 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combined nucleobase and backbone modifications enhance DNA duplex stability and preserve biocompatibility†
Afaf H.
El-Sagheer‡§
ab and
Tom
Brown‡§
a
aSchool of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: tb2@soton.ac.uk; A.H.El-Sagheer@soton.ac.uk; Fax: +44 (0)2380 592991; Tel: +44 (0)2380 592974
bChemistry Branch, Dept. of Science and Mathematics, Faculty of Petroleum and Mining Engineering, Suez University, Suez, 43721, Egypt
First published on 17th October 2013
Abstract
DNA strands containing a triazole linkage flanked on its 3′-side by an aminoethylphenoxazine nucleobase analogue (G-clamp) have been prepared by solid-phase synthesis followed by CuAAC-mediated click oligonucleotide ligation. The stability of the doubly modified DNA duplexes and DNA–RNA hybrids is greatly increased, whereas a single base pair mismatch located at or adjacent to the modifications is strongly destabilising, making triazole G-clamp a potent mismatch/point mutation sensor. A DNA strand containing this unnatural combination was successfully amplified by PCR to produce unmodified copies of the original template, with deoxyguanosine inserted opposite to the G-clamp-triazole nucleotide analogue. This study shows for the first time that a polymerase enzyme can read through a combined backbone/nucleobase modification surprisingly well. These favourable properties suggest new applications for oligonucleotides containing the G-clamp triazole modification in biotechnology, nanotechnology, diagnostics and therapeutics.
Introduction
Templated chemical ligation of alkyne and azide-functionalised oligonucleotides using the CuAAC reaction1,2 has recently been used to assemble long DNA strands up to 300 bases in length containing a triazole mimic of a DNA phosphodiester linkage.3 The base sequence of DNA strands containing this artificial linkage can be copied by DNA polymerases during PCR with high fidelity,3 and can also be transcribed by RNA polymerase (Fig. 1).4 Remarkably, plasmids containing essential3 and non-essential5 triazole-containing genes are functional in E. coli despite the apparent dissimilarity of the artificial linkage to a normal phosphodiester DNA bridge. Biocompatibility is observed even when triazole linkers are placed just 4 base-pairs apart in opposite strands of DNA.5 The structural and thermodynamic properties of the biocompatible triazole linkage have been determined by NMR, ultraviolet melting analysis and circular dichroism, revealing a normal B-DNA duplex with reduced resistance to thermal denaturation.6 This relative instability did not detrimentally affect the biocompatibility of the triazole linkage, but increasing the thermodynamic stability of the modified DNA is likely to improve discrimination against mismatched base pairs in more complex biological systems, leading to increased fidelity and sequence discrimination. The lowering of melting temperature caused by the triazole DNA backbone has also been observed in RNA.7 This suggests that its incorporation into antisense oligonucleotides,8 miRNA probes,9 and siRNA constructs10,11 is likely to lead to reduced potency. This could limit its use in biomedical and diagnostic applications where strategically placed triazole linkages otherwise confer favourable properties on therapeutic oligonucleotides, protecting them against degradation in vivo.12 Other triazole backbones have been synthesised,12–14 but none give rise to duplexes of the same stability as canonical DNA.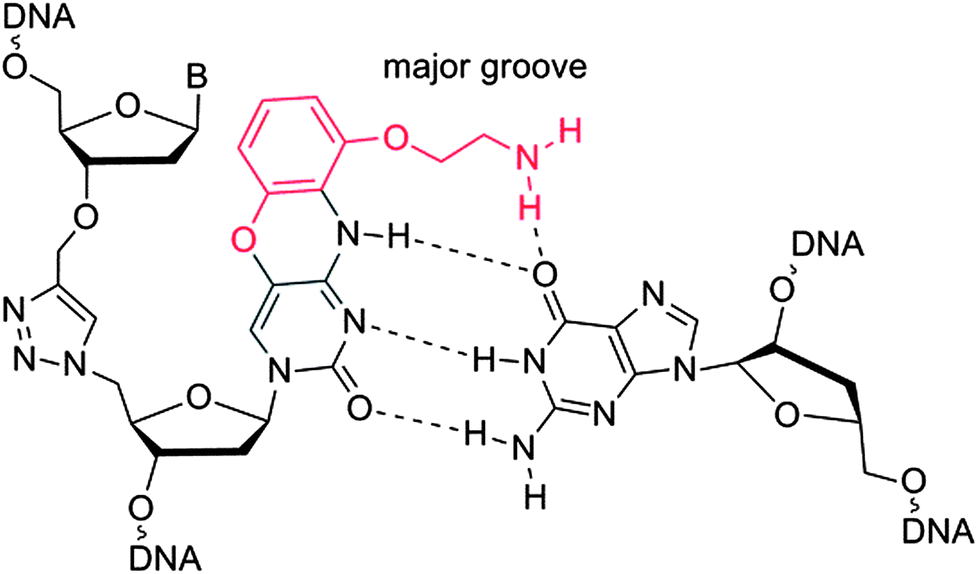 | ||
| Fig. 1 Triazole G-clamp base paired with guanine in complementary DNA. The additional steric bulk of the G-clamp nucleobase relative to cytosine is in red. | ||
In order to address this we have been investigating approaches to stabilise triazole-containing oligonucleotides, bearing in mind that any modification must also permit read-through by DNA polymerases. Many chemical adaptations have been developed to increase the thermal stability of nucleic acid duplexes,15 but with biocompatibility as the major criterion we focused on those that would be expected to produce minimal structural perturbation.
We now report the synthesis and properties of a triazole-containing DNA analogue that forms more stable duplexes than natural DNA. This is achieved by the incorporation of the aminoethylphenoxazine analogue of 2′-deoxycytidine (G-clamp) adjacent to the triazole linkage. This cytosine analogue increases duplex stability by a combination of increased intra-helical base stacking and additional hydrogen bonding to guanine (Fig. 1).16 It has been shown to be effective in a DNA17,18 and PNA19 context, and analogues have also been developed to recognise 8-oxoguanine in DNA.20 G-clamp is essentially deoxycytidine with an additional aminoethoxyphenoxy group protruding into the major groove. We reasoned that this structural change might be accommodated by polymerase enzymes without causing mutagenesis, and we have investigated this concept.
Results and discussion
In theory increased duplex stability might be achieved by inserting the G-clamp in the DNA strand at either the 5′- or 3′-side of the triazole linkage. However, placing the modification at the 5′-side would require the synthesis of a novel monomer for use in oligonucleotide synthesis. This could take the form of a resin-bound nucleoside consisting of a G-clamp nucleobase, deoxyribose sugar and 3′-propargyl group with an additional cleavable attachment point to the solid support; or more likely a 3′-propargyl G-clamp nucleoside-5′-phosphoramidite monomer for reverse direction oligonucleotide synthesis.21 Both of these are major undertakings, and it is evidently more straightforward to introduce the G-clamp at the 3′-side of the triazole linkage. It was apparent to us that this might be achieved using G-clamp phosphoramidite monomer18 combined with a variation of the methodology we have previously developed to synthesise triazole-linked DNA.3 This requires two oligonucleotides; one with a 5′-azide and the other with a 3′-propargyl group, followed by click ligation to join the two oligonucleotides together.The methodology that is generally used to synthesise 5′-azide oligonucleotides22 involves conversion of the 5′-OH group of the terminal deoxyribose sugar to azide whilst the oligonucleotide is attached to the solid support. This transformation (Fig. 2), which is accomplished using methyltriphenoxyphosphonium iodide, provides a general method to substitute the nucleobase on the 3′-side of the triazole linkage with unnatural analogues, in this case G-clamp. It has been shown that G-clamp is strongly stabilising at CpC steps in normal DNA,16 so we decided initially to synthesise and evaluate an oligonucleotide containing the equivalent triazole containing dinucleotide analogue (5-MeCtCC where CC = G-clamp and t = triazole). The 5-methylated analogue of cytosine was used for synthetic convenience.3
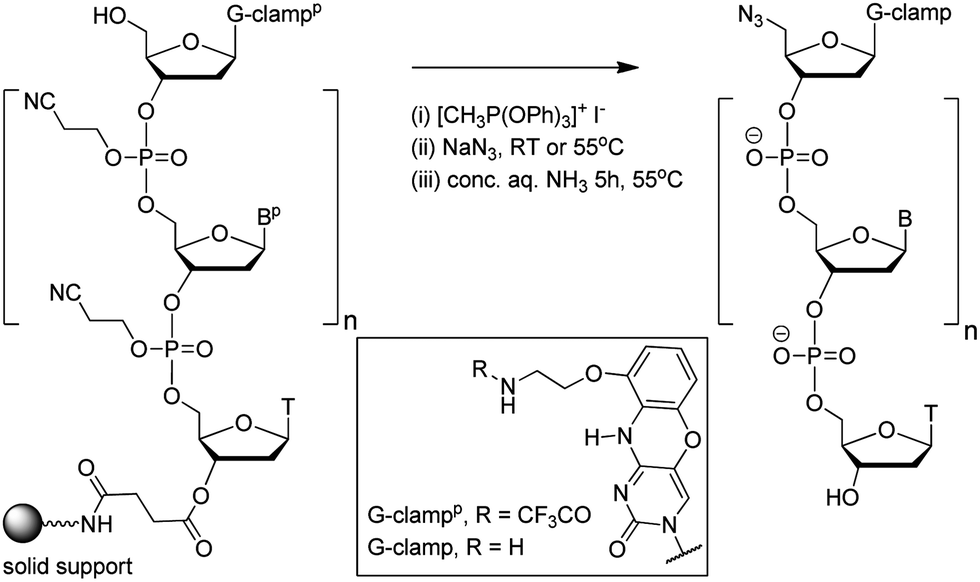 | ||
| Fig. 2 Conversion of the 5′-OH group of a G-clamp oligonucleotide to azide on the solid support. Structure of protected and unprotected G-clamp in inset. | ||
A 13-mer triazole G-clamp oligonucleotide (ODN-3, Table 1) was synthesised in order to provide a sensitive probe for duplex stability. To accomplish this, the G-clamp phosphoramidite monomer was incorporated at the 5′-end of oligonucleotide ODN-1 (Table 1) by standard solid-phase methods and the 5′-OH group was converted to azide on the solid support as described above (Fig. 2). This functional group conversion has not been previously carried out on 5′-G-clamp oligonucleotides and we were pleased to observe that it proceeded smoothly (Fig. 3A and B). After cleavage from the solid support and nucleobase/phosphate deprotection, ODN-1 was obtained in high purity. Ligation of ODN-1 to an excess of 3′-propargyl-functionalised ODN-26 by the CuAAC reaction1,2 yielded the triazole-containing 13-mer ODN-3. The reaction proceeded efficiently without the need for a complementary splint (Fig. 3C and D) and excess of 3′-propargyl ODN-2 was removed by HPLC purification. Ultraviolet melting studies were performed with ODN-3 hybridised to a series of matched and mismatched complementary DNA and RNA strands (Fig. 4, Table 2). The stability of the duplex formed by ODN-3 and its unmodified DNA complement (ODN-7) was compared to the equivalent unmodified canonical duplex with a central 5-methy cytosine (ODN-4/ODN-7) and to the duplex containing a triazole linkage and a cytosine base in place of G-clamp (ODN-6/ODN-7). ODN-6 was made by clicking ODN-2 with ODN-5 using the method described above.
| Code | Sequence 5′ to 3′ |
|---|---|
| a MeCY = 3′-propargyl-5-methyl dC, CZ = 5′-azido dC, CCZ = 5′-azido G-clamp, t = triazole linkage. F = amidohexylfluorescein. | |
| ODN-1 | dCCZTGCAGC |
| ODN-2 | dCGACGMeCY |
| ODN-3 | dCGACGMeCtCCTGCAGC |
| ODN-4 | dCGACGMeCCTGCAGC |
| ODN-5 | dCZTGCAGC |
| ODN-6 | dCGACGMeCtCTGCAGC |
| ODN-7 | dGCTGCAGGCGTCG |
| ODN-8 | dGCTGCAGACGTCG |
| ODN-9 | dGCTGCAGCCGTCG |
| ODN-10 | dGCTGCAGTCGTCG |
| ODN-11 | dGCTGCAAGCGTCG |
| ODN-12 | dGCTGCACGCGTCG |
| ODN-13 | dGCTGCATGCGTCG |
| ODN-14 | rGCUGCAGGCGUCG |
| ODN-15 | rGCUGCAGACGUCG |
| ODN-16 | rGCUGCAAGCGUCG |
| ODN-17 | dFTGTGTGCTGGCGATCTTA – splint for click reaction to synthesise PCR template |
| ODN-18 | dGCATTCGAGCAACGTAAGATCGMeCY |
| ODN-19 | dCCZAGCACACAATCTCACACTCTGGAATTCACACTGACAATACTGCCGACACACATAACC |
| ODN-20 | dGCATTCGAGCAACGTAAGATCGMeCtCCAGCACACAATCTCACACTCTGGAATTCACACTGACAATACTGCCGACACACATAACC – PCR template |
| ODN-21 | dGCATTCGAGCAACGTAAG – PCR primer |
| ODN-22 | dGGTTATGTGTGTCGGCAG – PCR primer |
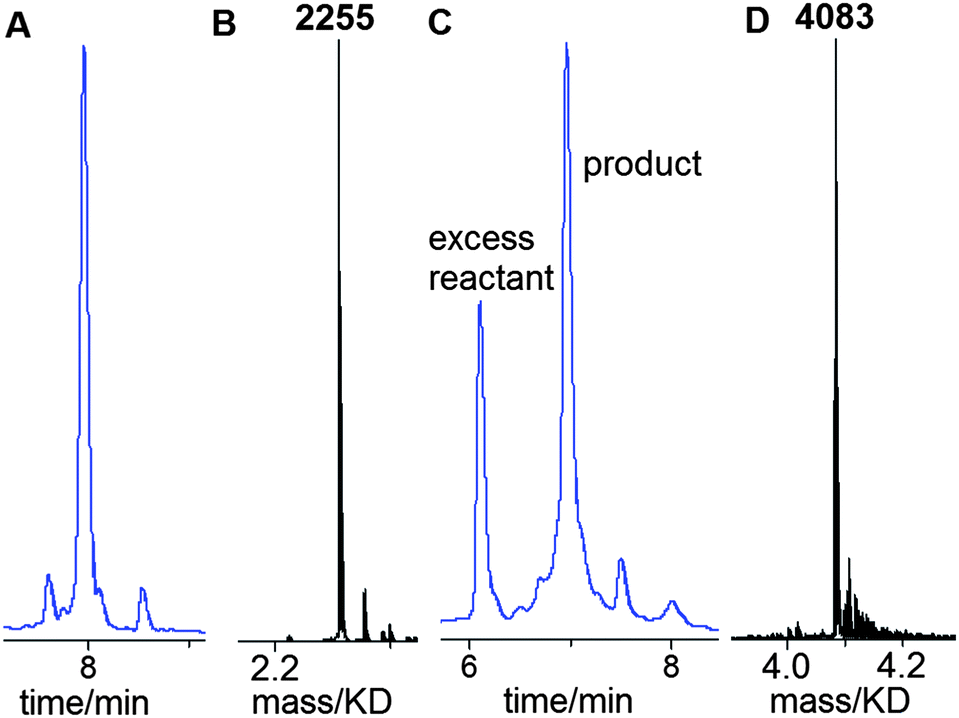 | ||
| Fig. 3 Efficient non-templated click ligation to synthesise triazole G-clamp ODN-3. (A) Reversed-phase HPLC and (B) ESI-mass spectrum of unpurified ODN-1 (5′-azido G-clamp 7-mer). (C) Reversed-phase HPLC and (D) ESI-mass spectrum of click reaction between ODN-1 and ODN-2 (3′-propargyl 6-mer) to give triazole G-clamp 13-mer (ODN-3). An excess of ODN-2 was used to drive this reaction to completion. | ||
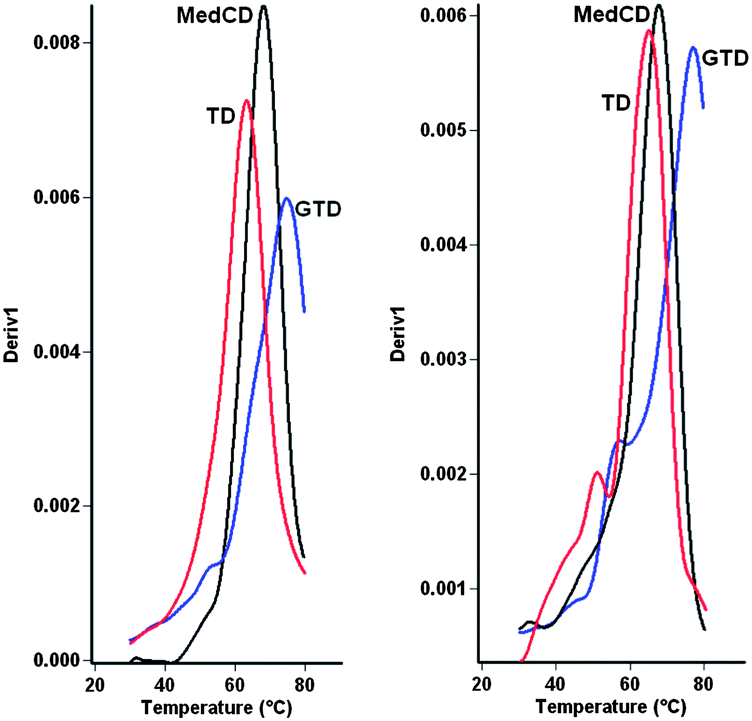 | ||
| Fig. 4 UV melting studies (derivatives of melting curves). Triazole G-clamp stabilises DNA duplexes and DNA–RNA hybrids. (A). DNA–DNA duplexes; MedCD = control duplex (ODN-4 + ODN-7, Tm = 68.5 °C), TD = triazole duplex (ODN-6 + ODN-7, Tm = 63.0 °C), GTD = G-clamp triazole duplex (ODN-3 + ODN-7, Tm = 75.0 °C). (B). DNA–RNA hybrid duplexes; MedCD = control duplex (ODN-4 + ODN-14, Tm = 68.1 °C), TD = triazole duplex (ODN-6 + ODN-14, Tm = 64.8 °C), GTD = G-clamp triazole duplex (ODN-3 + ODN-14, Tm = 77.5 °C). Duplexes contain a CC step around triazole site and 100% Watson–Crick base pairs. | ||
| ODN code | ODN code | Central base pairs* | MeCxC central linkage | T m | ΔTm |
|---|---|---|---|---|---|
a ΔTm = (Tm duplex − Tm control duplex). t = central triazole linkage in ODN in column 1 of the table. p = central phosphodiester linkage in ODN in column 1 of the table. CC = G-clamp, MeC = 5-methylcytosine, r = RNA. * Central base pairs = base pairs around triazole site proceeding from 5′-end of ODN-3, 4 or 6 in column 1. For example entry 1 in Table 2a, MeCG–CCG is derived from: 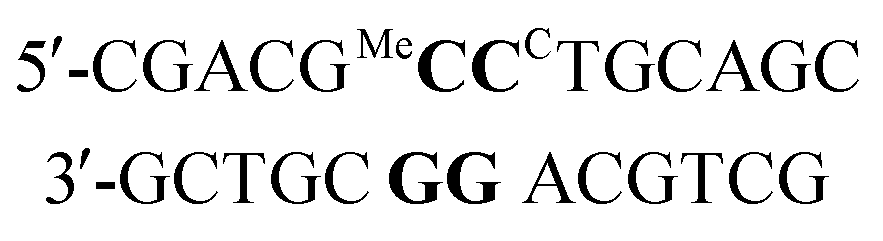 All oligonucleotide sequences are in Table 1. All oligonucleotide sequences are in Table 1.
|
|||||
| (a) Watson–Crick base pairing DNA duplexes | |||||
| ODN-3 | ODN-7 | MeCG–CCG | t | 75.0 | |
| ODN-4 | ODN-7 | MeCG–CG | p | 68.5 | −6.5 |
| ODN-6 | ODN-7 | MeCG–CG | t | 63.0 | −12.0 |
| (b) Watson–Crick base pairing DNA–RNA hybrid duplexes | |||||
| ODN-3 | rODN-14 | MeCG–CCG | t | 77.5 | |
| ODN-4 | rODN-14 | MeCG–CG | p | 68.1 | −9.4 |
| ODN-6 | rODN-14 | MeCG–CG | t | 64.8 | −12.7 |
| (c) G-clamp triazole DNA–DNA mismatch studies | |||||
| ODN-3 | ODN-7 | MeCG–CCG | t | 75.0 | — |
| ODN-3 | ODN-8 | MeCA–CCG | t | 55.3 | −19.7 |
| ODN-3 | ODN-9 | MeCC–CCG | t | 55.0 | −20.0 |
| ODN-3 | ODN-10 | MeCT–CCG | t | 55.1 | −19.9 |
| ODN-3 | ODN-11 | MeCG–CCA | t | 56.3 | −18.7 |
| ODN-3 | ODN-12 | MeCG–CCC | t | 53.0 | −22.0 |
| ODN-3 | ODN-13 | MeCG–CCT | t | 55.8 | −19.2 |
| Average = −19.9 | |||||
| (d) DNA–DNA mismatch studies | |||||
| ODN-4 | ODN-7 | MeCG–CG | p | 68.5 | — |
| ODN-4 | ODN-8 | MeCA–CG | p | 54.1 | −14.4 |
| ODN-4 | ODN-9 | MeCC–CG | p | 47.9 | −20.6 |
| ODN-4 | ODN-10 | MeCT–CG | p | 52.1 | −16.4 |
| ODN-4 | ODN-11 | MeCG–CA | p | 45.5 | −23.0 |
| ODN-4 | ODN-12 | MeCG–CC | p | 49.0 | −19.5 |
| ODN-4 | ODN-13 | MeCG–CT | p | 52.2 | −16.3 |
| Average = −18.4 | |||||
| (e) Triazole DNA–DNA mismatch studies | |||||
| ODN-6 | ODN-7 | MeCG–CG | t | 63.0 | — |
| ODN-6 | ODN-8 | MeCA–CG | t | 50.6 | −12.4 |
| ODN-6 | ODN-9 | MeCC–CG | t | 46.8 | −16.2 |
| ODN-6 | ODN-10 | MeCT–CG | t | 48.7 | −14.3 |
| ODN-6 | ODN-11 | MeCG–CA | t | 45.3 | −17.7 |
| ODN-6 | ODN-12 | MeCG–CC | t | 47.6 | −15.4 |
| ODN-6 | ODN-13 | MeCG–CT | t | 50.2 | −12.8 |
| Average = −14.8 | |||||
| (f) G-clamp triazole DNA–RNA mismatch studies | |||||
| ODN-3 | rODN-14 | MeCG–CCG | t | 77.5 | — |
| ODN-3 | rODN-15 | MeCA–CCG | t | 58.2 | −19.3 |
| ODN-3 | rODN-16 | MeCG–CCA | t | 53.0 | −24.5 |
| Average = −21.9 | |||||
| (g) DNA–RNA mismatch studies | |||||
| ODN-4 | rODN-14 | MeCG–CG | p | 68.1 | — |
| ODN-4 | rODN-15 | MeCA–CG | p | 53.3 | −14.8 |
| ODN-4 | rODN-16 | MeCG–CA | p | 49.5 | −18.6 |
| Average = −16.7 | |||||
| (h) Triazole DNA–RNA mismatch studies | |||||
| ODN-6 | rODN-14 | MeCG–CG | t | 64.8 | — |
| ODN-6 | rODN-15 | MeCA–CG | t | 51.1 | −13.7 |
| ODN-6 | rODN-16 | MeCG–CA | t | 44.9 | −19.9 |
| Average = −16.8 | |||||
Incorporation of the G-clamp increased the UV melting temperature (Tm) of the triazole DNA duplex by an impressive 6.5 °C compared to the unmodified duplex, and by 12 °C compared to the triazole duplex with cytosine in place of the G-clamp (Table 2a). Even greater stabilisation was observed when the triazole G-clamp ODN-3 was hybridised to an RNA complement. In this case the increase in Tm was 9.4 °C compared to the normal DNA–RNA hybrid, and 12.7 °C relative to the triazole duplex with cytosine instead of G-clamp (Table 2b). To dispel concerns that the stabilisation due to the triazole G-clamp is unique to a MeCtCC base stacking step, studies were also carried out to determine the melting temperatures of duplexes containing a TtCC step. The same trends in duplex stability were observed, i.e. triazole G-clamp > canonical DNA > triazole DNA (ESI†). For chemically modified DNA to be useful in a biological context it is essential that it is selective for its chosen target; i.e. duplexes containing mismatched base pairs must be destabilised. To evaluate this, duplexes containing a single methylated or unmethylated C.A or C.C mismatch on either side of the triazole linkage were studied. In the DNA–DNA series the average mismatch destabilisation was 19.9 °C for the triazole G-clamp duplex, 18.4 °C for the normal non-triazole duplex and 14.8 °C for the triazole duplex without G-clamp (Table 2c–e), indicating that triazole G-clamp is the most efficient sensor of DNA mispairing. In the context of DNA–RNA hybrid duplexes the beneficial effects of triazole G-clamp were even greater, with an average mismatch destabilisation of 21.9 °C compared to 16.7 °C for normal DNA and 16.8 °C for triazole DNA without the G-clamp (Table 2f–h). Overall these melting studies confirm that the stability of DNA can be greatly improved by incorporation of G-clamp on the 3′-side of the artificial triazole backbone. Moreover, potent mismatch discrimination can be achieved if the G-clamp nucleobase is directly involved in mispairing, and also when the base directly on the other side of the triazole linkage is mispaired. There is a marked improvement in mismatch discrimination for the combination of triazole and G-clamp compared to normal DNA and DNA containing triazole alone.
Having established that G-clamp is effective in stabilising triazole DNA duplexes, it was important to determine whether DNA polymerases can read through the combination of the triazole and G-clamp to faithfully produce complementary copies. The outcome of these investigations was uncertain as there are no previous reports of replication of DNA strands that contain G-clamp monomers, let alone examples of G-clamp combined with backbone modifications. To investigate the polymerase-compatibility of the duplex-stabilising modification we synthesised an 81-mer PCR template (ODN-20) containing triazole G-clamp using a complementary splint (ODN-17). The splint was employed to assist CuAAC-catalysed oligonucleotide ligation between 3′-propargyl ODN-18 and 5′-azide ODN-19. The click reaction was efficient and the template was purified and analysed by HPLC and mass spectrometry (Fig. 5A and B). We then carried out experiments to find out if the doubly-modified linkage can be read through in a linear fashion. This was successful (ESI†). PCR amplification was carried out (Fig. 5C) and the amplicon was purified by agarose gel electrophoresis, inserted into a sequencing vector and analysed by Sanger DNA sequencing (Fig. 5D). The sequencing data from over 20 clones confirmed that the region around the triazole linkage had been read through correctly (ESI†). Overall this series of experiments prove that the G-clamp triazole linkage can be formed efficiently by click ligation, and can be read through faithfully by DNA polymerase enzymes (Fig. 6). This is an important result as it suggests that the triazole G-clamp combination could be used for in vivo applications that involve replication of the modified DNA.3,5
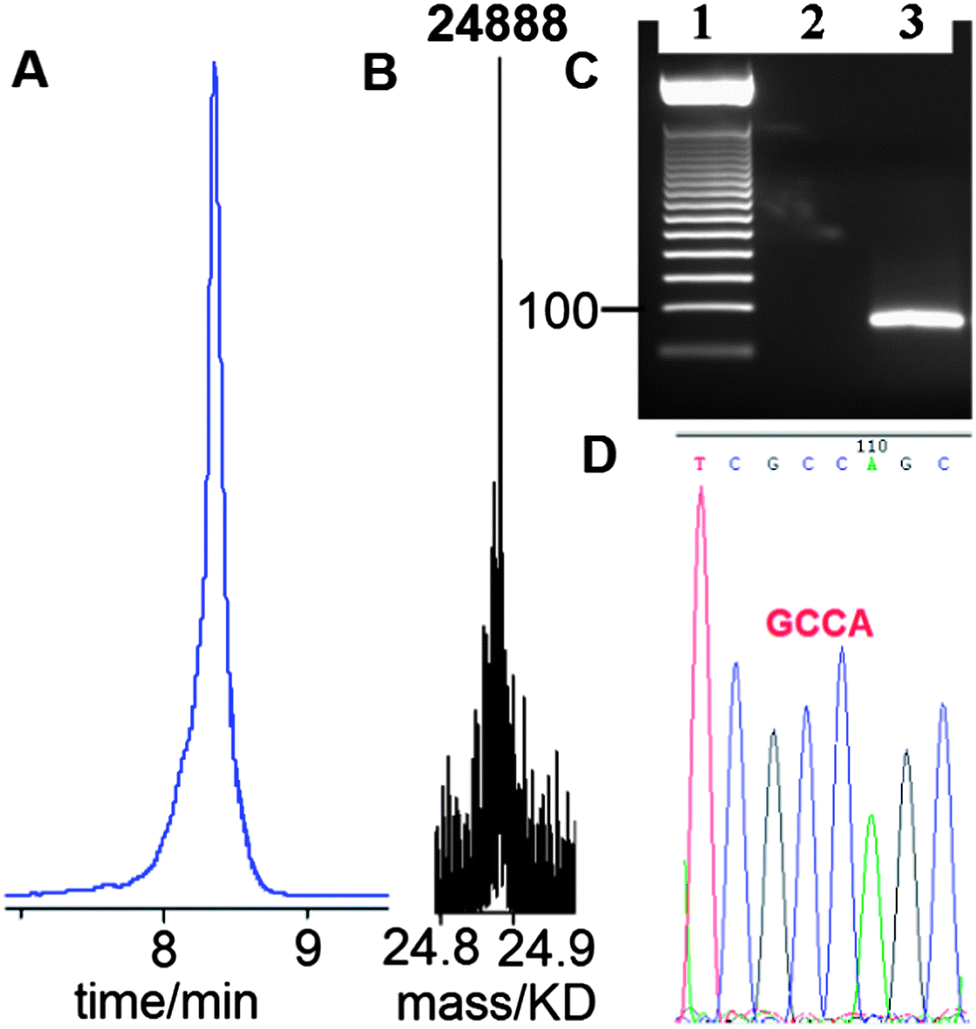 | ||
| Fig. 5 G-clamp triazole DNA template is efficiently chemically ligated and accurately amplified by PCR. (A) Reversed-phase HPLC, (B) ESI-mass spectrum of 81-mer ODN-20, and (C) 2% agarose gel for the PCR product using ODN-20 as a template. Lane 1; 50bp DNA ladder. Lane 2; control reaction with primers and without the template. Lane 3; PCR reaction using template ODN-20 (5 ng) and primers ODN-21 and ODN-22 (Table 1). (D) Sanger sequencing of cloned PCR template showing the correct sequence (dGCCA) around the site of the original triazole G-clamp. Details of DNA sequencing in ESI.† | ||
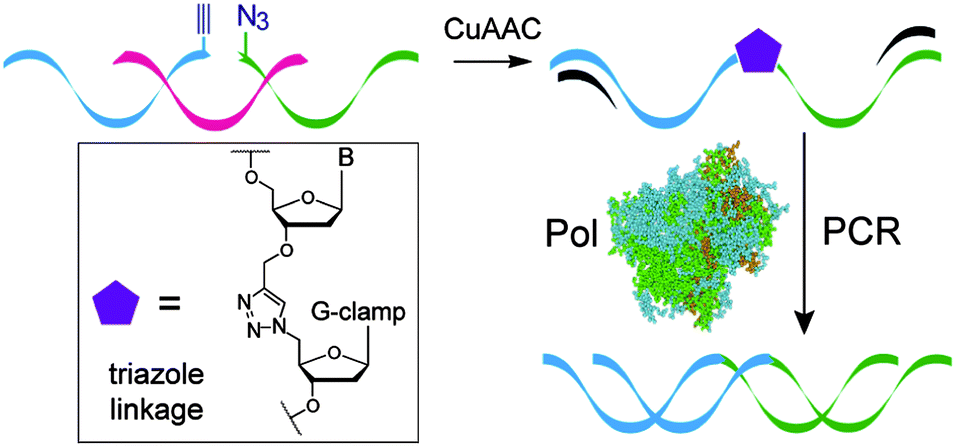 | ||
| Fig. 6 Formation of the G-clamp triazole linkage by the CuAAC reaction and faithful read-through by PCR. | ||
In the context of future therapeutic and diagnostic applications it is important to understand duplex forming properties of oligonucleotides containing multiple modifications of triazole G-clamp. It was unclear whether this would confer additional duplex stability or lead to a collapse of the duplex structure due to steric constraints. To this end we used double templated click ligation to synthesise a 13-mer oligonucleotide containing two units of triazole G-clamp and compared the stability of its fully complementary DNA duplex to equivalent unmodified and triazole modified duplexes (ESI†). Very large increases in Tm (12.2 °C and 22.7 °C respectively) were observed confirming the potential of triazole G-clamp as a modification in antisense oligonucleotides. With this result in mind future synthetic strategies will focus on producing the triazole G-clamp dinucleotide phosphoramidite for direct incorporation into oligonucleotides during solid-phase synthesis.
Conclusions
A simple strategy for the synthesis of oligonucleotides containing a G-clamp triazole linkage has been established. The G-clamp is introduced by standard solid-phase oligonucleotide synthesis and the triazole linkage is inserted in a CuAAC reaction between 3′-propargyl and 5′-azido G-clamp oligonucleotides.1,2 The click reaction works in templated and non-templated modes, although templation is recommended for the synthesis of long oligonucleotides to guarantee an efficient reaction. All the required reagents are available commercially, making this methodology widely accessible. G-clamp triazole-modified DNA has been shown to form duplexes which are more stable than the equivalent unmodified canonical DNA. Interestingly the presence of a single G-clamp nucleobase is sufficient to increase the duplex stability of triazole DNA significantly beyond that of unmodified DNA, and two additions lead to very large stabilisation. This is likely to be a consequence of increased base stacking and H-bonding, preventing fraying of the base pair adjacent to the triazole linkage.6 Importantly, duplexes containing the triazole G-clamp are greatly destabilised by the presence of mismatched base pairs close to the modification, demonstrating that triazole G-clamp is a powerful discriminatory sensor of Watson–Crick base pairing. The increased duplex stability and mismatch destabilisation suggests that the G-clamp triazole modification could be beneficial in antisense, siRNA and miRNA applications. All-triazole DNA backbone analogues23–27 are being developed for these applications using the click concept,28 but there are issues related to poor aqueous solubility.29 One solution to this problem is to develop chimeric oligonucleotides containing a combination of normal and triazole backbones. In this context the results presented here, demonstrating the high stability of the G-clamp triazole combination, are very encouraging. This principle of nucleobase compensation to achieve high duplex stability could be applied to other triazole DNA backbone analogues,12–14 but whether or not these would be compatible with accurate read-thorough by DNA polymerases remains to be established. When present in a DNA template, the G-clamp triazole linkage described here can be read through and accurately amplified by PCR, with the G-clamp nucleobase recognised as cytosine. Surprisingly the polymerase enzyme readily accommodates the combined backbone and nucleobase modifications. This study opens the way to the wider use of oligonucleotides containing triazole backbones in biotechnology, nanotechnology and various diagnostic and therapeutic applications.Experimental
Synthesis of 5′-azide G-clamp and dC oligonucleotides
G-clamp phosphoramidite monomer was obtained from Glen Research. Oligonucleotides were assembled on the 1.0 μmole scale (trityl-off) with 5′-dC or 5′-G-clamp as described in the ESI.† The protected oligonucleotide attached to the synthesis column was then treated with a 0.5 M solution of methyltriphenoxyphosphonium iodide in DMF (1.0 mL) which was periodically passed through the column via two 1 mL syringes over 15 min at room temperature. The column was then washed several times with dry DMF. To convert the 5′-iodo (dC or G-clamp) to 5′-azido (dC or G-clamp), sodium azide (15 mg) was suspended in dry DMF (1 mL), heated for 10 min at 70 °C then cooled down. The supernatant was taken up into a 1 mL syringe, passed back and forth through the column then left at room temperature overnight (for short oligonucleotides) or for 5 h at 55 °C (for long oligonucleotides). The column was then washed with DMF and acetonitrile and dried by passing a stream of argon gas through it. The resultant 5′-azide oligonucleotide was cleaved from the solid support and deprotected by exposure to concentrated aqueous ammonia solution for 60 min at room temperature followed by heating in a sealed tube for 5 h at 55 °C. Oligonucleotides were purified by HPLC as described in the ESI.†Synthesis of the G-clamp and normal triazole DNA templates
A solution of CuI click catalyst was prepared from tris-hydroxypropyltriazole ligand30 (14.0 μmol in 0.2 M NaCl, 90.0 μL), sodium ascorbate (20.0 μmol in 0.2 M NaCl, 40.0 μL) and CuSO4·5H2O (2.0 μmol in 0.2 M NaCl, 20.0 μL). This solution was added to the two reacting oligonucleotides (5′-azide and 3′-alkyne) and the complementary splint oligonucleotide, (100.0 nmol of each) in 0.2 M NaCl (100 μL). The mixture was kept at room temperature for 2 h. Reagents were then removed by NAP-10 gel-filtration and the ligated triazole DNA product was purified by HPLC as described in the ESI.† For the synthesis of the short (13-mer) triazole containing oligonucleotides, the overall conditions were the same except that a 1.5-fold excess of the alkyne oligonucleotide was used in a non-templated click ligation reaction (i.e. no splint oligonucleotide was added).Ultraviolet DNA melting studies
UV DNA melting was monitored on Cary 4000 Scan UV-Visible Spectrophotometer (Varian) at 3 μM concentration of each oligonucleotide in 10 mM phosphate buffer, 200 mM NaCl at pH 7.0. Spectra were recorded at 260 nm. The samples were initially denatured by heating to 85 °C (or 90 °C) at 10 °C min−1 then cooled to 20 °C at 1 °C min−1 and heated to 85 °C (or 90 °C) at 1 °C min−1. Eight successive melting curves were measured and Tm values were calculated from their derivatives using Cary Win UV Thermal application Software.PCR amplification of the G-clamp triazole template
GoTaq DNA polymerase was used to generate a PCR product from the 81-mer template (ODN-20) which includes a G-clamp triazole linkage. Reagents and conditions: 4 μL of 5× buffer (Promega green PCR buffer)¶ was used in a total reaction volume of 20 μL with 5 ng of the DNA template, 0.5 μM of each primer, 0.2 mM dNTP and 0.5 unit of GoTaq polymerase. The reaction mixture was loaded onto a 2% agarose gel in 1× TBE buffer. PCR cycling conditions: 95 °C (initial denaturation) for 2 min then 25 cycles of 95 °C (denaturation) for 15 s, 54 °C (annealing) for 20 s and 72 °C (extension) for 30 s. This was followed by leaving the PCR reaction mixture at 72 °C for 5 min. The PCR amplicon was extracted from the gel using a QIAquick Gel Extraction kit. It was then inserted into a vector for automated Sanger DNA sequencing (ESI†).Acknowledgements
This research was funded by the BBSRC sLOLA grant BB/J001694/1 “Extending the boundaries of nucleic acid chemistry.”Notes and references
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- A. H. El-Sagheer, A. P. Sanzone, R. Gao, A. Tavassoli and T. Brown, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11338–11343 CrossRef CAS PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Commun., 2011, 47, 12057–12058 RSC.
- A. P. Sanzone, A. H. El-Sagheer, T. Brown and A. Tavassoli, Nucleic Acids Res., 2012, 40, 10567–10575 CrossRef CAS PubMed.
- A. Dallmann, A. H. El-Sagheer, L. Dehmel, C. Mugge, C. Griesinger, N. P. Ernsting and T. Brown, Chem.–Eur. J., 2011, 17, 14714–14717 CrossRef CAS PubMed.
- D. Mutisya, C. Selvam, S. D. Kennedy and E. Rozners, Bioorg. Med. Chem. Lett., 2011, 21, 3420–3422 CrossRef CAS PubMed.
- E. Uhlmann and A. Peyman, Chem. Rev., 1990, 90, 543–584 CrossRef CAS.
- R. W. Carthew and E. J. Sontheimer, Cell, 2009, 136, 642–655 CrossRef CAS PubMed.
- C. V. Pecot, G. A. Calin, R. L. Coleman, G. Lopez-Berestein and A. K. Sood, Nat. Rev. Cancer, 2011, 11, 59–67 CrossRef CAS PubMed.
- B. L. Davidson and P. B. McCray, Nat. Rev. Genet., 2011, 12, 329–340 CrossRef CAS PubMed.
- A. M. Varizhuk, D. N. Kaluzhny, R. A. Novikov, A. O. Chizhov, I. P. Smirnov, A. N. Chuvilin, O. N. Tatarinova, G. Y. Fisunov, G. E. Pozmogova and V. L. Florentiev, J. Org. Chem., 2013, 78, 5964–5969 CrossRef CAS PubMed.
- S. Chandrasekhar, P. Srihari, C. Nagesh, N. Kiranmai, N. Nagesh and M. M. Idris, Synthesis, 2010, 3710–3714 CrossRef CAS PubMed.
- V. Madhuri and V. A. Kumar, Nucleosides, Nucleotides Nucleic Acids, 2012, 31, 97–111 CAS.
- J. Kurreck, Eur. J. Biochem., 2003, 270, 1628–1644 CrossRef CAS.
- J. A. Ortega, J. R. Blas, M. Orozco, A. Grandas, E. Pedroso and J. Robles, Org. Lett., 2007, 9, 4503–4506 CrossRef CAS PubMed.
- K. Y. Lin and M. D. Matteucci, J. Am. Chem. Soc., 1998, 120, 8531–8532 CrossRef CAS.
- W. M. Flanagan, J. J. Wolf, P. Olson, D. Grant, K. Y. Lin, R. W. Wagner and M. D. Matteucci, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3513–3518 CrossRef CAS.
- S. Rapireddy, R. Bahal and D. H. Ly, Biochemistry, 2011, 50, 3913–3918 CrossRef CAS PubMed.
- O. Nakagawa, S. Ono, Z. Li, A. Tsujimoto and S. Sasaki, Angew. Chem., Int. Ed., 2007, 46, 4500–4503 CrossRef CAS PubMed.
- C. D. Claeboe, R. Gao and S. M. Hecht, Nucleic Acids Res., 2003, 31, 5685–5691 CrossRef CAS PubMed.
- G. P. Miller and E. T. Kool, Org. Lett., 2002, 4, 3599–3601 CrossRef CAS PubMed.
- T. Fujino, N. Yamazaki and H. Isobe, Tetrahedron Lett., 2009, 50, 4101–4103 CrossRef CAS PubMed.
- H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Org. Lett., 2008, 10, 3729–3732 CrossRef CAS PubMed.
- A. Nuzzi, A. Massi and A. Dondoni, QSAR Comb. Sci., 2007, 26, 1191–1199 CAS.
- R. Lucas, R. Zerrouki, R. Granet, P. Krausz and Y. Champavier, Tetrahedron, 2008, 64, 5467–5471 CrossRef CAS PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- T. Fujino, Y. Miyauchi, N. Tsunaka, K. Okada and H. Isobe, Heterocycles, 2013, 87, 1023–1028 CrossRef CAS.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Oligonucleotide synthesis, DNA sequencing, UV melting, studies on oligonucleotides containing a TtC step, mass spectra of oligonucleotides, linear copying of template oligonucleotides, click ligation to synthesise oligonucleotide with two G clamp-triazole modifications. See DOI: 10.1039/c3sc51753e |
| ‡ TB and AHE-S are joint main authors. |
| § Current address: Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Rd, Oxford, OX1 3TA, UK. Email: tom.brown@chem.ox.ac.uk; afaf.el-sagheer@chem.ox.ac.uk; Tel: +44 (0)1865 275413. |
| ¶ The 5× Promega green PCR buffer (pH 8.5) which was provided with GoTaq DNA polymerase contains 7.5 mM MgCl2 to give a final Mg2+ concentration of 1.5 mM. The buffer contains Tris·HCl, KCl and two dyes (blue and yellow) that separate during electrophoresis to monitor the migration process. |
| This journal is © The Royal Society of Chemistry 2014 |