 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective formation of biphasic thin films of metal–organic frameworks by potential-controlled cathodic electrodeposition†
Minyuan
Li
and
Mircea
Dincă
*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: mdinca@mit.edu; Tel: +1 617-253-4154
First published on 5th September 2013
Abstract
Cathodic electrodeposition lends itself to the formation of biphasic metal–organic framework thin films at room temperature from single deposition baths using potential bias as the main user input. Depending on the applied potential, we selectively deposit two different phases as either bulk mixtures or bilayer films.
Non-traditional applications of metal–organic frameworks (MOFs), such as thin-film sensors and electrochemical devices1,2 have lagged behind gas storage and separation3–6 partly because the former require the integration of MOFs with surfaces and electrodes.7–21 Such applications could further benefit from the development of techniques that allow the formation of complex composites containing at least two MOF phases. Although the need for reliable means to deposit such materials as thin films or membranes has been recognized and addressed with several independent techniques,9–11,14,16,18,19,22–24 only a few have thus far enabled the deposition of biphasic MOF films,14,19,23,25–28 and no method exists to date that allows the controlled deposition of multiphasic MOF thin films made from lattice-mismatched crystalline phases. Herein, we demonstrate that biphasic films made from two structurally unrelated materials from the Zn-BDC (H2BDC = benzene-1,4-dicarboxylic acid) phase space can be deposited selectively as either mixed films or sandwich-type bilayer surface structures using a potential-controlled cathodic electrodeposition method.
Electrochemical methods are best suited to address the challenge of interfacing MOFs with electrode surfaces.29 Generally, electrodeposition methods find wide utility in industrial settings, because they enable film deposition only at the electrode–solution interface, do not require line-of-sight instrumental setups, as physical vapor deposition techniques do for instance, and can therefore be used to build conformal coatings on electrodes of virtually any geometry and surface area. Furthermore, because any exposed electrode surface is electrochemically active, the deposition of non-conductive, electrode-passivating films such as MOFs allows for in situ repairing of defects such as cracks and pinholes. Finally, the electrochemical nature of the process offers additional advantages in that the deposition progress can be monitored by the amount of passed charge, giving control over film thickness. Indeed, because of scalability, ease of processing, and the ability to work at room temperature, electrochemical synthesis based on anodic dissolution of the metal component—Cu, Al, Zn18,30—is the method of choice for the large-scale production of some of the commercially available MOFs. However, because the anode provides the metal ions for the MOF and is necessarily corroded during this process, anodic methods offer limited choices in terms of electrode surfaces, have thus far yielded only single-phase MOFs, and may not be best suited for the formation of more complex films. To address these challenges, we recently reported that electrodes can be used as chemically inert spectators and only as sources of electrons when employed in cathodic MOF electrodeposition schemes.31 We initially surmised that aqueous reduction of oxoanions such as NO3−, which produces hydroxide, would raise the pH of the solution near the cathode and induce crystallization of MOFs in an electrolysis bath containing the respective ligand and metal precursor by slowly deprotonating the ligand. We showed that this method was indeed effective for the deposition of Zn4O(BDC)3 (MOF-5), which was crystallized selectively on the surface of a fluorine-tin-oxide (FTO) electrode upon biasing a solution containing Zn(NO3)2 and H2BDC at a sufficiently negative potential.
Our initial results hinted at a general method whereby any conductive surface could function as the electron reservoir and, more importantly, any electrochemical half-reaction involving the reduction of a probase molecule would generate a base, increase the local pH near the electrode, and induce MOF crystallization (see Scheme 1). In fact, we surmised that nitrate may be a poor choice as a probase, because it typically requires large reduction overpotentials in both aqueous solutions32 and in our system (see also Fig. S1†). To circumvent this problem, we aimed to replace it with one of the reactions in eqn (I–III), which were potential candidates for inducing cathodic electrodeposition of MOFs. In particular, the reduction of triethylammonium, Et3NH+, to H2 and triethylamine, Et3N, was chosen because H2 is a relatively inert and insoluble molecule that would not interfere with MOF formation. Moreover, the pH buffering pair Et3NH+/Et3N could add another addressable handle to our deposition protocol. The latter was especially attractive, because previous studies had suggested that phase selection in MOF synthesis can be highly dependent on pH.23,33 We hypothesized that we could selectively deposit multiple MOF phases by simply controlling the Et3NH+ concentration and dialing the electrochemical deposition potential. Because the rate of an electrochemical reaction is proportional to the current density, which in turn is logarithmically dependent on overpotential,34 we expected that increasing the deposition potential would generate Et3N faster and increase the local pH at the electrode surface, thus modulating the phase of the deposited MOF. Finally, Et3N had been employed previously as a base in the original synthesis of MOF-535 and therefore had good precedent in constructing such materials.
| 2H2O + 2e− → H2 + 2HO− | (I) |
| 2R3NH+ (R = alkyl) + 2e− → H2 + 2R3N | (II) |
| 2H2BDC + 2e− → H2 + 2HBDC− | (III) |
 | ||
| Scheme 1 General scheme for the cathodically induced electrochemical deposition of MOFs, involving the reduction of probase (P), the generation of base equivalents (B), the deprotonation of ligands (H2BDC), and MOF crystallization from BDC2− and metal ions (Zn2+). | ||
To minimize the overpotential required for the reduction of Et3NH+, our probase, we chose Pt, a well-known catalyst for proton reduction and an otherwise inert metal, as our working electrode. As shown in the cyclic voltammogram (CV) in Fig. 1, the onset of proton reduction from a solution of Et3NHCl in DMF occurred at approximately −0.5 V (vs. Ag/Ag(cryptand)+) and reached a peak at Ep,c = −1.05 V when using a Pt button electrode and a scan rate of 100 mV s−1. Et3N was therefore formed below the reduction potential for Zn deposition, which occurred at approximately −1.0 V (see also Fig. S2†). Thus, MOF crystallization may occur in the absence of Zn plating, in contrast to what had been observed when using NO3− as a probase.31 Addition of H2BDC to the Et3NHCl/DMF solution only increased the peak current for H+ reduction, as expected for an increased proton concentration, but did not shift Ep,c, confirming that the electrochemical event is indeed proton reduction (Fig. 1).
 | ||
| Fig. 1 Cyclic voltammograms of a dilute solution of Et3NHCl (15 mM, red) and a dilute solution of Et3NHCl and H2BDC (15 mM each, blue) in DMF, highlighting the cathodic peak for proton reduction. | ||
Attempts to electrodeposit MOFs from electrolysis baths containing Zn(NO3)2, H2BDC, and concentrated Et3NH+ (≥300 mM) at −1.00 V (see also Fig. S3†) yielded a white crystalline film that adhered to the Pt gauze working electrode. As shown in Fig. 2, powder X-ray diffraction (PXRD) of this white film revealed a pattern that matched that of the anionic framework (Et2NH2)2Zn3(BDC)4,36 where Et3NH+ ions likely replace the Et2NH2+ charge-balancing ions present in the reported structure (see also Fig. S4 and S5†). No Zn plating was observed, and the only crystalline deposit under these conditions was (Et3NH)2Zn3(BDC)4. Scanning electron micrographs (SEMs) of this phase revealed distinctive feather-like crystallites of ∼5 μm width and sub-micron thickness (Fig. 2). Surprisingly, Zn plating was not observed even at more negative potentials if the concentration of Et3NH+ was maintained at or above 300 mM, and the only phase deposited under these conditions even at −1.50 V was (Et3NH)2Zn3(BDC)4 (see also Fig. S6–S8†). This contrasted with previous depositions from Zn(NO3)2 and H2BDC solutions devoid of Et3NH+, which at −1.60 V produced composites of MOF-5 and Zn metal on FTO.31 We reasoned that two effects may come into play when large concentrations of Et3NH+ are present in the electrodeposition bath: (1) any Zn that could plate is etched away by the Et3NH+ acid according to eqn (IV), and (2) the presence of a large concentration of Et3NH+ effectively buffers the pH and may never allow the accumulation of enough Et3N to induce the formation of a different crystalline phase such as MOF-5.
| 2Et3NH+ + Zn → Zn2+ + H2 + 2Et3N | (IV) |
 | ||
| Fig. 2 Analytical and microscopy data for films deposited from concentrated Et3NH+ solutions (≥300 mM). (a) Experimental PXRD patterns for films deposited at −1.00 V and −1.50 V, and expected patterns for Zn, MOF-5, and (Et3NH)2Zn3(BDC)4,36 showing negligible Zn plating and exclusive formation of (Et3NH)2Zn3(BDC)4. (b) A SEM of the film deposited at −1.50 V (see also Fig. S7 and S8†). The scale bar represents a length of 5 μm. (c) A portion of the crystal structure of the anionic framework [Zn3(BDC)4]2−, omitting guest DMF molecules and Et3NH+ ions for clarity.36 Green, red, and grey spheres represent Zn, O, and C atoms, respectively. H atoms are also omitted for clarity. | ||
This remarkable shift from one MOF phase to another depending on the concentration of Et3NH+ presented the intriguing prospect of selectively depositing two different MOF phases from a single solution of low/intermediate Et3NH+ concentrations, with phase selection depending on the applied deposition potential. We surmised that lowering the Et3NH+ concentration would have no effect on the phase deposited at more positive potential, that is (Et3NH)2Zn3(BDC)4, but would reduce the buffering capacity, thereby enabling the accumulation of Et3N, the increase in pH, and perhaps the formation of a second crystalline MOF phase at a more negative potential.
Indeed, bulk electrolysis at −1.10 V of a solution containing only 100 mM of Et3NH+ gave exclusively (Et3NH)2Zn3(BDC)4, while deposition at −1.50 V from an identical electrolysis bath gave a mixed film composed of MOF-5, Zn metal, and (Et3NH)2Zn3(BDC)4, as identified by PXRD in Fig. 3. This represents the first demonstration of the simultaneous deposition of biphasic MOF films from a single precursor solution. Shifting the deposition potential cathodically to −1.70 V virtually eliminated the deposition of (Et3NH)2Zn3(BDC)4 and gave mainly a composite film of Zn and MOF-5. SEMs of this composite did not show clear crystallite features and suggested a finely dispersed mixture of MOF-5 and Zn (see also Fig. S9–S11†). Importantly, similar Zn/MOF-5 composites deposited on FTO31 displayed high porosity, with a surface area of 917 m2 g−1 (2140 m2 g−1 adjusted for the MOF-5 component) (see also Fig. S12 and S13†), attesting that films grown by cathodic deposition maintain porosity and should be effective in the many applications proposed for MOFs that require surface attachment. This prospect is currently under investigation in our laboratory in the context of gas separation membranes.
 | ||
| Fig. 3 Analytical and microscopy data for films deposited from dilute Et3NH+ solutions (100 mM). (a) PXRD patterns expected for MOF-5 and (Et3NH)2Zn3(BDC)4,36 and experimentally observed for films deposited at −1.10 V, −1.50 V, and −1.70 V, showing the deposition of biphasic MOF films. (b and c) Scanning electron micrographs of the films deposited at −1.70 V and −1.50 V, respectively. In the −1.70 V reaction, the feather-like structures, prevalent in the samples deposited at −1.50 V, were far less prominent. Scale bars correspond to a length of 10 μm. | ||
Encouraged by the phase control enabled by potential modulation, we sought to deposit not just heterostructured mixed films as above, but also bilayer structures by sequential electrolyses at two different potentials. Notably, the electrolysis of a 100 mM Zn(NO3)2, 50 mM H2BDC, and 100 mM Et3NHCl solution in DMF at −1.10 V for 6 h followed by deposition at −1.50 V for only 5 min gave films whose PXRD patterns revealed the presence of both (Et3NH)2Zn3(BDC)4 and MOF-5 (Fig. 4). Prolonged electrolysis at this potential, up to 20 min, shifted the relative ratio of (Et3NH)2Zn3(BDC)4 to MOF-5 with a significant increase in the latter, observable by PXRD. SEMs of these films revealed remarkable bilayer structures, wherein the Zn/MOF-5 composite, grown at −1.50 V, was deposited underneath the distinctive feather-like crystalline layer of (Et3NH)2Zn3(BDC)4 grown at −1.10 V. This was anticipated because the insulating (Et3NH)2Zn3(BDC)4 film was expected to passivate the Pt surface, but suggests that the anionic phase can be penetrated by the precursors required to form MOF-5. More importantly, the deposition of bilayer/biphasic MOF films had not been demonstrated before outside the more laborious layer-by-layer approach,9,26,28 and is enabled here from a single precursor solution through simple potential modulation.
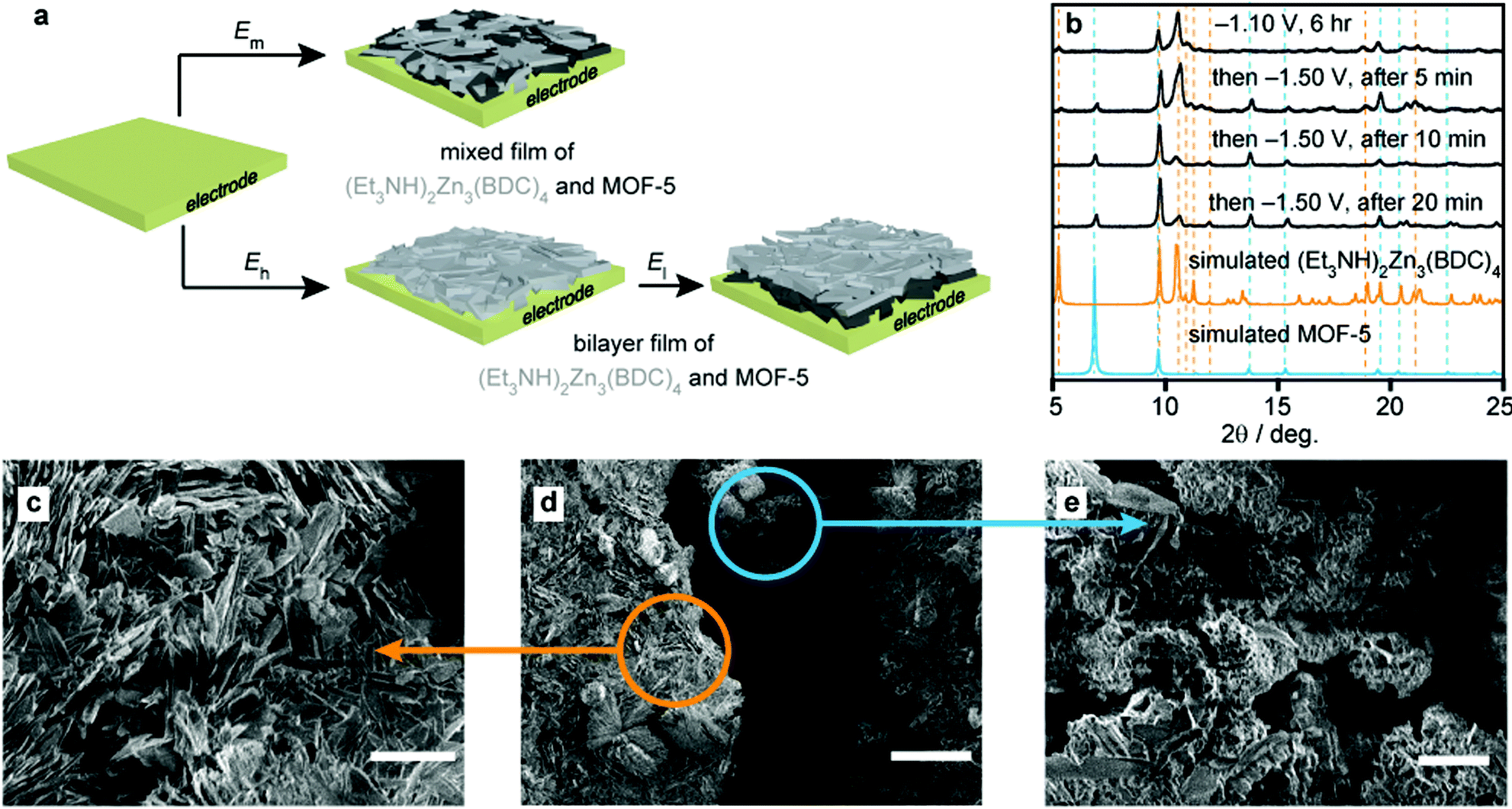 | ||
| Fig. 4 Deposition of mixed and bilayer MOF films under potentiostatic control. (a) Schematic illustration of the formation of a biphasic mixed film at (cathodic) potential, El. El < Em < Eh. (b) Expected PXRD patterns for MOF-5 and (Et3NH)2Zn3(BDC)4,36 and observed patterns after sequential electrodeposition at −1.10 V (top) followed by at −1.50 V for 5, 10, and 20 min intervals. (c–e) Scanning electron micrographs of a film produced by sequential growth at −1.10 V and −1.50 V, displaying the characteristic feather-like morphology of (Et3NH)2Zn3(BDC)4 in the top layer (c, orange arrow), and the small crystallites associated with the Zn/MOF-5 composite in the layer closer to the electrode surface (e, blue arrow). Scale bars correspond to lengths of 10 μm (c), 50 μm (d), and 5 μm (e). | ||
When the second deposition step was held at more cathodic potentials, such as −1.70 V, similar bilayer constructs could be observed. However, the rapid growth of the bottom layer bulges and ruptures the top MOF layer (Fig. S17†) even with the deposition time kept short at this more negative potential. Although the exact mechanisms that allow selective deposition of one phase over another at various potentials are still unclear, we propose that more negative potentials allow the accumulation of base equivalents and in turn promote the formation of the μ4-oxo atom required for the nucleation of {Zn4O} clusters and MOF-5 (see eqn (V)), where L is a coordinating ligand and 3 ≤ x ≤ 5. At sufficiently negative potentials, NO3− reduction also takes place and may be necessary‡ for MOF-5 formation.37 Furthermore, Zn metal may be required for MOF-5 formation either for mediating NO3− reduction or for templating the crystallization of MOF-5. These are fascinating mechanistic questions worthy of more in-depth examination. Regardless, these results demonstrate that the formation of MOF heterostructures—an important goal for the technological implementation of MOFs—is enabled by cathodic electrodeposition with unprecedented selectivity and minimal user input.
| LxZn(OH) + Et3N → [LxZnO]− + Et3NH+ | (V) |
Conclusions
Numerous applications proposed for MOFs depend critically on the development of selective, generalizable, and scalable methods for their growth as thin films, membranes, or composites. We expect that the versatility of the cathodic deposition approach, demonstrated here, will enable the formation of complex MOF architectures for using these emerging materials in a range of important applications. Many challenges remain before these techniques can be translated to industrial settings, not least of which are concerns regarding the orientation of the crystallites, which could be addressed by surface functionalization for instance,7,13 and the extension of this method to other metal–ligand systems, both of which are currently being addressed in our lab. Overall, the results presented herein provide a potential roadmap for a large-scale fabrication methodology for heterostructured multiphasic and multilayered MOF thin films and membranes.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0006937. M.D. thanks 3 M and MIT's Research Support Committee for Non-Tenured Faculty Funds and the MISTI Hayashi Fund for travel support. This work made use of the MRSEC Shared Experimental Facilities at MIT, which are supported in part by the NSF under Award DMR-0819762. Grants from the NSF (CHE-9808061, DBI-9729592) also provided instrument support to the DCIF at MIT. M.L. gratefully acknowledges the NSF Graduate Research Fellowship Program for support under Grant no. 1122374. We thank Dr A. F. Cozzolino and Dr C. R. Wade for assistance with the NMR measurements and K. Armburst for assistance with coulometry.Notes and references
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- M. D. Allendorf, A. Schwartzberg, V. Stavila and A. A. Talin, Chem.–Eur. J., 2011, 17, 11372–11388 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- M. Shah, M. C. McCarthy, S. Sachdeva, A. K. Lee and H.-K. Jeong, Ind. Eng. Chem. Res., 2012, 51, 2179–2199 CrossRef CAS.
- A. J. Brown, J. R. Johnson, M. E. Lydon, W. J. Koros, C. W. Jones and S. Nair, Angew. Chem., 2012, 124, 10767–10770 CrossRef.
- B. Liu, M. Tu and R. A. Fischer, Angew. Chem., Int. Ed., 2013, 52, 3402–3405 CrossRef CAS PubMed.
- A. Carné-Sánchez, I. Imaz, M. Cano-Sarabia and D. Maspoch, Nat. Chem., 2013, 5, 203–211 CrossRef PubMed.
- G. Xu, T. Yamada, K. Otsubo, S. Sakaida and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 16524–16527 CrossRef CAS PubMed.
- D. Witters, N. Vergauwe, R. Ameloot, S. Vermeir, D. De Vos, R. Puers, B. Sels and J. Lammertyn, Adv. Mater., 2012, 24, 1316–1320 CrossRef CAS PubMed.
- G. Lu, O. K. Farha, W. Zhang, F. Huo and J. T. Hupp, Adv. Mater., 2012, 24, 3970–3974 CrossRef CAS PubMed.
- Y. Ikezoe, G. Washino, T. Uemura, S. Kitagawa and H. Matsui, Nat. Mater., 2012, 11, 1081–1085 CAS.
- C. Hou, J. Peng, Q. Xu, Z. Ji and X. Hu, RSC Adv., 2012, 2, 12696–12698 RSC.
- D. Bradshaw, A. Garai and J. Huo, Chem. Soc. Rev., 2012, 41, 2344–2381 RSC.
- R. Ameloot, F. Vermoortele, W. Vanhove, M. B. J. Roeffaers, B. F. Sels and D. E. De Vos, Nat. Chem., 2011, 3, 382–387 CrossRef CAS PubMed.
- F. Jeremias, S. K. Henninger and C. Janiak, Chem. Commun., 2012, 48, 9708–9710 RSC.
- A. Schoedel, C. Scherb and T. Bein, Angew. Chem., Int. Ed., 2010, 49, 7225–7228 CrossRef CAS PubMed.
- R. Ameloot, L. Stappers, J. Fransaer, L. Alaerts, B. F. Sels and D. E. De Vos, Chem. Mater., 2009, 21, 2580–2582 CrossRef CAS.
- A. Bétard and R. A. Fischer, Chem. Rev., 2012, 112, 1055–1083 CrossRef PubMed.
- R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565–571 CrossRef CAS PubMed.
- P. Falcaro, A. J. Hill, K. M. Nairn, J. Jasieniak, J. I. Mardel, T. J. Bastow, S. C. Mayo, M. Gimona, D. Gomez, H. J. Whitfield, R. Riccò, A. Patelli, B. Marmiroli, H. Amenitsch, T. Colson, L. Villanova and D. Buso, Nat. Commun., 2011, 2, 237 CrossRef PubMed.
- M. Kubo, W. Chaikittisilp and T. Okubo, Chem. Mater., 2008, 20, 2887–2889 CrossRef CAS.
- H. Lu and S. Zhu, Eur. J. Inorg. Chem., 2013, 1294–1300 CrossRef CAS.
- M. N. Shah, M. A. Gonzalez, M. C. McCarthy and H.-K. Jeong, Langmuir, 2013, 29, 7896–7902 CrossRef CAS PubMed.
- K. Hirai, S. Furukawa, M. Kondo, H. Uehara, O. Sakata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 8057–8061 CrossRef CAS PubMed.
- O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus, M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer and C. Wöll, J. Am. Chem. Soc., 2007, 129, 15118–15119 CrossRef CAS PubMed.
- Y. Yoo and H.-K. Jeong, Cryst. Growth Des., 2010, 10, 1283–1288 CAS.
- O. Shekhah, K. Hirai, H. Wang, H. Uehara, M. Kondo, S. Diring, D. Zacher, R. A. Fischer, O. Sakata, S. Kitagawa, S. Furukawa and C. Wöll, Dalton Trans., 2011, 40, 4954–4958 RSC.
- I. Gurrappa and L. Binder, Sci. Technol. Adv. Mater., 2008, 9, 043001 CrossRef.
- A. Martinez Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489–3498 CAS.
- M. Li and M. Dincă, J. Am. Chem. Soc., 2011, 133, 12926–12929 CrossRef CAS PubMed.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- H. Kim, S. Das, M. G. Kim, D. N. Dybtsev, Y. Kim and K. Kim, Inorg. Chem., 2011, 50, 3691–3696 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2000 Search PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS PubMed.
- A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigby and J. E. Warren, CrystEngComm, 2005, 7, 548–550 RSC.
- S. Hausdorf, J. Wagler, R. Mossig and F. O. R. L. Mertens, J. Phys. Chem. A, 2008, 112, 7567–7576 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional CVs, PXRD patterns, SEMs, graphic representations of the anionic framework—[Zn3(BDC)4]2−, a N2-sorption isotherm at 77 K, and a 1H NMR spectrum. See DOI: 10.1039/c3sc51815a |
| ‡ We note that the use of other zinc precursors (triflate, chloride, or perchlorate) could inhibit MOF-5 formation during electrodeposition under similar reaction conditions. |
| This journal is © The Royal Society of Chemistry 2014 |