size of the ring, or the nature of the amino acids associated with the lasso fold stabilization were recently overhauled by the identification of new members of this RiPP family. In this work we demonstrate the isolation of four genome-mining-predicted lasso peptides featuring the unprecedented amino acids serine or alanine at position 1 of the core peptide. By a mutational approach we were able to predict the lasso fold for four peptides (caulonodins IV to VII). This prediction was confirmed for caulonodin V by the full elucidation of its 3D-structure via NMR and for caulonodin VI by the determination of long range NOE-contacts. Furthermore, the substrate specificity of the biosynthetic machinery for the atypical position 1 was probed. Additionally, utilizing the recent growth of functional lasso peptide precursor sequences we were able to identify a conserved motif in the C-terminal part of the leader peptide through bioinformatics analysis. Employing an extensive in vivo analysis for substitution tolerance of the biosynthetic machinery in this conserved region confirmed the significance of several residues, indicating that the predicted motif is very likely a general leader peptide recognition sequence specific for lasso peptide maturation.
Introduction
The constant search for novel compounds for anti-cancer treatment, as antiviral drugs or antibacterial agents against multi-resistant strains presents one of the great challenges in the 21st century. The discovery and characterization of new natural compounds is the basis for this endeavor. These molecules can possess a wide variety of intrinsic bioactivities or may be useful as scaffolds for drug design and epitope grafting.1,2 While selective screenings for certain bioactivities have been performed in the past by bacterial or fungal fermentations, nowadays the rational approaches gain more ground.3,4 In particular, genome mining driven predictions and isolations of new compound become more common5,6 as sequencing costs drop dramatically. Thousands of bacterial genomes have been already sequenced, providing large amounts of data to be analyzed in upcoming research.A vast group of natural compounds that are readily accessible to genome mining approaches are the ribosomally assembled and post-translationally modified peptides (RiPPs).5,7 The gene clusters responsible for the production of these compounds contain several processing enzymes that transform a precursor peptide, consisting of a leader and a core peptide into the mature product.8 Therefore, their gene clusters are often smaller than those encoding non-ribosomal peptide synthetases (NRPS) or poly ketide synthases (PKS) with products of comparable complexity. There are nearly two dozen families of RiPPs classified by their chemical modifications and biochemical routes to these modifications.7 One family of RiPPs that is rather simple from a chemical point of view, but possesses an intriguing 3D-structure as its distinctive feature, are the lasso peptides.9
These RiPPs with a knotted structure10–12 are produced from an approx. 50 amino acid (aa) precursor peptide A, by a reaction mechanism involving two interdependent enzymes.13 The first enzyme B was shown to be a cysteine protease, while protein C shows cyclase activity.14 The latter reaction is mediated via an AMP-activation of an acidic side chain, which then attacked by the newly generated N-terminus of the core peptide leads to macrolactam ring formation. The C-terminal part of the peptide is in the meantime threaded through the ring in a presumable prefolding reaction that precedes the ring closure.8 For both reactions catalyzed by the B and the C protein, a complex formation as well as ATP hydrolysis is essential.13 The energy consumption of protein B suggests its involvement in precursor binding and prefolding. After the assembly, export might occur by an ATP-binding cassette (ABC) transporter encoded from the lasso gene cluster (as for microcin J25 and capistruin) or by another endogenous transport system, conferring immunity to the producer strain.15–17 The functions of lasso peptides in the producer strains remain elusive, although the recent discovery of a specific isopeptidase gave rise to the idea that lasso peptides may act as scavenging or signaling molecules.18 This family of RiPPs can be further divided into classes according to the presence (classes I and III) or absence (class II) of conserved cysteine residues involved in the formation of up to two disulfide bonds.19,20
Since the discovery of the first lasso peptides anantin21,22 and microcin J2523 in the early 1990s and the first reported lasso structure24 in 1994, several paradigms were established for this class of RiPPs. These include the extraordinary stability against temperature, the necessity for bulky side chain amino acids in the C-terminal tail, the restriction of ring size to eight or nine amino acids and the presence of a glycine or cysteine at the first position of the core peptide.25,26 Three of these doctrines were recently overhauled. Heat sensitive lasso peptides were recently discovered27 and it was shown that small amino acids are capable of entrapping the tail under certain circumstances.28 Furthermore, three lasso peptides with a ring size of only seven amino acids have just been reported.29 While the paradigm of the first amino acid to be Gly or Cys was still sustained, several mutagenesis approaches have shown that although the tolerance for substitution at this initial position is highly restricted,30,31 the biosynthetic machinery of the caulosegnins is capable of processing detectable amounts of G1X variants.28 To investigate the possibility of naturally occurring non-Gly1 class II lasso peptides, clusters from a recent expanded genome mining approach in proteobacteria were inspected more closely.26 These clusters have a simple ABC gene organization but feature precursors that do not fit the criterion of Gly at position 1 of the core peptide. Two of these clusters, which have been previously identified as potential lasso peptide biosynthetic gene clusters,32 are present in the genome of Caulobacter sp. K31. In total, this strain carries three lasso peptide biosynthetic gene clusters, from which only one, produces three regular Gly1 type lasso peptides,26 whereas the other clusters suggest different N-terminal residues.
Materials and methods
Strains and general methods
Caulobacter sp. K31 was purchased from the German Collection for Microorganisms and Cell Cultures (DSMZ). E. coli TOP10, which was used for cloning, and E. coli BL21(DE3), which was used for heterologous expression, were purchased from Invitrogen. Oligonucleotides and carboxypeptidase Y were purchased from Sigma Aldrich. Trypsin was purchased from Promega. Restriction enzymes, Phusion polymerase and T4 DNA Ligase were purchased from New England Biolabs. The identity of constructed plasmids and mutants was determined with DNA Sanger Sequencing and was performed by GATC Biotech AG (Konstanz).Cloning of the caulonodin biosynthesis clusters
Genomic DNA of Caulobacter sp. K31 was used to amplify the Caul19xxAABC and the Caul22xxAABC gene cluster with PCR (for primers see ESI Table S1†). PCR was performed with Phusion Polymerase following the instructions of the manufacturer. PCR was subjected to gel electrophoresis and amplicons with the correct size were excised, purified and subsequently digested with NdeI and XhoI. pET41a vector was digested in the same way and the fragments were ligated with T4 DNA Ligase. Transformation was performed with TOP10 cells for cloning and BL21(DE3) for expression.Engineering of the clusters and construction of caulonodin and rhodanodin mutants
The Caul19xxAABCpET41a plasmid and the Caul22xxAABCpET41a plasmid were used to engineer the clusters by deletion of the intergenic region between the second precursor and the gene encoding the B-protein and incorporation of a ribosomal binding site. Both these plasmids were further modified to create four single precursor plasmids by deletion of one of the precursors: Caul1984A1rbsBCpET41a, Caul198xA2rbsBCpET41a, Caul2238A1rbsBCpET41a, Caul2239A2rbsBCpET41a. The cluster engineering was entirely done with site-directed-ligation-independent-mutagenesis (SLIM).33These single precursor constructs were used for mutagenesis to create lasso peptide variants and for the investigation of the lasso precursor. These follow up mutagenic modifications were either done with a modified protocol of the site directed mutagenesis with inverse PCR34 or with the SLIM protocol. The rhotA_RBS_BC pET41a plasmid26 was used to create the rhodanodin variant. Primers for each mutant or mutant group are shown in ESI Table S1.†
Heterologous expression of the WT and mutated caulonodin and rhodanodin constructs
E. coli BL21(DE3) cells were transformed with one of the caulonodin or rhodanodin plasmids or mutants thereof and grown overnight at 37 °C in LB medium containing kanamycin (50 μg mL−1). M9 minimal medium supplemented with M9 vitamin mix (ESI Table S2†) and containing kanamycin (50 μg mL−1) was inoculated with the overnight culture to an OD600 of 0.01. The culture was grown at 37 °C to an OD600 of 0.45. Then the temperature was shifted to 20 °C, the cells were grown for 45 min to adapt to the new growth conditions and the expression was induced with the addition of IPTG to a final concentration of 0.1 mM. Expression was continued for 3 days and the cells were harvested by centrifugation. Pellets were extracted as stated in the purification section and extracts were subsequently applied to high-performance liquid chromatography (HPLC) for purification or analyzed by a high-performance liquid chromatography mass spectrometry (HPLC-MS) system.Purification of the caulonodins IV to VII
The cell pellet (6000 rpm, 20 min) of a single precursor expression culture was extracted with methanol for 1 h at RT. The obtained solvent after extraction was evaporated to dryness with a rotational evaporator at 40 °C or in case of the variants removed via lyophilization.Crude extracts of the caulonodin fermentations were resuspended in 20% acetonitrile and subsequently applied to a preparative reverse phase (RP) HPLC system (1100 series Agilent) using a C18HTec column (250 × 21 mm) with a gradient of water/0.1% trifluoroacetic acid (solvent A), acetonitrile/0.1% trifluoroacetic acid (solvent B) with a flow rate of 18 mL min−1. The gradient for all four caulonodins was as follows: a linear increase from 10% B to 60% B in 30 min followed by the washing increasing to 95% B in 2 min and holding 95% B for 5 min. Retention times (Rt) of the produced caulonodins IV to VII were 19.4, 16.0, 16.1 and 18.5 min respectively. The yield of caulonodin IV was approx. 3 mg L−1 culture. Caulonodin VI was produced with a yield of around 10 mg L−1 culture, while only about 0.7 mg L−1 culture of caulonodin VII were obtained.
Further purification of caulonodin V for NMR spectroscopic investigation was performed with an semi preparative scale HPLC system (1260 series Agilent Technologies) with a fraction collector using a Nucleodur C18ec column (250 × 5 mm) at a column temperature of 25 °C and a flow rate of 0.8 mL min−1 with the following gradient using the same solvents as before: linear increase from 25% B to 32.5% B in 15 min followed by the washing performed in the same manner as the preparative scale. Caulonodin V had a Rt of 12.0 min. The final yield of caulosegnin V was 4 mg L−1 culture.
Mass spectrometric analysis
Mass spectrometric analysis of the caulonodin and rhodanodin extracts and variant extracts was performed with a high-resolution LTQ-FT ultra instrument (Thermo Fisher Scientific) connected to a micropore 1100 HPLC system (Agilent) using sample amounts of up to 100 μL. UV absorption spectra were recorded at 215 nm. Separation was achieved using a CC Nucleosil 300-8 C18 column (125 × 2 mm) (Macherey-Nagel) applying the following gradient of water/0.1% trifluoroacetic acid (solvent A) and acetonitrile/0.1% trifluoroacetic acid (solvent B) at a column temperature of 40 °C and a flow rate of 0.2 mL min−1: starting with a linear increase from 5% to 50% B in 30 min, a subsequent increase from 50% to 95% B in 2 min and holding 95% B for additional 5 min.To quantify the products of a wild type or mutant fermentation, UV-peak areas were integrated and relative production was determined by comparison between mutant and wild type. Fermentations were carried out in triplicates for each mutant and the averages as well as standard deviations were calculated.
Collision-induced dissociation fragmentation studies within the linear ion trap were done using online HPLC-MS. In most cases the doubly charged ions were selected for fragmentation, as they were the dominant species in the spectra. The energy for fragmentation was set to 35 for every measurement performed.
Mass spectrometric analysis after the thermal stability and protease assays of the purified caulonodins was performed with a low-resolution 1100 series MSD (Hewlett-Packard) coupled with a micropore 1260 HPLC system (Agilent Technologies).
For each of the caulonodins an adapted gradient was used. For caulonodin V a linear increase from 24% B to 31.5% B in 15 min was applied. Caulonodin VI assays were analyzed with a gradient from 23% B to 30.5% B. For caulonodin VII the gradient was further adjusted starting from 28% B and finishing with 35.5% B. The washing steps were applied analog to the purification.
Heat stability assays of the caulonodins and variants
To investigate the thermal stability of the four new caulonodin lasso peptides, a solution of 10 μg purified lasso peptide was incubated at 95 °C for up to 4 h. Samples were cooled to 4 °C and were subsequently analyzed via low-resolution HPLC-MS using the individual caulonodin gradients.Heat stability of the variants was investigated by incubating 30 μL of the respective extract at 95 °C for 1 h. The samples were cooled and analyzed via high-resolution HPLC-MS or further treated with carboxypeptidase Y. As a reference 30 μL of the respective untreated extract were analyzed.
Protease assays of the caulonodins and variants
Stability against proteolytic degradation of the caulonodins and the branched-cyclic variants were investigated by incubating 10 μg of the purified lasso peptide before and after heat treatment (95 °C, 1 h) with carboxypeptidase Y. The carboxypeptidase Y assays were performed with 0.5 U carboxypeptidase Y in a buffer containing 50 mM MES and 1 mM CaCl2 at pH = 6.75 for 4 h or 16 h at 25 °C.Variants were not isolated and therefore 30 μL of the pellet extract were mixed with 30 μL of a carboxypeptidase Y (1.5 U) containing buffer (50 mM MES, 1 mM CaCl2, pH = 6.75) and incubated for 2 h at 25 °C. To stop the digest 50 μL of water were added and the samples were frozen and stored at −20 °C until applied to high or low-resolution HPLC-MS analysis.
NMR-spectroscopic methods
Samples for NMR measurements contained 4.0 mg of caulonodin V in 200 μL H2O–D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or 4.5 mg caulonodin VI in 200 μL H2O–D2O (9:1), respectively in Wilmad 3 mm tubes (Rototec Spintec). Spectra were recorded on a Bruker Avance 600 MHz spectrometer equipped with an inverse triple resonance 1H–13C–15N probe with z-gradient. The effect of temperature on the structures was surveyed by recording 1H spectra at variable temperatures. Thus, all 2D spectra for the structure determination of caulonodin V were recorded at 298 K, while for caulonodin VI 288 and 300 K were chosen for DQF-COSY, TOCSY and NOESY spectra. For sequential assignment DQF-COSY,35 TOCSY36 and NOESY37 experiments were performed in phase-sensitive mode using states-TPPI.38 TOCSY spectrum was recorded with a mixing time of 140 ms, while NOESY spectra were observed at 150 and 300 ms mixing times. Water suppression was fulfilled using excitation sculpting39 with gradients for DQF-COSY, TOCSY and NOESY. 1D spectra were acquired with 65536 data points, while 2D spectra were collected using 4096 points in the F2 dimension and 512 increments in the F1 dimension. For 2D spectra 32 transients were used, with relaxation delay of 3.0 s. Chemical shifts were referenced to H2O signal, which was calibrated in turn using 2,2-dimethyl-2silapentane-5-sulfonat (DSS) as internal standard in a different sample at the same temperature. All spectra were processed with Bruker TOPSPIN 3.1. NOE cross-peaks were analyzed within the program Sparky.40
:1) or 4.5 mg caulonodin VI in 200 μL H2O–D2O (9:1), respectively in Wilmad 3 mm tubes (Rototec Spintec). Spectra were recorded on a Bruker Avance 600 MHz spectrometer equipped with an inverse triple resonance 1H–13C–15N probe with z-gradient. The effect of temperature on the structures was surveyed by recording 1H spectra at variable temperatures. Thus, all 2D spectra for the structure determination of caulonodin V were recorded at 298 K, while for caulonodin VI 288 and 300 K were chosen for DQF-COSY, TOCSY and NOESY spectra. For sequential assignment DQF-COSY,35 TOCSY36 and NOESY37 experiments were performed in phase-sensitive mode using states-TPPI.38 TOCSY spectrum was recorded with a mixing time of 140 ms, while NOESY spectra were observed at 150 and 300 ms mixing times. Water suppression was fulfilled using excitation sculpting39 with gradients for DQF-COSY, TOCSY and NOESY. 1D spectra were acquired with 65536 data points, while 2D spectra were collected using 4096 points in the F2 dimension and 512 increments in the F1 dimension. For 2D spectra 32 transients were used, with relaxation delay of 3.0 s. Chemical shifts were referenced to H2O signal, which was calibrated in turn using 2,2-dimethyl-2silapentane-5-sulfonat (DSS) as internal standard in a different sample at the same temperature. All spectra were processed with Bruker TOPSPIN 3.1. NOE cross-peaks were analyzed within the program Sparky.40
Structure calculations for caulonodin V were performed with the program CYANA 2.1.41 The internal linkage was realized by setting the distance constraints between N of Gly1 and Cδ of Glu9 to be 1.33 Å. NOE cross-peaks observed in the 150 ms mixing time NOESY experiment were converted into distance constraints manually. In this way, 144 unambiguous distance constraints were obtained, 48 for the backbone, 22 for long-range interactions, and 74 for the side-chains. Thus, there was an average of 8.0 distance constraints per residue. In addition, constraints of torsion angles ϕ and χ1 were determined by analyzing the vicinal coupling constants 3JHNα and 3Jαα. Only unambiguous coupling constants were used. Thus, 3JHNα ≥ 9 Hz was observed for Ser1, Ile2, Asp4, Gly6, Thr15 and Tyr16. The torsion angles ϕ of these residues were restrained to −120° ± 30°. For the residues Gly3, Ser5, Leu7–Gln14 and Trp17 3JHNα < 9 Hz was detected. Thus their torsion angles ϕ were restrained to −70° ± 30°. Stereospecific assignment of the following prochiral β-methylene protons were fulfilled by measuring 3Jαβ and analyzing patterns of the intraresidual NOE interactions dαβ and dNβ:42 Ser1, Ile2 and Ser10 in (g2g3); Glu9, Tyr16 and Trp17 in (t2g3). For g2g3 and t2g3 conformations around the Cα–Cβ bond the torsion angle χ1 was constrained in the range of −60 ± 30°and 60 ± 30° respectively.
The above mentioned constraints were used in the simulated annealing protocol for calculation in CYANA 2.1 program. The calculation initiated with 50 random conformers and the resultant structures were engineered by the program package Sybyl 7.343 to include the covalent linkage between the nitrogen of Ser1 and Cδ of Glu9, followed by energy minimization under NMR constraints using TRIPOS force field within Sybyl. Thus, on the basis of low energies and minimal violations of the experimental data, a family of 15 structures was chosen. These 15 energy-minimized conformers show an average root-mean-square deviation of 0.03 Å and are kept to represent the solution structure of caulonodin V (PDB accession code 2mlj).
Results and discussion
Caulobacter K31 as a rich source of lasso peptides
Previous genome mining studies26,32,44 identified three potential lasso peptide biosynthetic gene clusters in Caulobacter sp. K31 (Fig. 1a). These feature a plain ABC gene organization and therefore are lacking a transport protein, while in close proximity several genes are located that are typical for proteobacterial lasso clusters, namely a GntR type regulator upstream and a TonB dependent receptor isopeptidase cluster downstream. All three clusters contain at least two genes encoding precursor peptides, raising the number of putative class II lasso peptide products derived from this strain to seven. The cluster 519x AAABC features three precursor peptides and has already been studied for production of lasso peptides in a recent B-protein-based genome mining approach focused on proteobacteria.26 The two remaining clusters, 198x AABC and 22xx AABC, feature two precursor peptides each. While the processing enzymes share high degrees of homology to those of the characterized cluster, the precursors do not fit the general criteria defined for lasso peptides, having unusual residues at the possible cleavage site between leader and lasso sequence. For the two precursors of cluster 22xx AABC the sequence at the cleavage site is TR/AG and TR/SG, respectively. If the location of the highly conserved Thr45 remained at the penultimate position of the leader peptide, this would lead to lasso peptides containing Ser or Ala at position 1. Applying the same assumption to the two precursors of cluster 198x AABC (cleavage site TQ/SF and TR/SI), results in two possible lasso peptides both with Ser at position 1 of the core peptide (Fig. 1b). Although more than 20 years after the discovery of the first lasso peptides21–23 no instances have been reported where another amino acid than Gly or Cys was present at position 1 of a lasso peptide. Based on the similarity of the three lasso clusters present in Caulobacter sp. K31 and the fact that one of them is functional, we were encouraged to investigate the two clusters with the unprecedented N-terminal residues, Ala and Ser, for lasso peptide production with our established methods. | ||
| Fig. 1 (a) Overview of the three putative lasso peptide biosynthetic gene clusters, (b) the four precursors from the two uncharacterized clusters, (c) the four constructs used for heterologous expression, (d) high-resolution HPLC-MS analysis of the four constructs shown as UV traces and mass signals. | ||
Heterologous production of four new lasso peptides from Caulobacter K31
In recent publications it was shown that the incorporation of an artificial ribosomal binding site between the genes encoding the precursor peptide and the downstream located biosynthetic genes greatly enhances the heterologous production of lasso peptides in E. coli.45 Furthermore, the design of single precursor constructs was shown to be beneficial for the production and subsequent purification of lasso peptides.28,29 Accordingly, four constructs were created for the heterologous expression of the four potential new lasso peptides from Caulobacter sp. K31 (Fig. 1c). Fermentations were carried out in M9 minimal medium for 3 days at 20 °C and pellet extracts were analyzed by high-resolution Fourier transform mass spectrometry system coupled to a HPLC system. In all four cases, masses were detected that correspond to the predicted full length lasso peptides, which have a serine or alanine at position 1 (Fig. 1d). The following mass fragmentation (MS2) analyses confirmed the identity of these peptides and in two of four cases prove the internal backbone to side chain cyclization of residue 1 (Ser or Ala) to Glu9 (Fig. 2). These four new lasso peptides, termed caulonodins IV to VII, are the first examples for native class II lasso peptides that do not start with a glycine but instead feature an alanine or serine residue at position 1. | ||
| Fig. 2 MS2 spectra of (a) caulonodin IV, (b) caulonodin V, (c) caulonodin VI, (d) caulonodin VII; fragments of the b and c-series highlighted in red, fragments of the y and z-series highlighted in blue; peptide bonds with high frequency of breakage due to Pro are marked in red. | ||
This essentially redefines the prerequisites for class II lasso peptides, which considered glycine to be the only suitable amino acid for the N-terminus of the core peptide. Therefore, the experimental confirmation of these peptides facilitates further genome mining for lasso peptides with any amino acid at the N-terminal position. Applying this new criterion on the results of a recent genome mining study that focused on proteobacteria raises the number of suitable precursor peptides from 7426,29 to a total of 97 out of the 98 identified precursors.
Heat and protease stability of caulonodins IV to VII
Each peptide could be produced and purified from its respective single precursor construct in sufficient amounts to allow subsequent characterization. Nonetheless, the production of the four peptides significantly differed. While the fermentation of caulonodin VII yielded 0.7 mg L−1 culture, caulonodins IV and V were produced with approx. 3 to 4 mg L−1. Caulonodin VI was produced best with a yield of 10 mg L−1 of culture.All peptides were investigated for their heat stability by incubation at 95 °C and subsequent treatment with carboxypeptidase Y to distinguish lasso peptide and unfolded branched-cyclic peptide as previously described.27
Caulonodin IV showed an unusual behavior upon thermal denaturation. Instead of the formation of a clear second peak with an identical mass, which was observed for other heat sensitive lasso peptides, it showed a decrease of the UV signal (ESI Fig. S1a†). A closer inspection of the sample revealed the formation of precipitate, indicating a strong increase in hydrophobicity of the thermally denatured peptide lowering its solubility. Therefore, it was not possible to investigate its stability against carboxypeptidase Y.
The HPLC elution profile of caulonodin VI significantly changed after heat treatment at 95 °C. A second peak with the same mass arose as well as other peaks with masses fitting to hydrolysis products of the lasso peptide (ESI Fig. S1b†). Close monitoring of this behavior with short time intervals revealed the step by step process of unfolding and subsequent hydrolysis between Asp17 and Pro18, followed by an addition of water at an unknown position. The unfolded full length peptide as well as the truncated variants were susceptible to carboxypeptidase Y.
The Caulonodins V and VII showed no change in the chromatographic behavior even after prolonged exposure (up to 4 h) to 95 °C. Nonetheless, proteolytic digestions revealed sensitivity against carboxypeptidase Y in both cases leading to the assumption that unfolding has happened and that the chromatographic behavior of the lasso peptides and their respective branched-cyclic analogues are very similar (ESI Fig. S1c and d†).
Identification of the sterically demanding amino acids for maintenance of the lasso fold
Several mutagenesis approaches have shown that the tolerance for substitutions in the sequence of a lasso peptide is rather high with the exceptions of some hot spots i.e. the amino acids involved in the macrolactam ring formation (positions 1 and 7/8/9) and the so-called plug amino acids involved in the maintenance of the lasso topology.27–31,46 Therefore, mutagenesis of possible plug amino acids and subsequent production and stability investigations can give strong hints for involvement of certain amino acids in the entrapment of the tail. From experience with other nine ring lasso peptides, the residues needed for maintenance of the lasso fold must be rather large (K, R, F, Y or W), which leaves up to three possible residues for each of the four peptides (ESI Table S3†). Hence, a set of mutants was created for each lasso peptide, replacing the possible plugs with alanine and other residues to investigate their influence on production and stability of the resulting variants.Analysis of the production and stability of the six caulonodin IV variants strongly suggests that Phe17 plays the major role in stabilization of the lasso fold, since the F17A substitution was not tolerated while the F17W variant was produced on wild type level (Fig. 3 ESI Table S4†) and was stable against thermal denaturation and subsequent carboxypeptidase Y digestion (ESI Fig. S2a†).
 | ||
| Fig. 3 Results of the mutational analysis of the plug amino acids for caulonodin IV–VII. Produced amounts of the lasso peptide variants according to UV signal integral in comparison to wild-type; color coding according to legend, asterisk marks heat stable mutants; + = detected by MS, − = not detected by MS. | ||
For caulonodin V, the alanine scan of the possible plugs indicated that either Tyr16 or Trp17 is responsible for the entrapment of the tail, since both variants were only detectable in trace amounts while a control mutant T15A was produced at 3% of wild type level. The subsequent exchange of these two possible plug residues with the smaller amino acid Phe (Y16F, W17F) also led to barely detectable amounts of lasso peptide. Only W17Y was produced at approx. 8% of wild type level (Fig. 3 ESI Table S4†). Therefore, it is very likely that both Tyr16 and Trp17 significantly contribute to the stabilization of the lasso fold and are positioned on opposite sides of the macrolactam ring.
The mutational investigation of caulonodin VI showed that while the production is lowest for the Y16A variant and Y16W is produced on wild type level (Fig. 3 ESI Table S4†), the incorporation of Trp at position 16 did not confer heat stability. Instead, the R15W variant, although only produced in small amounts, was stable against thermally induced unfolding. Subsequent carboxypeptidase Y digestion proved the resistance conferring lasso fold of this variant (ESI Fig. S2b†). On the basis of these observations it is not entirely clear if Arg15 or Tyr16 is the sterically demanding, fold-stabilizing amino acid directly below the ring. Since both residues seem to significantly participate in the fold maintenance, a structure suggestion is given where they are positioned on opposite sides of the ring (ESI Fig. S3†). This would also be in agreement with mutational studies of other lasso peptides, where the exchange of the upper plug residue with alanine could completely abolish or at least strongly diminish the peptide production.27,28,30,31
The plug scan for caulonodin VII revealed both variants R14A and R15A to be produced, although with a partially strongly decreased yield. The W16A variant was not produced, which suggests that Trp16 could be the most important residue for the lasso fold maintenance (Fig. 3 and Table S4†). The exchange to Tyr (W16Y) was tolerated well, but the stability was not significantly lowered. On the other hand, the incorporation of a Trp at position 15 (R15W) conferred heat stability to the peptide although the production was very low (ESI Fig. S2c†). These results suggest a very similar fold to caulonodin VI. Therefore both Arg15 and Trp16 are most likely to participate in the overall stabilization by being located on opposite sides of the macrolactam ring.
With this relatively small set of mutants it was possible to identify the possible plug amino acids of all four new caulonodins with reasonable certainty and as such to predict their fold (ESI Fig. 3†).
NMR structure elucidation of caulonodin V and fold determination of caulonodin VI
Although the mutational study provides a good idea of the three dimensional fold for all four new caulonodins, an unambiguous elucidation of the structure can only be achieved by NMR spectroscopic or X-ray crystallographic methods. Therefore, two caulonodins were selected and subjected to 1D and 2D-NMR experiments.NMR spectra were recorded on a sample of 4.0 mg of caulonodin V dissolved in 200 μL of H2O–D2O (9:1) leading to a concentration of 10.1 mM. The samples were prepared following standard procedures (see material and methods for Experimental details). The 1H spectra of caulonodin V at temperatures between 283 and 303 K with 5 K increments are shown in ESI Fig. S4.† Spectra are presented in the region 10.6–6.6 ppm for clarity and labels for the signal assignments of the amide protons are attached. Neat and well resolved single set of signals were observed and a diverse distribution of the temperature response of these signals revealed a stable lasso fold of this peptide.47 The best signal resolution was observed at 298 K and a full signal assignment (see ESI Table S5†) of the 1H signals was obtained by standard procedures.48 A combination of DQF-COSY and NOESY produced sequential assignments (i.e. all αH and NH and their sequence in the backbone), and a combination of DQF-COSY and TOCSY allowed the determination of the side chains. One pure conformation was observed and full assignment of 1H signals was thus obtained (NOESY spectrum: ESI Fig. S5, 1H chemical shifts: ESI Table S5†). Strong NOE contacts between the NH of Ser1 and the γH of Glu9 were observed, showing an internal linkage between these two residues. Inspection of 1H spectra between 283 and 303 K (ESI Fig. S4†) revealed almost no temperature dependence of the NH of Glu9 and a very weak dependence of those of Tyr16 and Trp17. Furthermore, a large number of long-range NOE contacts were observed (ESI Fig. S5†). These are the connectivity between Asp4–Tyr14, Ser5–Tyr16, Gly6–Tyr16, Leu7–Tyr16, Arg8–Tyr16, Glu9–Tyr16, Ser10–Tyr16, Ser1–Trp17, Glu9–Trp17, Leu7–Trp17, Ile2–Pro18, Ser10–Ser13 and Ser10–Gln14. All these short distances identified in the NOESY spectrum are in favor of a lasso structure of caulonodin V.
Observed NOE cross-peaks were converted into distance constraints manually and used for structure calculation as well as torsion angle constraints from unambiguous coupling constants. With these constraints, 15 minimum energy structures were obtained that are in minimal violation with the experimental data (for details see methods and ESI Table S5†).
The family of 15 structures shown in Fig. 4 represents the lasso fold of caulonodin V in aqueous solution at 25 °C (for Ramachandran plot see ESI Fig. S6†). The extra cyclic part of the peptide is threaded through the ring and thus divided into two parts, the seven membered loop and the two residue tail. This loop is up to date the second largest after microcin J25 and might therefore be suitable for epitope grafting applications.49 In particular, the residues M11-S12-S13-Q14 form a slightly bent linear sequence that is applicable for the presentation of a peptide epitope. The two amino acids Tyr16 and Trp17 are located directly above and below the ring respectively. The structure is therefore in agreement with the postulate, which predicted Tyr16 and Trp17 to play the most important roles in stabilizing the structure, acting as the plug amino acids on opposite sides of the ring. Furthermore, the structure of caulonodin V is the first one of a lasso peptide with a 9 aa glutamate-mediated ring (29 atoms). Although the structure is stabilized in a sandwich-like manner, as it is the case for astexin-1 and microcin J25, the fold seems less stable due to the enlarged ring.
 | ||
| Fig. 4 3D structure of caulonodin V determined by NMR-spectroscopy. (a) Overlay of the 15 minimum energy structures, only backbone shown; (b) schematic representation of the structure; (c) relaxed eyes stereo view of the structure, backbone atoms of the aa in the ring in yellow, in the tail in blue, side chain atoms of the macrolactam ring forming aa in red, of the sterically fixating aa in green and other aa in grey; (d) interaction of steric locks in the tail and the ring, backbone and side chains shown as sticks colored by atoms, ring surface shown in orange, plug surfaces colored by atoms. | ||
To acquire more evidence for the suggested structures of the caulonodins from the second biosynthetic machinery caulonodin VI was subjected to 1D and 2D NMR experiments. The 1H spectra in temperature range 283–303 K (ESI Fig. S7†) show a chemical shift dispersion of 2.5 ppm for the amide protons, which denotes a stable secondary structure of caulonodin VI in aqueous solution. The 2D spectra assured the signal assignments of the backbone and partially the side chain protons of Ala1, Gly2, Thr3, Gly4, Val5, Leu7, Glu9, Thr10, Gln12, Ile13, Lys14, Arg15, Tyr16, Asp17, and Ala19 (ESI Table S7†). Thus, long-range NOEs between NH of Ala1 and γCH2 of Glu9 were observed verifying the isopeptide bond between these two amino acids. Further long-range NOEs (ESI Fig. S8†) were detected between the backbone NH of Arg15 and the αH of Gly2, the NH of Tyr16 and the βCH2 of Leu7, the 2,6H and the 3,5H of Tyr16 and the αH of Gly2, the 2,6H and the 3,5H of Tyr16 and the γCH2 of Leu7, the 2,6H and the 3,5H of Tyr16 and the βCH2 of Glu9, and the 2,6H and the 3,5H of Tyr16 and the γCH2 of Glu9. These contacts reveal Arg15 and Tyr16 to be the most likely residues to serve as plugs for caulonodin VI, which was already suggested by the results of the mutational analysis. However, a full structure determination was not possible, due to the strong overlay of the side chain signals. In particular, due to the reduced thermal stability of caulonodin VI, it was not possible to unambiguously assign any signals to Asn11 and the overlay in the region 0.8 to 2.5 ppm prevented full assignment of the side chain hydrogens including βH for 9 of the 19 aa. For a full 3D structure determination concerning a lasso peptide an average of about 8 to 10 unambiguous constraints per residue are needed, which is strongly dependent on clear side chain assignments. A structural calculation on the basis of the observed data would produce a 3D structure of insufficient quality and high uncertainty and is therefore not advised.
Elucidation of substrate specificity at the N-terminus
For the four new caulonodins the amino acid at position 1 is especially interesting since these are the first examples of class II lasso peptides featuring a different residue than glycine at the N-terminus. Hence, a set of mutants was created substituting the initial Ser or Ala of the best produced peptide of each cluster (caulonodin V and VI) with other amino acids of comparable size (Gly, Ala/Ser, Cys, Val and Thr) and a large one (Phe). In general agreement with previous mutagenesis studies,27,28,30,31 the tolerance for substitution at this position was very limited (Fig. 5 ESI Table S4†). Only the Ser to Ala substitution and vice versa was tolerated very well. The glycine substitution lead to a strong decrease in production, indicating that the specificity of the biosynthetic machinery for the first amino acid has shifted from glycine towards the slightly larger amino acids alanine and serine. Despite the chemical and sterical similarity (Cys, Val, Thr), the other variants were barely detected. As expected, the same severe impact on production was observed for the Phe substitution, which was designed as a negative control. Although the substrate specificity of the maturation machinery for position 1 in these two clusters is still very high, it represents so far the first example of a biosynthetic machinery producing lasso peptides with two amino acids (Ala and Ser) at the initial position in similar amounts and tolerating a third amino acid (Gly) with moderately to strongly reduced production. | ||
| Fig. 5 Results of the mutational analysis of the specificity for position 1 in (a) the caulonodin V system and (b) the caulonodin VI system. Produced amounts of the lasso peptide variants according to UV signal integral in comparison to wild-type; color code as in Fig. 3; insets show column diagram with standard deviation error bars; + = detected by MS. | ||
Identification of a lasso peptide leader motif and mutational analysis of the caulonodin leader peptides
Since Caulobacter sp. K31 features only two putative secondary metabolite clusters identified by antiSMASH,50,51 it is remarkable that it contains three lasso peptide biosynthetic gene clusters with a total of seven products. These three clusters therefore present an interesting starting point for comparative bioinformatic studies. It is apparent, that the two clusters investigated in this study are very similar to each other (protein identities of over 40%), while the previously studied three precursor cluster differs significantly (protein identities of below 31%). The precursor peptides show an overall low identity of only 9% and a similarity of 16%. Interestingly these residues are mainly located in the leader peptide in a range of ten amino acids in front of the cleavage site (ESI Fig. S9†). To verify the importance of this region five deletion mutants of the precursor genes coding for caulonodin V were created omitting six or seven amino acid long parts of the leader sequence of the precursor peptide. The initial methionine and the last two amino acids of the leader peptide containing the previously investigated highly conserved Thr-2 were left untouched (Fig. 6a).27–29,45 The results showed that only deletion of the first seven amino acids led to an amount of produced lasso peptide detectable by UV (Fig. 6b ESI Table S4†). All other deletions almost completely abolished the production. It therefore seems that the C-terminal 14 residues of the leader sequence play a major role in the interaction with the biosynthetic machinery and the processing of the precursor into the mature lasso peptide. A potential general recognition site for the processing of lasso peptides might be located in this region.52 It is known from other RiPPs like cyanobactins and lanthipeptides, that recognition sequences often directly precede or follow the core peptide sequence.53,54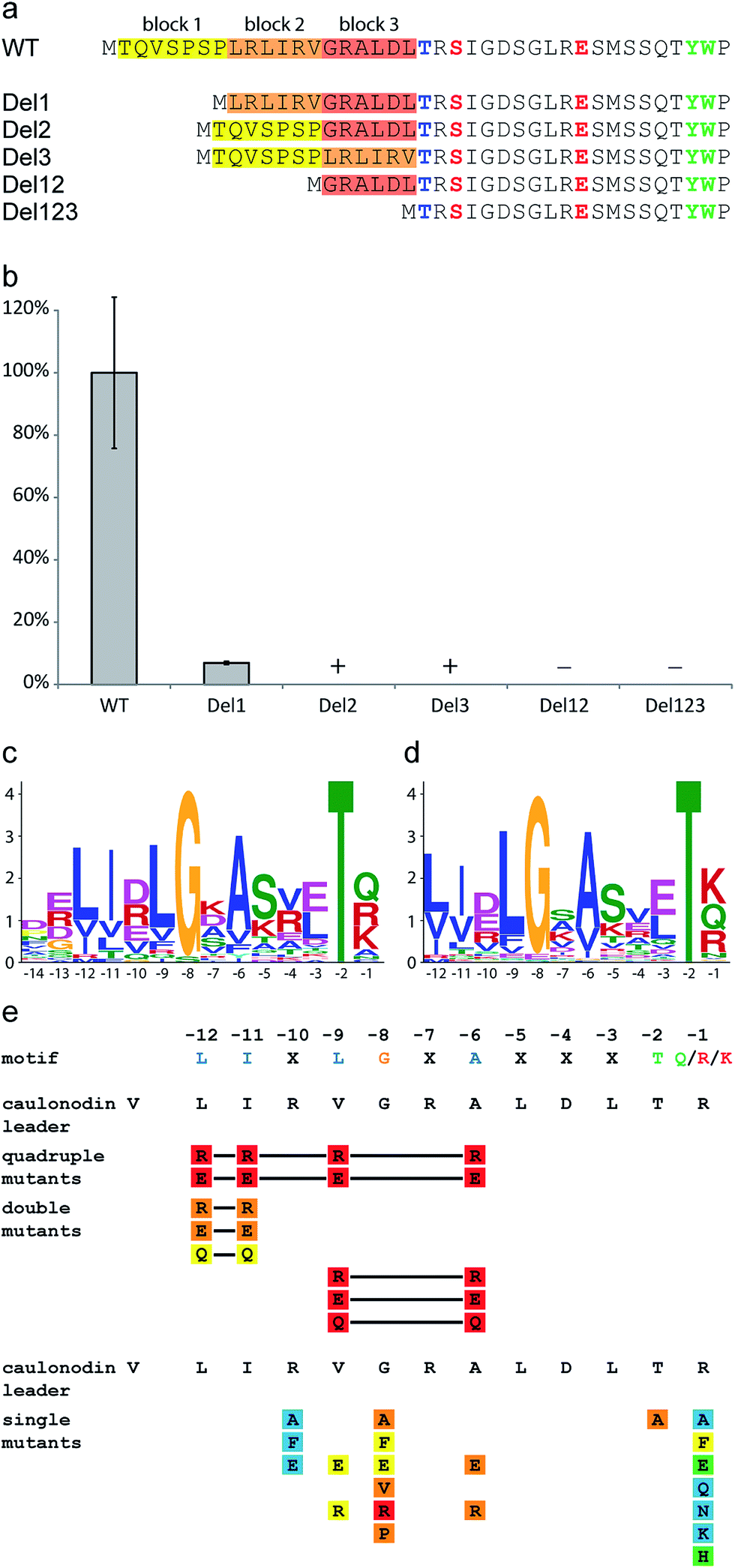 | ||
| Fig. 6 Effects of the deletion, single and multiple substitution mutations of the lasso peptide leader sequence on caulonodin V production. (a) Scheme showing constructed deletions of caulonodin V leader peptide and (b) their influence on peptide production compared to WT, according to UV signal; + detected by MS, − = not detected by MS; (c) motif identified in 27 leader peptides by the MEME algorithm using all sequences from the 30 functional precursors and (d) all non-identical putative precursors from recently identified lasso gene clusters26 as query; (e) influence of substitutions in the precursor on production of caulonodin V; color code as in Fig. 3. | ||
Recently, several new lasso peptides have been isolated by genome mining approaches and 30 lasso peptides and their respective gene clusters are known. Although they are mainly produced by proteobacteria, three examples are from an actinomycetal origin6,55 and therefore present the basis for a broader view. A bioinformatic analysis of the precursor peptides from all known functional lasso clusters with the MEME algorithm56 unveiled a motif located in the C-terminal part of the leader peptide ranging from position −12 to −1 (Fig. 6c) with the consensus sequence LIxLGxAxxxTx. The same motif was identified, when all putative precursors identified from a recent genome mining study26 were used as the query (Fig. 6d). Interestingly, this motif was not observed in the precursors of the prototypical lasso peptide microcin J25, capistruin and burhizin. On the other hand it was not only found in 24 proteobacteria but also in the three actinomyces species and it may therefore present a more or less general recognition motif in the leader sequence of lasso peptide precursors (alignment shown in ESI Fig. S10†).
In this motif, several residues are highly conserved, namely Gly-8 and Thr-2 as well as hydrophobic residues at positions −12, −11, −9 and −6. Furthermore there is some conservation of the residue −1 (Gln/Lys/Arg), which predominantly contains an amino function and the residue −10 (Asp/Arg/Glu), which is in most cases a charged residue, even though the polarity seems not to be conserved. The residues −7, −5, −4 and −3 are quite variable, although residue −5 is in general rather hydrophilic, while the residues −4 and −3 alternate between hydrophilic and hydrophobic.
To investigate the importance of certain residues in this newly discovered motif, 29 single or multiple substitution mutants were created for all highly and moderately conserved residues in the precursor gene coding for caulonodin V (Fig. 6e ESI Table S4†). The importance of the four hydrophobic residues at positions −12, −11, −9 and −6 was assessed with quadruple, double and certain single mutants, revealing a general importance of this hydrophobic patch for the in vivo maturation. Furthermore, these substitutions highlighted that Ala-6 is the most important residue of the four, since its single residue substitutions were barely detected. The previously reported importance of Thr-245 was confirmed by the significantly decreased production of a T-2A mutant. The newly discovered conserved Gly-8 showed an almost comparable significance since all substitutions were processed in very small amounts (Ala, Phe, Glu, Val, Pro) or not at all (Arg). The two other residues probed in this approach R-10 and R-1 showed only minor influence on effective maturation. All three R-10 variants were produced on wild type level, rendering this position completely variable. The processing of the R-1 variants shows a preference of the maturation machinery towards medium or small residues (Ala, Glu, Gln, Asn, Lys, His) over large amino acids (Phe). Since this is the ultimate position of the leader peptide, it is very likely that besides Thr-2, this residue is also crucial for the recognition by the protease function containing protein B. Together with the previous studies the protease might recognize two medium sized residues best, while large residues disturb the interaction.
To further emphasize the general relevance of the identified motif, a mutational approach was designed to show an enhancing effect on the production of a lasso peptide upon restoration of the correct motif. For this, the precursor of the lasso peptide rhodanodin from Rhodanobacter thiooxidans LCS2 was chosen,26 in which the motif was also identified (ESI Fig. S10†), but the central Gly-8 was substituted by Ser. This was especially intriguing as lasso peptide precursors found in other Rhodanobacter species conformed to the conserved motif. The exchange mutation S-8G led to a more than 10–fold production increase (ESI Table S4†), highlighting the significance of this residue within the motif.
It was shown that several conserved residues, which are clustered in the identified motif, have a strong impact on the effective processing of the precursor peptide to the mature lasso peptide in vivo. Some of these residues, especially Gly-8 and Ala-6 can be considered as equally important for the enzyme recognition as the previously identified Thr-2.
Conclusion
In this work, we applied genome mining techniques to broaden the scope for new lasso peptides beyond previously established boundaries by looking for unusual precursor peptide sequences. Four of these sequences are present in two clusters from the strain Caulobacter sp. K31, which also features a standard lasso peptide biosynthesis cluster that was recently characterized. Utilizing the engineered clusters in an E. coli expression system, it was possible to isolate and characterize four new lasso peptides, the caulonodins IV to VII, featuring Ser or Ala at position 1, breaking the paradigm of class II lasso peptides. A mutational study focusing on the identification of the plug amino acids, made it possible to predict the folds of all four new caulonodins with reasonable certainty. These predictions were tested for two instances, caulonodin V and VI. The 3D fold and structure elucidation by NMR techniques confirmed the postulated folds for both caulonodins and unveiled a lasso structure of caulonodin V with a seven membered loop and a two residue tail. The substrate specificity and the tolerance for substitution of the atypical amino acid at position 1 was probed for both biosynthesis clusters in a mutational approach revealing a shifted specificity from Gly to Ser and Ala. Both residues were accepted equally well, while the general tolerance for substitution at this key position is very limited.The second major part of this study focused on the leader peptide and its influence on effective processing by the biosynthetic enzymes. An initial deletion study pointed towards the C-terminal part as an important region of the leader peptide. A subsequent bioinformatics analysis using the MEME algorithm uncovered a motif with several highly conserved residues. These residues were analyzed for their influence on in vivo processing of the precursor peptide. Substitutions of several residues had a significant impact on production comparable to Thr-2 exchanges, revealing their importance for effective processing. Since these residues are clustered in an area of the precursor close to the cleavage site, it is likely that they form a recognition motif for the post-translational modification enzymes. Hence, this motif may directly interact with the lasso peptide processing machinery consisting of proteins B and C in a similar fashion as it was shown for other RiPPs like lanthipeptides by shifting the equilibrium of the enzymes from an inactive to an active state.53,57 Future studies focusing on the interaction of soluble lasso peptide processing enzymes with the precursors or co-crystallization experiments may provide further insight into the mechanism of this interaction.
Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft and the LOEWE program of the state of Hesse is gratefully acknowledged.References
- A. G. Poth, L. Y. Chan and D. J. Craik, Biopolymers, 2013, 100, 480–491 CrossRef PubMed.
- Y. Ji, S. Majumder, M. Millard, R. Borra, T. Bi, A. Y. Elnagar, N. Neamati, A. Shekhtman and J. A. Camarero, J. Am. Chem. Soc., 2013, 135, 11623–11633 CrossRef CAS PubMed.
- C. D. Deane and D. A. Mitchell, J. Ind. Microbiol. Biotechnol., 2014, 41, 315–331 CrossRef CAS PubMed.
- M. Hedvat, L. Emdad, S. K. Das, K. Kim, S. Dasgupta, S. Thomas, B. Hu, S. Zhu, R. Dash, B. A. Quinn, R. A. Oyesanya, T. P. Kegelman, U. K. Sokhi, S. Sarkar, E. Erdogan, M. E. Menezes, P. Bhoopathi, X. Y. Wang, M. G. Pomper, J. Wei, B. Wu, J. L. Stebbins, P. W. Diaz, J. C. Reed, M. Pellecchia, D. Sarkar and P. B. Fisher, Anti-Cancer Agents Med. Chem., 2012, 12, 1143–1155 CrossRef CAS.
- J. E. Velasquez and W. A. van der Donk, Curr. Opin. Chem. Biol., 2011, 15, 11–21 CrossRef CAS PubMed.
- R. D. Kersten, Y. L. Yang, Y. Q. Xu, P. Cimermancic, S. J. Nam, W. Fenical, M. A. Fischbach, B. S. Moore and P. C. Dorrestein, Nat. Chem. Biol., 2011, 7, 794–802 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- T. J. Oman and W. A. van der Donk, Nat. Chem. Biol., 2010, 6, 9–18 CrossRef CAS PubMed.
- M. O. Maksimov and A. J. Link, J. Ind. Microbiol. Biotechnol., 2014, 41(2), 333–344 CrossRef CAS PubMed.
- K. A. Wilson, M. Kalkum, J. Ottesen, J. Yuzenkova, B. T. Chait, R. Landick, T. Muir, K. Severinov and S. A. Darst, J. Am. Chem. Soc., 2003, 125, 12475–12483 CrossRef CAS PubMed.
- K. J. Rosengren, R. J. Clark, N. L. Daly, U. Goransson, A. Jones and D. J. Craik, J. Am. Chem. Soc., 2003, 125, 12464–12474 CrossRef CAS PubMed.
- M. J. Bayro, J. Mukhopadhyay, G. V. Swapna, J. Y. Huang, L. C. Ma, E. Sineva, P. E. Dawson, G. T. Montelione and R. H. Ebright, J. Am. Chem. Soc., 2003, 125, 12382–12383 CrossRef CAS PubMed.
- K. P. Yan, Y. Li, S. Zirah, C. Goulard, T. A. Knappe, M. A. Marahiel and S. Rebuffat, ChemBioChem, 2012, 13, 1046–1052 CrossRef CAS PubMed.
- S. J. Pan, J. Rajniak, W. L. Cheung and A. J. Link, ChemBioChem, 2012, 13, 367–370 CrossRef CAS PubMed.
- T. A. Knappe, U. Linne, S. Zirah, S. Rebuffat, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2008, 130, 11446–11454 CrossRef CAS PubMed.
- K. Kuznedelov, E. Semenova, T. A. Knappe, D. Mukhamedyarov, A. Srivastava, S. Chatterjee, R. H. Ebright, M. A. Marahiel and K. Severinov, J. Mol. Biol., 2011, 412, 842–848 CrossRef CAS PubMed.
- K. Adelman, J. Yuzenkova, A. La Porta, N. Zenkin, J. Lee, J. T. Lis, S. Borukhov, M. D. Wang and K. Severinov, Mol. Cell, 2004, 14, 753–762 CrossRef CAS PubMed.
- M. O. Maksimov and A. J. Link, J. Am. Chem. Soc., 2013, 135, 12038–12047 CrossRef CAS PubMed.
- S. Duquesne, D. Destoumieux-Garzon, S. Zirah, C. Goulard, J. Peduzzi and S. Rebuffat, Chem. Biol., 2007, 14, 793–803 CrossRef CAS PubMed.
- T. A. Knappe, U. Linne, X. Xie and M. A. Marahiel, FEBS Lett., 2010, 584, 785–789 CrossRef CAS PubMed.
- D. F. Wyss, H. W. Lahm, M. Manneberg and A. M. Labhardt, J. Antibiot., 1991, 44, 172–180 CrossRef CAS.
- W. Weber, W. Fischli, E. Hochuli, E. Kupfer and E. K. Weibel, J. Antibiot., 1991, 44, 164–171 CrossRef CAS.
- R. A. Salomon and R. N. Farias, J. Bacteriol., 1992, 174, 7428–7435 CAS.
- D. Frechet, J. D. Guitton, F. Herman, D. Faucher, G. Helynck, B. Monegier du Sorbier, J. P. Ridoux, E. James-Surcouf and M. Vuilhorgne, Biochemistry, 1994, 33, 42–50 CrossRef CAS.
- S. Rebuffat, A. Blond, D. Destoumieux-Garzon, C. Goulard and J. Peduzzi, Curr. Protein Pept. Sci., 2004, 5, 383–391 CrossRef CAS.
- J. D. Hegemann, M. Zimmermann, S. Zhu, D. Klug and M. A. Marahiel, Biopolymers, 2013, 100, 527–542 CrossRef PubMed.
- M. Zimmermann, J. D. Hegemann, X. Xie and M. A. Marahiel, Chem. Biol., 2013, 20, 558–569 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2013, 135, 210–222 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, S. Zhu, H. Steuber, K. Harms, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2014, 53(8), 2230–2234 CrossRef CAS PubMed.
- O. Pavlova, J. Mukhopadhyay, E. Sineva, R. H. Ebright and K. Severinov, J. Biol. Chem., 2008, 283, 25589–25595 CrossRef CAS PubMed.
- T. A. Knappe, U. Linne, L. Robbel and M. A. Marahiel, Chem. Biol., 2009, 16, 1290–1298 CrossRef CAS PubMed.
- K. Severinov, E. Semenova, A. Kazakov, T. Kazakov and M. S. Gelfand, Mol. Microbiol., 2007, 65, 1380–1394 CrossRef CAS PubMed.
- J. Chiu, D. Tillett, I. W. Dawes and P. E. March, J. Microbiol. Methods, 2008, 73, 195–198 CrossRef CAS PubMed.
- A. Hemsley, N. Arnheim, M. D. Toney, G. Cortopassi and D. J. Galas, Nucleic Acids Res., 1989, 17, 6545–6551 CrossRef CAS PubMed.
- M. Rance, O. W. Sorensen, G. Bodenhausen, G. Wagner, R. R. Ernst and K. Wuthrich, Biochem. Biophys. Res. Commun., 1983, 117, 479–485 CrossRef CAS.
- A. Bax and D. G. Davis, J. Magn. Reson., 1985, 65, 355–360 CAS.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546–4553 CrossRef CAS PubMed.
- D. Marion, M. Ikura, R. Tschudin and A. Bax, J. Magn. Reson., 1989, 85, 393–399 CAS.
- T. L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112, 275–279 CrossRef CAS.
- T. D. Goddard and D. J. Kneller, SPARKY 3, University of California, San Francisco Search PubMed.
- T. Herrmann, P. Guntert and K. Wuthrich, J. Mol. Biol., 2002, 319, 209–227 CrossRef CAS.
- G. Wagner, Prog. Nucl. Magn. Reson. Spectrosc., 1990, 22, 101–139 CrossRef CAS.
- Tripos, Tripos Inc., 1699 South Hanley Rd. St. Louis, MO 631444, 7.3 edn, 2006 Search PubMed.
- M. O. Maksimov, I. Pelczer and A. J. Link, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15223–15228 CrossRef CAS PubMed.
- S. J. Pan, J. Rajniak, M. O. Maksimov and A. J. Link, Chem. Commun., 2012, 48, 1880–1882 RSC.
- S. J. Pan and A. J. Link, J. Am. Chem. Soc., 2011, 133, 5016–5023 CrossRef CAS PubMed.
- X. Xie and M. A. Marahiel, ChemBioChem, 2012, 13, 621–625 CrossRef CAS PubMed.
- K. Wüthrich, NMR of proteins and nucleic acids, Wiley, New York, 1986 Search PubMed.
- T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, U. Linne, F. Sasse, H. Kessler, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2011, 50, 8714–8717 CrossRef CAS PubMed.
- K. Blin, M. H. Medema, D. Kazempour, M. A. Fischbach, R. Breitling, E. Takano and T. Weber, Nucleic Acids Res., 2013, 41, W204–W212 CrossRef PubMed.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed.
- W. L. Cheung, S. J. Pan and A. J. Link, J. Am. Chem. Soc., 2010, 132, 2514–2515 CrossRef CAS PubMed.
- G. C. Patton, M. Paul, L. E. Cooper, C. Chatterjee and W. A. van der Donk, Biochemistry, 2008, 47, 7342–7351 CrossRef CAS PubMed.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS PubMed.
- J. Inokoshi, M. Matsuhama, M. Miyake, H. Ikeda and H. Tomoda, Appl. Microbiol. Biotechnol., 2012, 95, 451–460 CrossRef CAS PubMed.
- T. L. Bailey, M. Boden, F. A. Buske, M. Frith, C. E. Grant, L. Clementi, J. Ren, W. W. Li and W. S. Noble, Nucleic Acids Res., 2009, 37, W202–W208 CrossRef CAS PubMed.
- T. J. Oman, P. J. Knerr, N. A. Bindman, J. E. Velasquez and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 6952–6955 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Heat and protease stability assays, 3D fold schemes, 1H-NMR and 2D NOE spectra of caulonodin V, Ramachandran plot, 1H-NMR and 2D NOE spectra of caulonodin VI, caulonodin precursor alignment, MEME leader alignment, a list of all used oligonucleotide primers, contents of M9 vitamin mix, overview of possible plug amino acids, detailed production amounts of all created mutants, 1H chemical shift assignment and structural statistics of caulonodin V and a partial 1H chemical shift assignment of caulonodin VI. See DOI: 10.1039/c4sc01428f |
| This journal is © The Royal Society of Chemistry 2014 |