 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An organic cation as a silver(I) analogue for the arylation of sp2 and sp3 C–H bonds with iodoarenes†
Carlos
Arroniz
ab,
J. Gabriel
Denis
a,
Alan
Ironmonger
b,
Gerasimos
Rassias‡
b and
Igor
Larrosa
*a
aSchool of Biological and Chemical Sciences, Queen Mary University of London, Joseph Priestley Building, Mile End Road, E1 4NS, London, UK. E-mail: i.larrosa@qmul.ac.uk; Tel: +44 (0) 20 7882 8404
bGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, Hertfordshire, UK
First published on 26th June 2014
Abstract
Reactions promoted by stoichiometric amounts of silver salts suffer from high cost, limited availability and raise environmental concerns. This manuscript describes studies leading to the discovery of a general replacement for silver with an inexpensive and convenient organic salt in palladium catalyzed direct C(sp2)–H and C(sp3)–H arylation reactions.
One of the fundamental goals in chemical research is the development of broadly applicable, cost-effective, practical and sustainable synthetic methods. In this context, the development of C–H bond arylation processes has attracted significant interest as more environmentally-friendly alternatives to traditional cross-couplings. These new transformations use readily available starting materials, thus avoiding the preparation and use of organometallic reagents as coupling partners and the associated generation of stoichiometric amounts of metallic waste.1
In 2005, Daugulis and Zaitsev developed a method for the direct arylation of anilides with iodoarenes using a catalytic system involving Pd(OAc)2 in combination with stoichiometric AgOAc.2 This powerful system and its various modifications have since been successfully applied to a great variety of C–H arylation processes involving iodoarene coupling partners (Scheme 1).3,4 However, to date one of the major drawbacks of these methodologies is the requirement for stoichiometric silver additives.5 Silver(I) salts are a uniquely appropriate partner for Pd, Au, Cu, Rh, Ru and Pt catalysts, particularly when halide abstraction is required in the catalytic process.6 However, due to the ability of silver salts to also act as oxidants and Lewis acids, sometimes undesired side reactions are observed.7 Furthermore, when used as stoichiometric reagents, these expensive silver salts considerably increase the overall cost of the process and generate significant amounts of metal waste. Therefore those transformations are prohibitively expensive for industry. Importantly, whereas there are alternatives to Ag salts in their role of oxidants, the only other well-known halide scavengers (Pb, Tl and Hg salts) are highly toxic and rarely used for this purpose.
 | ||
| Scheme 1 Selected examples of the general scope of current protocols for C–H arylation using iodoarenes and stoichiometric silver salts. | ||
Recent studies on the use of Ag salts as additives in Au-catalyzed reactions have shown that, in addition to abstracting a halogen, Ag may play additional roles essential to the catalytic process and form mixed Au–Ag active species.8 In the case of C–H arylation reactions, it is possible that Ag salts may also be acting as terminal oxidants, assisting in the oxidative addition step or facilitating the C–H activation. A recent mechanistic study on the role of additives in palladium acetate-catalyzed ortho-C–H bond functionalizations suggested that Ag interacts with Pd in a cooperative manner for an enhanced heterobimetallic C–H activation.9 We now report studies that demonstrate that the role of silver salts in several C–H arylation processes with iodoarenes is exclusively that of iodide capture. Furthermore, we have identified that in these processes silver salts can be conveniently replaced with a more redox stable and inexpensive quaternary ammonium salt while maintaining the high reactivity and broad substrate class scope displayed by the previous Pd/Ag systems. To the best of our knowledge, this is the first example of a silver replacement of general applicability to a wide range of C–H arylation processes.
Our initial efforts were directed at assessing the role of Ag salts in a common C–H arylation methodology: the ortho-arylation of benzoic acid 1a with iodoarene 2a (Table 1). Reaction under standard, Ag salt-mediated, conditions led to formation of biaryl product 3aa in 87% yield (entry 1).4ac Replacement of AgOAc with KOAc led to a reduction in yield to 9%, roughly consistent with two turnovers of the Pd catalyst (entry 2). A qualitative colorimetric test suggested the presence of PdI2 in the reaction mixture.10 Replacing Pd(OAc)2 with PdI2 led to no reaction (entry 3). Finally, using PdI2 in combination with AgOAc fully restored the high reactivity of the system (entry 4). These results suggest that in these reactions Pd(OAc)2 is a competent catalyst for the C–H arylation process, but is poisoned by iodide after two turnovers (Scheme 2). Thus, the silver salt is required for regenerating catalytically active Pd(OAc)2 from inactive PdI2via the formation and precipitation of the highly insoluble AgI salts, and not for the C–H arylation itself.

 | ||
| Scheme 2 Poisoning and regeneration of Pd catalyst in C–H arylation. | ||
Having established that Ag salts are not intrinsically required for the C–H arylation process, we hypothesised that a more benign acetate salt could equally facilitate regeneration of the Pd catalyst if the acetate counter-cation produced a highly insoluble iodide salt within the reaction medium. To examine the validity of this hypothesis we investigated the replacement of AgOAc with a variety of acetate salts (Table 2). The addition of NaOAc, CsOAc or Cu(OAc)2 proved to be ineffective for the regeneration of the catalyst (entries 1–3).11 To our delight, a survey of quaternary ammonium salts (entries 4–7) revealed that tetramethylammonium acetate could be used to regenerate the catalyst (entry 5), leading to the product in 45% yield (ca. 9 catalyst turnovers). Further, NMe4OAc could be formed in situ by the equimolar combination of the more readily available and inexpensive NMe4Cl and KOAc (entry 8). An experiment using unreactive PdI2 demonstrated that, like AgOAc, NMe4OAc salts are able to regenerate catalytically active Pd species in situ (entry 9). Increasing the equivalents of base and iodide abstractor gave an improved yield (entries 10 and 11), with 2.05 equiv. of NMe4Cl affording similar yields of product to those obtained using AgOAc (compare entry 11 with entry 1 in Table 1).12
|
|
|||
|---|---|---|---|
| Entry | Pd cat. | Additive (equiv.) | 3aa (%) |
| a Unless otherwise noted, all reactions were carried out using 0.5 mmol of 1a and 3 equiv. of 2a. b Yields were determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. c Reaction run for 48 h. NMe4Cl and KOAc added in two portions. | |||
| 1 | Pd(OAc)2 | NaOAc (1) | 9 |
| 2 | Pd(OAc)2 | Cu(OAc)2 (1) | 9 |
| 3 | Pd(OAc)2 | CsOAc (1) | 10 |
| 4 | Pd(OAc)2 | NH4OAc (1) | 4 |
| 5 | Pd(OAc)2 | NMe4OAc (1) | 45 |
| 6 | Pd(OAc)2 | NEt4OAc (1) | 9 |
| 7 | Pd(OAc)2 | NBu4OAc (1) | 12 |
| 8 | Pd(OAc)2 | NMe4Cl (1) + KOAc (1) | 44 |
| 9 | PdI2 | NMe4Cl (1) + KOAc (1) | 42 |
| 10 | Pd(OAc)2 | NMe4Cl (1.25) + KOAc (1.25) | 62 |
| 11c | Pd(OAc)2 | NMe4Cl (2.05) + KOAc (1.8) | 85 |

The effect of NMe4 salts on the catalytic activity is remarkable, in particular when compared with the ineffective ammonium salts bearing much larger (NBu4 and NEt4) or smaller (NH4) cations. This may be due to a more favourable cation/anion radii ratio, leading to a more effective crystal packing of the corresponding iodide salt.13 The solubility of NMe4I in AcOH was determined to be lower than 0.3 mg mL−1, which compares favourably to the higher solubilities of NH4I (4.7 mg mL−1), NEt4I (6.5 mg mL−1) and NBu4I (647 mg mL−1).
Having found a suitable cheap and benign replacement for Ag(I) salts, we explored the effects of substitution in both the benzoic acid and the iodoarene coupling partners (Scheme 3). Gratifyingly, both electron-poor and electron-rich iodoarenes led to the corresponding biaryl products in yields similar to, or higher than, those reported for the same AgOAc mediated process (3ab–ae).4ac Varied substitution was also possible on the benzoic acid coupling partner, with high yields observed for ortho, meta and/or para substituted benzoic acids (3ba–ha).
 | ||
| Scheme 3 Scope of the silver-free C–H arylation of benzoic acids with iodoarenes. All reactions were carried out on 0.5 mmol scale. Yields are of the isolated pure material. | ||
We envisaged that formation of unreactive PdI2 may be a common intermediate in the majority of Pd(II)-catalysed C–H arylation reactions with iodoarene coupling partners. Therefore, we explored the general applicability of NMe4OAc as an organic analogue for Ag(I)-salts to a wide variety of C–H arylation reactions (Scheme 4). It is noteworthy that simply by adjusting a few reaction parameters the system could be applied to other classes of substrates. Using free amides instead of carboxylic acids as the directing group, the NMe4-mediated protocol furnished the biaryl products in good yield (3ia–jd). Other common directing groups for C–H arylation, pyridine and benzoquinoline, were also successfully used under silver-free conditions (3kg–lg). We then explored the application of our arylation protocol to other heteroarenes in the absence of a directing group. Remarkably, this protocol also allowed the arylation of N-methylindole and 2-chlorothiophene in good yields (3ma–ng). These reactions proceeded with high C2 and C5 regioselectivity, respectively (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 by 1H NMR). Finally, the C–H arylation of electron-poor arenes was also tested and, to our delight, silver-free couplings with pentafluorobenzene could be successfully achieved (3oa–od).
:5 by 1H NMR). Finally, the C–H arylation of electron-poor arenes was also tested and, to our delight, silver-free couplings with pentafluorobenzene could be successfully achieved (3oa–od).
 | ||
| Scheme 4 Generality of the silver-free C–H arylation of arenes with iodoarenes. All reactions were carried out on 0.5 mmol scale. Yields are of the isolated pure material. | ||
To further highlight the applicability and significance of the new catalytic system, we then examined C(sp3)–H bond arylation reactions (Scheme 5). Importantly, functionalization at the benzylic position of 8-methylquinoline proceeded well under silver-free conditions for iodoarenes containing both electron-donating and electron-withdrawing groups (3pa–pj).
 | ||
| Scheme 5 Scope of the C(sp3)–C(sp2) coupling. All reactions were carried out on 0.5 mmol scale. Yields are of the isolated pure material. | ||
The development of NMe4-promoted reactions offers major economical and practical advantages over existing Ag-mediated methods. Firstly, the method provides a new avenue to reduce the cost of a number of chemical processes; NMe4Cl is ca. 50 times cheaper than AgOAc,14 and is produced in large scale (>5000 tons per year). Secondly, the synthetic advancement is also favourable from an environmental point of view; while AgOAc and AgI are both classed as very toxic to the aquatic environment, NMe4OAc and its by-product NMe4I15 are only classed as irritants. Moreover, the tolerance of the system covers a broad range of chemical motifs and the procedure is operationally trivial; the reaction proceeds smoothly under air with no special precautions needed. Overall, the methodology greatly increases the potential for C–H bond direct arylations to be exploited in pharmaceutical and agrochemical industries, where the value of a synthetic process is mainly assessed by the viability in terms of cost and environmental impact.
In order to demonstrate the utility of the new silver-free coupling for process chemistry, we scaled-up the reaction more than 3000 times in a 5 L reactor. Further optimization of the method allowed the arylation of 1.6 mol (218 g) of o-toluic acid 1c with 2 equiv. of iodobenzene (2b) and 2 mol % Pd(OAc)2 (Scheme 6). After an acid–base work-up, the reaction afforded 295 g of product 3cb in 87% yield with a 97% a/a purity by HPLC suitable for most applications. The purity could be upgraded to >99.9% if required by a simple re-slurry process in n-heptane.10 The analogous reaction using AgOAc would have required 267 g of the silver salt, worth £840, and would have generated 376 g of waste in the form of AgI.16 Excluding costs associated with waste streams, this new process represents savings of ca. 70%. We are confident that our NMe4-promoted methodology could be readily carried out on multi-kilogram scale.
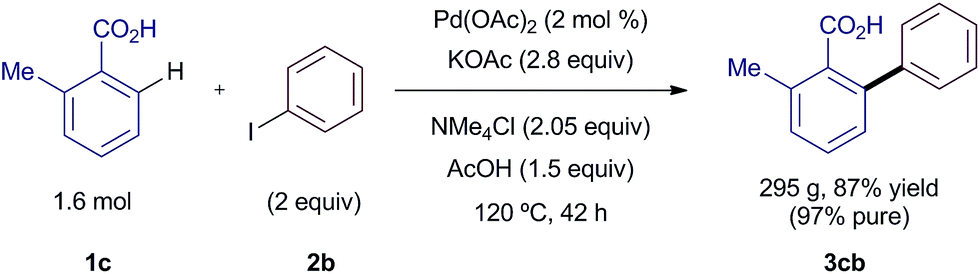 | ||
| Scheme 6 Scale-up of the silver-free arylation reaction. See ESI† for full details. | ||
Our study helps to define the mechanism of previously reported Ag-promoted systems.3,4 The comparable reactivity, and required reaction times, observed between the NMe4-mediated method and those using silver salts suggests that the role of Ag(I) is purely that of a halophile in the catalyst regeneration step. Therefore, other functions attributed to Ag(I) such as terminal oxidant, activator of iodoarenes or assistant in the C–H activation step can most likely be ruled out for this type of arylation processes. Based on these results, a proposed general catalytic cycle for the reactions here reported is depicted in Scheme 7. We postulate a Pd(II/IV) cycle where the C–H activation of the arene would take place first to form an aryl Pd(II) complex I. This intermediate may then undergo oxidative addition to Pd(IV) species II which, after reductive elimination, would release the biaryl product and the catalyst in the form of PdIOAc (III). This intermediate could undergo ligand exchange with NMe4OAc to form Pd(OAc)2, disproportionate to PdI2 and Pd(OAc)2 or could also initiate a second catalytic cycle which would result in unreactive PdI2. Finally, PdI2 would react with NMe4OAc, regenerating catalytically active Pd(OAc)2.
 | ||
| Scheme 7 Proposed general catalytic cycle. | ||
Conclusions
In summary, we have described the discovery of an organic analogue of silver(I)-salts, and develop reaction conditions that allow the Pd-catalysed C–H coupling reaction of a variety of classes of substrates: benzoic acids, benzamides, 2-phenylpyridines, benzoquinolines, indoles, thiophenes, polyfluorobenzenes and 8-methylquinolines with diverse substituted iodoarenes under silver-free conditions. The new method is easily scalable and benefits from the use of inexpensive and readily available reactants, an operationally simple procedure, high functional group tolerance and low-toxicity waste. We believe that this discovery will provide a pathway for facilitating application of numerous existing, and new, direct C–H bond functionalizations to industry, in addition to opening the door to other novel silver-free transition metal catalysed processes.Acknowledgements
We gratefully acknowledge GlaxoSmithKline and the Engineering and Physical Sciences Research Council for generous support via ImpactQM KTA, the European Research Council for a Starting Research Grant (to I.L.) and MINECO-Spain for a FPI Fellowship (to J.G.D.). We thank Stephen Hermitage (GSK) for helpful discussions.Notes and references
- For recent reviews on direct arylation reactions and Pd-catalysed C–H activation methodologies, see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) Topics in Current Chemistry: C–H Activation, ed. J.-Q. Yu and Z. Shi, 1st edn, Springer, Berlin Heidelberg, 2010 Search PubMed; (f) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (g) C. S. Yeung and M. V. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (h) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (i) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (j) L. McMurray, F. O. O'Hara and M. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (k) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- O. Daugulis and V. G. Zaitsev, Angew. Chem., Int. Ed., 2005, 44, 4046 CrossRef CAS PubMed.
- For reviews: (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (b) O. Daugulis, Top. Curr. Chem., 2010, 292, 57 CrossRef CAS; (c) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657 CrossRef CAS PubMed; (c) K. Kobayashi, A. Sugie, M. Takahashi, K. Masui and A. Mori, Org. Lett., 2005, 7, 5083 CrossRef CAS PubMed; (d) D. Shabashov and O. Daugulis, Org. Lett., 2006, 8, 4947 CrossRef CAS PubMed; (e) A. Lazareva and O. Daugulis, Org. Lett., 2006, 8, 5211 CrossRef CAS PubMed; (f) K. Kobayashi, M. S. M. Ahmed and A. Mori, Tetrahedron, 2006, 62, 9548 CrossRef CAS PubMed; (g) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed; (h) G. L. Turner, J. A. Morris and M. F. Greaney, Angew. Chem., Int. Ed., 2007, 46, 7996 CrossRef CAS PubMed; (i) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879 CrossRef CAS PubMed; (j) D. Shabashov and O. Daugulis, J. Org. Chem., 2007, 72, 7720 CrossRef CAS PubMed; (k) N. Arai, T. Miyaoku, S. Teruya and A. Mori, Tetrahedron Lett., 2008, 49, 1000 CrossRef CAS PubMed; (l) N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926 CrossRef CAS PubMed; (m) E. F. Flegeau, M. E. Popkin and M. F. Greaney, Org. Lett., 2008, 10, 2717 CrossRef CAS PubMed; (n) C. Qin and W. Lu, J. Org. Chem., 2008, 73, 7424 CrossRef CAS PubMed; (o) D. Shabashov, J. R. M. Maldonado and O. Daugulis, J. Org. Chem., 2008, 73, 7818 CrossRef CAS PubMed; (p) V. S. Thirunavukkarasu, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2008, 47, 9462 CrossRef CAS PubMed; (q) F. Yang, Y. Wu, Z. Zhu, J. Zhang and Y. Li, Tetrahedron, 2008, 64, 6782 CrossRef CAS PubMed; (r) R. B. Bedford, R. L. Webster and C. J. Mitchell, Org. Biomol. Chem., 2009, 7, 4853 RSC; (s) M. P. Huestis and K. Fagnou, Org. Lett., 2009, 11, 1357 CrossRef CAS PubMed; (t) T. Nishikata, A. R. Abela and B. H. Lipshutz, Angew. Chem., Int. Ed., 2010, 49, 781 CrossRef CAS PubMed; (u) S. A. Ohnmacht, A. J. Culshaw and M. F. Greaney, Org. Lett., 2010, 12, 224 CrossRef CAS PubMed; (v) O. René and K. Fagnou, Org. Lett., 2010, 12, 2116 CrossRef PubMed; (w) G.-W. Wang, T.-T. Yuan and D.-D. Li, Angew. Chem., Int. Ed., 2011, 50, 1380 CrossRef CAS PubMed; (x) W. Li, Z. Xu, P. Sun, X. Jiang and M. Fang, Org. Lett., 2011, 13, 1286 CrossRef CAS PubMed; (y) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef CAS PubMed; (z) F. Chen, Q.-Q. Min and X. Zhang, J. Org. Chem., 2012, 77, 2992 CrossRef CAS PubMed; (a a) D.-D. Li, T.-T. Yuan and G.-W. Wang, J. Org. Chem., 2012, 77, 3341 CrossRef CAS PubMed; (a b) M. Wasa, K. S. L. Chan, X.-G. Zhang, J. He, M. Miura and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570 CrossRef CAS PubMed; (a c) C. Arroniz, A. Ironmonger, G. Rassias and I. Larrosa, Org. Lett., 2013, 15, 910 CrossRef CAS PubMed; (a d) J.-C. Wan, J.-M. Huang, Y.-H. Jhan and J.-C. Hsieh, Org. Lett., 2013, 15, 2742 CrossRef CAS PubMed; (a e) R. Feng, J. Yao, Z. Liang, Z. Liu and Y. Zhang, J. Org. Chem., 2013, 78, 3688 CrossRef CAS PubMed; (a f) P. Ricci, K. Kraemer, X. C. Cambeiro and I. Larrosa, J. Am. Chem. Soc., 2013, 135, 13258 CrossRef CAS PubMed; (a g) S. Islam and I. Larrosa, Chem.–Eur. J., 2013, 19, 15093 CrossRef CAS PubMed; (a h) Z. Huang and G. Dong, J. Am. Chem. Soc., 2013, 135, 17747 CrossRef CAS PubMed; (a i) W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430 CrossRef CAS PubMed; (a j) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109 CrossRef CAS PubMed; (a k) L. C. M. Castro and N. Chatani, Chem.–Eur. J., 2014, 20, 4548 CrossRef PubMed; (a l) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed.
- Some success has been achieved in avoiding silver salts for specific substrates via bidentate-chelation assistance: (a) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (b) M. Ye, G.-L. Gao, A. J. F. Edmunds, P. A. Worthington, J. A. Morris and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19090 CrossRef CAS PubMed; (c) L. Huang, Q. Li, C. Wang and C. Qi, J. Org. Chem., 2013, 78, 3030 CrossRef CAS PubMed; (d) M. Ye, A. J. F. Edmunds, J. A. Morris, D. Sale, Y. Zhang and J.-Q. Yu, Chem. Sci., 2013, 4, 2374 RSC; (e) D. S. Roman and A. B. Charette, Org. Lett., 2013, 15, 4394 CrossRef CAS PubMed.
- (a) M. Naodovic and H. Yamamoto, Chem. Rev., 2008, 108, 3132 CrossRef CAS PubMed; (b) J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149 CrossRef CAS PubMed; (c) M. Alvarez-Corral, M. Muñoz-Dorado and I. Rodríguez-García, Chem. Rev., 2008, 108, 3174 CrossRef CAS PubMed; (d) Y. Yamamoto, Chem. Rev., 2008, 108, 3199 CrossRef CAS PubMed; (e) J.-M. Weibel, A. Blanc and P. Pale, Silver in Organic Chemistry, ed. M. Hamata, Wiley, New Jersey, 2010 Search PubMed.
- (a) C. Nevado and A. M. Echavarren, Chem.–Eur. J., 2005, 11, 3155 CrossRef CAS PubMed; (b) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (c) G. Rassias, N. G. Stevenson, N. R. Curtis, J. M. Northall, M. Gray, J. C. Prodger and A. J. Walker, Org. Process Res. Dev., 2010, 14, 92 CrossRef CAS; (d) M. Vellakkaran, M. M. S. Andappanb and N. Kommu, Green Chem., 2014, 16, 2788 RSC.
- (a) D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS PubMed; (b) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS PubMed; (c) A. Homs, I. Escofet and A. M. Echavarren, Org. Lett., 2013, 15, 5782 CrossRef CAS.
- M. Anand, R. B. Sunoj and H. F. Schaefer III, J. Am. Chem. Soc., 2014, 136, 5535 CrossRef CAS PubMed.
- See ESI† for details.
- Recently, Yu and co-workers have identified a viable alternative for catalyst regeneration in iodination reactions by the effective combination of a specific amide auxiliary and CsOAc as iodide scavenger, see: (a) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326 CrossRef CAS PubMed Earlier studies on halogenation of C–H bonds showed that Pd(OAc)2 can be obtained from PdI2 with IOAc. This iodonium(I) reagent is not suitable for silver-free arylations as it has to be preformed from AgOAc or PhI(OAc)2in situ, see: ; (b) D.-H. Wang, X.-S. Hao, D.-F. Wu and J.-Q. Yu, Org. Lett., 2006, 8, 3387 CrossRef CAS PubMed; (c) T.-S. Mei, R. Giri, N. Maugel and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 5215 CrossRef CAS PubMed; (d) T.-S. Mei, D.-H. Wang and J.-Q. Yu, Org. Lett., 2010, 12, 3140 CrossRef CAS PubMed.
- The reaction performed best under neat conditions but some organic solvents are also tolerated. For example, running the reaction 1 M in dioxane, EtOAc or PhCl led to 64%, 53% and 46%, respectively. See Table S1 in the ESI† for more details.
- J. Palomo and P. N. Pintauro, J. Membrane Sci., 2003, 215, 103 CrossRef CAS.
- Based on Sigma-Aldrich catalogue prices (April 2014), NMe4Cl costs £10.5 mol−1 whereas AgOAc costs £526 mol−1.
- NMe4I was isolated from the reaction mixture by filtration and characterized, see ESI.†.
- In addition, work-up of reaction mixtures is more difficult when silver is present in the system due to its poor filtration properties. Also, recycling of metal catalysts is hampered by the presence of other metals in the waste, see: (a) E. Jimenez-Nuñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (b) H. Schmidbaur and A. Schier, Z. Naturforsch., 2011, 66b, 329 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental procedures, characterization data and NMR spectra. See DOI: 10.1039/c4sc01215a |
| ‡ Present address: Department of Chemistry, University of Patras. 26500, Patras, Greece. |
| This journal is © The Royal Society of Chemistry 2014 |