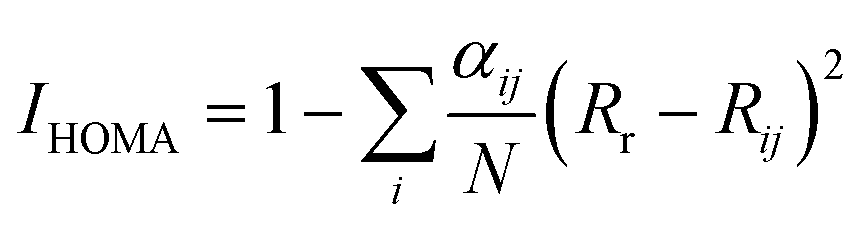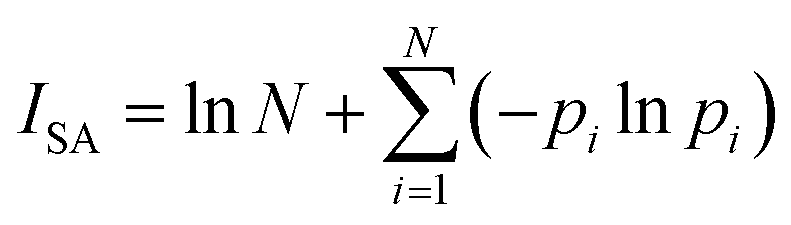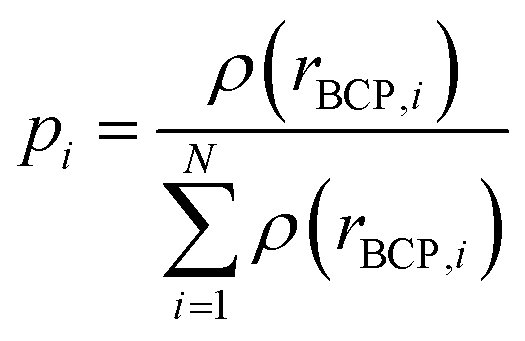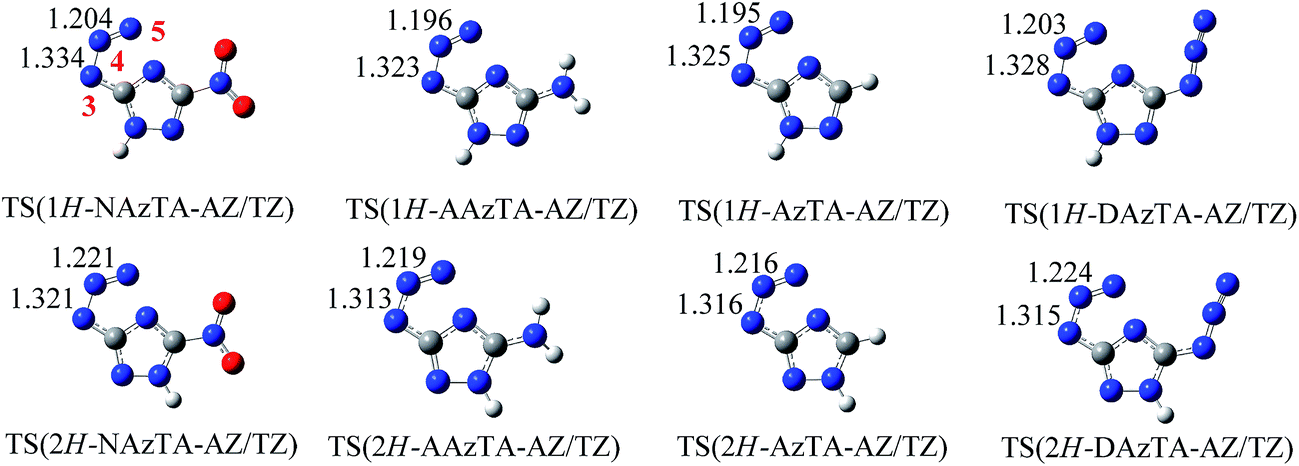DOI:
10.1039/C4RA14560G
(Paper)
RSC Adv., 2015,
5, 9503-9509
1H/2H and azide/tetrazole isomerizations and their effects on the aromaticity and stability of azido triazoles†
Received
14th November 2014
, Accepted 23rd December 2014
First published on 23rd December 2014
Abstract
Isomerization is one of the most curious aspects of organic chemistry. In this work, 1H/2H and azide/tetrazole (AZ/TZ) isomerizations in 3-nitro-5-azido-1,2,4-triazole (NAzTA), 3-amino-5-azido-1,2,4-triazole (AAzTA), and 5-azido-1,2,4-triazole (AzTA) were studied using the density functional theory method. The 1H/2H tautomerization proceeds most easily for AAzTA while hardest for NAzTA, suggesting that –NH2 facilitates the process, while –NO2 gives an opposite effect. The AZ/TZ isomerization takes place most easily for AzTA, and then AAzTA and NAzTA in sequence, revealing that this kind of isomerization more easily proceeds in the molecule with only the –N3 group and substitution of –NH2 or –NO2 prevents the process. The AZ/TZ isomerization is much easier than the 1H/2H tautomerization. For the azide isomers, the triazole ring has higher aromaticity in the 1H-tautomer than in the 2H-form, i.e., tautomerization from 1H to 2H isomer makes the stability decrease. For the tetrazole isomers, both the triazole ring and the tetrazole ring have high aromaticity. One more aromatic ring in the tetrazole isomers implies an increase in the stability by the isomerization from an azide to tetrazole.
1. Introduction
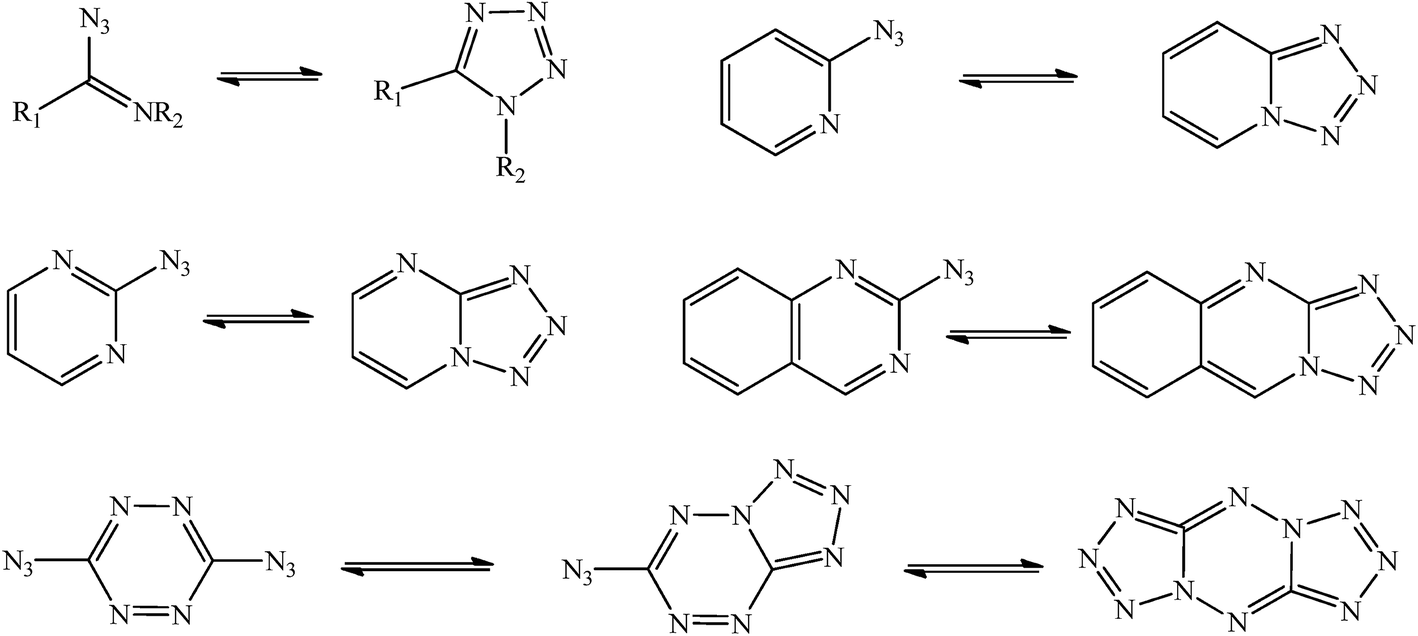
Azide/tetrazole (AZ/TZ) isomerization is a common phenomenon in azido compounds1,2 and has been studied experimentally or theoretically3–6 for, such as, azidoazomethine, 2-azidopyridine, 2-azidopyrimidine, 2-azidoquinazoline, and 3,6-di(azido)-1,2,4,5-tetrazine (Fig. 1). The tetrazole isomers, formed by a 1,5-dipolar cyclization, are potential replacements for the hazardous azides. However, there is no research to compare the stability of the azide and its isomeric tetrazole6 and the reports on the AZ/TZ isomerization in azido triazoles are few. In this work, three 1,2,4-triazoles, i.e., 3-nitro-5-azido-1,2,4-triazole (NAzTA), 3-amino-5-azido-1,2,4-triazole (AAzTA), and 3-azido-1,2,4-triazole (AzTA) were studied to explore the substituent effects (–NO2 and –NH2) on the AZ/TZ isomerization and to test the stability of the azide and its isomeric tetrazole. In addition, based on their similar structures with 3-amino-5-nitro-1,2,4-triazole (ANTA), these compounds should also have the similar chemical behavior with ANTA. Studies7,8 revealed ANTA has two stable existent forms, i.e., 1H-ANTA and 2H-ANTA, and the 1H/2H tautomerization is one of the reaction channels in thermal reactions of ANTA. Therefore, the 1H/2H tautomerization has also been considered for the title compounds.
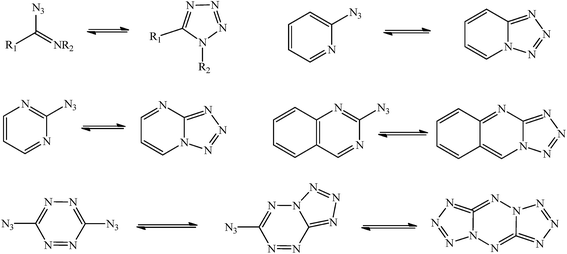 |
| | Fig. 1 Azide/tetrazole isomerization in azidoazomethine, 2-azidopyridine, 2-azidopyrimidine, 2-azidoquinazoline, and 3,6-di(azido)-1,2,4,5-tetrazine. | |
Aromaticity is one of the most important concepts in contemporary organic chemistry and it is also a useful quantity in rationalizing structure, stability, and reactivity of many molecules. The relationship between structure, aromaticity and stability has been investigated by many researches.9–12 Generally, higher aromaticity corresponds to higher stability. That is to say, investigation on the aromaticity is helpful for understanding and comparing the stability of compounds to some extent. Many criteria, such as the harmonic oscillator model of aromaticity (IHOMA), shannon aromaticity index (ISA), iso-chemical shielding surface (ICSS) in the Z direction (ICSSZZ), multi-center bond order (I), aromatic stabilization energy (ASE), etc, have been put forward to quantify this property. In this work, three criteria, IHOMA, ISA, and ICSSZZ, were adopted to rationalize the aromaticity of the isomers of title compounds. From the viewpoint of aromaticity, the effects of the 1H/2H and AZ/TZ isomerizations on the stability were investigated.
The work reveals the effects of –NO2, –NH2, and –N3 on the chemical behavior of NAzTA, AAzTA, and AzTA and demonstrates the effects of 1H/2H and AZ/TZ isomerizations on the stability. The results allow for better understanding these compounds as well as the differences among them and help to further cognize the two isomerizations in organic compounds.
2. Theoretical methods
In literatures, various theoretical methods, e.g., W1,13 MP2,14 B3LYP,14 etc., have been used to study the structure and tautomerism of organic compounds. Investigation on ANTA showed that B3LYP can produce similar results with MP2 and reveal the tautomerism of ANTA effectively.14 Considering the similar structures of the title compounds with ANTA, B3LYP functional was chosen in this research. The azide tautomers (AZ) of NAzTA, AAzTA, and AzTA in gas and solution phases were studied using the B3LYP functional15,16 of density functional theory and the 6-31G*,17 6-311++G**,18,19 and AUG-cc-PVDZ20 basis sets. The CPCM continuum solvent model21 was adopted to simulate the liquid surroundings.
The activation energy (Ea) of the isomerism reaction was calculated by the following equation:
where
ETS and
ER are the total energies after the correction of zero-point energy (ZPE) for the transition state (TS) and reactant, respectively. The TS was located by scanning the potential energy surface and the reliability of the TS was verified by analyzing the optimized structures, frequencies, and intrinsic reaction coordinate (IRC).
Aromaticity of various isomers was quantified by IHOMA, ISA, and ICSSZZ. The generalized IHOMA can be written as:
| |
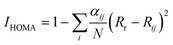 | (2) |
where
N is the total number of the atoms considered,
j denotes the atom next to the atom
i,
α and
Rr are the constants for each type of atom pair given in
ref. 22.
ISA was calculated as follows:
| |
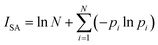 | (3) |
| |
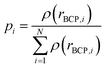 | (4) |
where
pi is the normalized electron density at the bond critical points (BCP) of the
ith bond.
N is the total number of the BCPs in the ring.
ρ(
rBCP,i) is the electron density at the position
rBCP,i of the
ith BCP.
Calculations of ICSSZZ at the ring centers were performed using the gauge-independent atomic orbital (GIAO) method23,24 at the B3LYP/6-311++G** level. ICSS is the isosurface of nucleus-independent chemical shift (NICS), it can clearly exhibit the distribution of NICS in a 3D space and present a more intuitive picture of aromaticity than NICS does. Just like NICSZZ has conspicuous advantages over NICS,25–27 ICSSZZ is more physically meaningful than ICSS.28 The absolute magnetic shielding value at the point 1 Å above the ring center (NICSZZ(1)) was recommended to be a better measure of the π electron delocalization,29 similarly, the ICSSZZ(1) was used as the magnetic criterion to weigh the aromaticity. In this paper, the values of IHOMA, ISA, and ICSSZZ(1) were obtained using the Multiwfn program.28
3. Results and discussion
3.1. Azide tautomers of NAzTA, AAzTA, AzTA
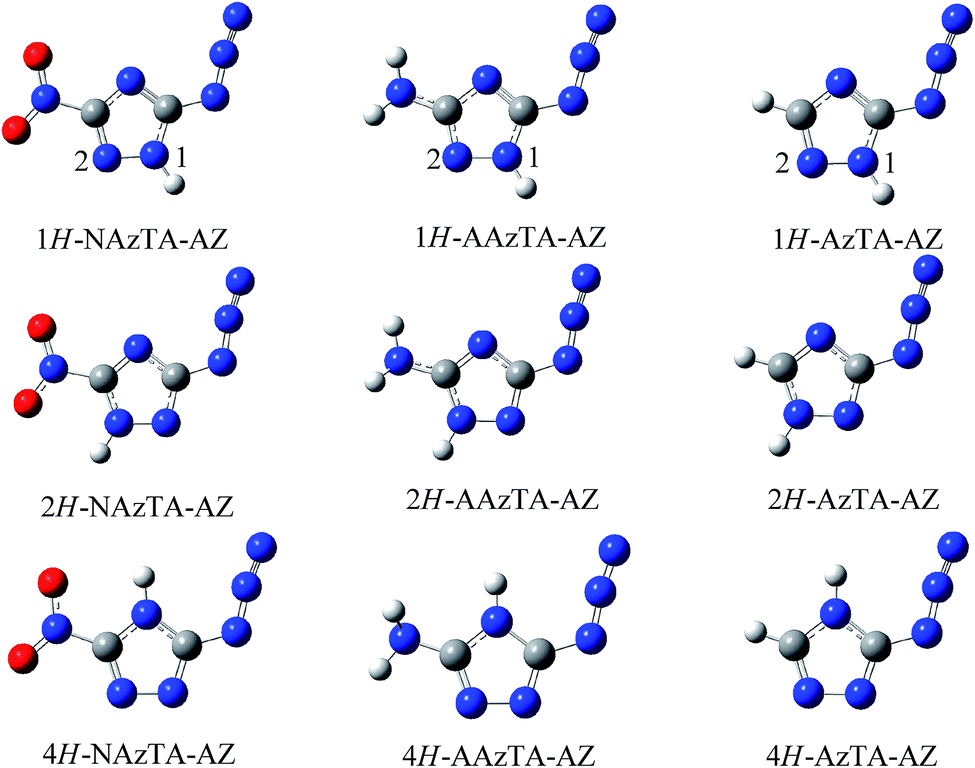
3.1.1. Main existent form in gas phase. The optimized geometries at the B3LYP/6-311++G** level are shown in Fig. 2. The atomic cartesian coordinates of the optimized structures at various levels are presented in the ESI.† –NO2 and –N3 groups are coplanar with the triazole ring plane, while the –NH2 group deviates from the plane. The inside and outside N–N bonds of –N3 in all conformers keep the double and triple characteristics, respectively, as was found in other azido compounds.30,31 The energy differences (ΔE(1) and ΔE(2)) between the tautomers at different levels are listed in Table 1. The ZPE-corrected total energies of different azide tautomers E(1H-AZ), E(2H-AZ), and E(4H-AZ) are listed in Table 1s in the ESI.† It is found that the values of energies of three tautomers depend on the basis sets, while the order is consistent, i.e., E(1H-AZ) < E(2H-AZ) < E(4H-AZ). Generally, the conformer with the lowest energy possesses the highest stability, hence, the relative stability of three tautomers decreases in the order of 1H-AZ > 2H-AZ > 4H-AZ. ΔE(1)s are small under all theoretical levels (less than 6 kJ mol−1), and ΔE(2)s are relatively big and range from 39–60 kJ mol−1. These may indicate that for the title compounds, 1H-tautomer is the main existent form, 2H-tautomer is the next one, while 4H-tautomer rarely exist.
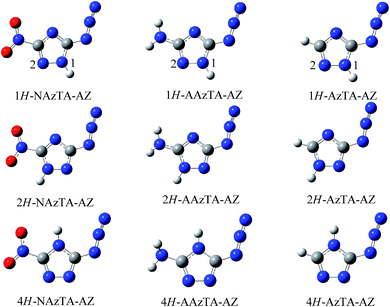 |
| | Fig. 2 Three azide tautomers (AZ) of NAzTA, AAzTA, and AzTA. | |
Table 1 Energy differences (ΔE, kJ mol−1) between tautomers for all title compounds at different levelsa
| |
NAzTA |
AAzTA |
AzTA |
| ΔE(1) = E(2H-AZ) − E(1H-AZ), ΔE(2) = E(4H-AZ) − E(1H-AZ). |
| B3LYP/6-31G* |
ΔE(1) |
1.72 |
3.77 |
2.71 |
| ΔE(2) |
42.66 |
54.63 |
49.43 |
| B3LYP/6-311++G** |
ΔE(1) |
3.30 |
5.63 |
3.23 |
| ΔE(2) |
45.42 |
58.57 |
51.04 |
| B3LYP/AUG-cc-PVDZ |
ΔE(1) |
0.61 |
4.81 |
1.75 |
| ΔE(2) |
39.79 |
55.12 |
46.83 |
3.1.2. Relative abundance of tautomers in gas phase. The relative populations of 1H-, 2H- and 4H-AZs in gas phase were evaluated using the equation: ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnK, where ΔG denotes the difference in the Gibbs free energies of two isomers and K is the equilibrium constant between them. Table 2 collects the obtained abundance values of 1H- and 2H-AZs. The populations of 4H-AZs can be easily obtained from those of 1H- and 2H-AZs. Clearly, for each compound in gas phase, 1H-AZ has the largest molar ratio, 2H-AZ has a slightly lower abundance, and 4H-AZ has a negligible population since the sum of molar ratios of 1H- and 2H-AZs is equal to 1 even at 800 K. The close molar ratios of 1H-AZ and 2H-AZ suggest that both 1H- and 2H-AZs of each title compound can be observed in gas phase, as the above analysis shown. In addition, with the increase in temperature, the population of 1H-AZ increases and that of 2H-AZ decreases, e.g., for NAzTA, the ratio of 1H-AZ increases from 0.519 to 0.567 and that of 2H-AZ decreases from 0.481 to 0.432 from 200 K to 800 K.
lnK, where ΔG denotes the difference in the Gibbs free energies of two isomers and K is the equilibrium constant between them. Table 2 collects the obtained abundance values of 1H- and 2H-AZs. The populations of 4H-AZs can be easily obtained from those of 1H- and 2H-AZs. Clearly, for each compound in gas phase, 1H-AZ has the largest molar ratio, 2H-AZ has a slightly lower abundance, and 4H-AZ has a negligible population since the sum of molar ratios of 1H- and 2H-AZs is equal to 1 even at 800 K. The close molar ratios of 1H-AZ and 2H-AZ suggest that both 1H- and 2H-AZs of each title compound can be observed in gas phase, as the above analysis shown. In addition, with the increase in temperature, the population of 1H-AZ increases and that of 2H-AZ decreases, e.g., for NAzTA, the ratio of 1H-AZ increases from 0.519 to 0.567 and that of 2H-AZ decreases from 0.481 to 0.432 from 200 K to 800 K.
Table 2 Molar ratios of 1H- and 2H-tautomers obtained at the B3LYP/6-311++G** level
| T/K |
NAzTA |
AAzTA |
AzTA |
| 1H-AZ |
2H-AZ |
1H-AZ |
2H-AZ |
1H-AZ |
2H-AZ |
| 200 |
0.519 |
0.481 |
0.509 |
0.491 |
0.532 |
0.468 |
| 298 |
0.533 |
0.467 |
0.527 |
0.474 |
0.557 |
0.443 |
| 400 |
0.545 |
0.455 |
0.546 |
0.454 |
0.576 |
0.424 |
| 500 |
0.553 |
0.447 |
0.563 |
0.437 |
0.589 |
0.411 |
| 600 |
0.559 |
0.441 |
0.577 |
0.423 |
0.598 |
0.402 |
| 700 |
0.564 |
0.436 |
0.588 |
0.412 |
0.606 |
0.394 |
| 800 |
0.567 |
0.432 |
0.598 |
0.402 |
0.611 |
0.388 |
3.1.3. Main existent form in liquid phase. To investigate the solvent effects on the structure and stability of tautomers, the geometries were optimized in five solvents with dielectric constant (ε) of 2.2706 (benzene), 8.9300 (dichloromethane), 20.4930 (acetone), 32.6130 (methanol), and 78.3553 (water) using the CPCM continuum solvent model. There is a basic agreement between the geometries in gas and liquid phases, while the total energies (Es) of the tautomers in liquid phase are quite different from those in gas phase. The energies of three tautomers in liquid phase (Es in Table 2s in the ESI†) decrease in the order of 4H-AZ > 1H-AZ > 2H-AZ, e.g., for ANTA in water, the E of 1H-AZ, 2H-AZ and 4H-AZ are −502.19185, −502.19914 and −502.19103 a.u., respectively. 4H-AZs possess the highest energies, suggesting they have the lowest stability and may barely exist in solution as in gas phase. The 2H-AZs have the lowest energies, indicating that they have the highest stability and largest population, which is different from the situation in gas phase. The findings that 1H-tautomer is the most stable form in gas phase while 2H-tautomer is the most stable one in solutions are similar with that of ANTA.14,32,33 In addition, the energy difference between 1H-AZ and 2H-AZ (ΔE = E(1H-AZ) − E(2H-AZ) in Table 3) increases with the increasing ε, implying that 2H-AZs are more stable in the polar solvents. Taking NAzTA for example, the energy difference between 1H-AZ and 2H-AZ varies from 8.27 (in benzene) to 13.42 kJ mol−1 (in water).
Table 3 Energy differences between 1H- and 2H- azide tautomers (ΔE = E(1H-AZ) − E(2H-AZ), kJ mol−1) predicted at the B3LYP/6-311++G** level for NAzTA, AAzTA, and AzTA in different solutions
| |
Benzene |
Dichloromethane |
Acetone |
Methanol |
Water |
| NAzTA |
8.27 |
12.08 |
12.92 |
13.15 |
13.42 |
| AAzTA |
0.95 |
6.09 |
7.27 |
7.61 |
7.96 |
| AzTA |
0.76 |
3.86 |
4.54 |
4.75 |
4.94 |
3.2. Isomerization reaction
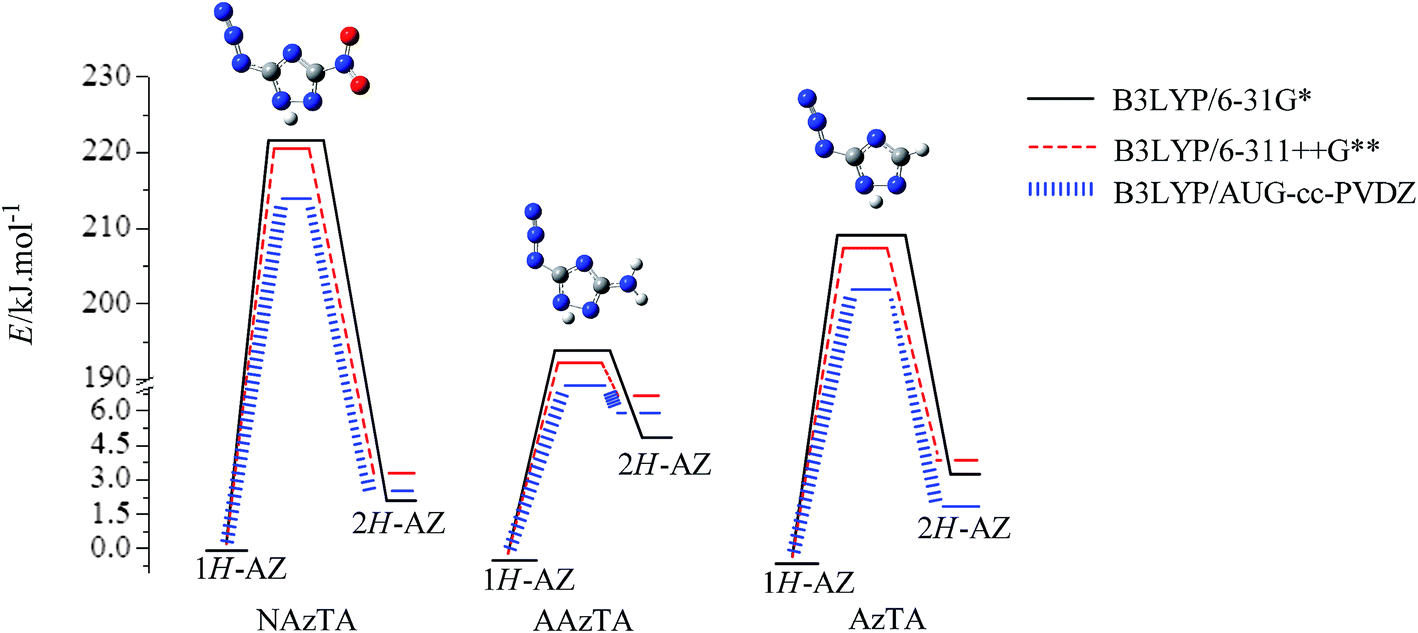
3.2.1. 1H/2H tautomerism. 1H/2H tautomerisms in NAzTA, AAzTA, and AzTA were investigated at the B3LYP/6-31G*, B3LYP/6-311++G**, and B3LYP/AUG-cc-PVDZ levels. 1H-AZ isomerizes to 2H-AZ by a direct 1,2-hydrogen transfer between two adjacent nitrogen atoms (labeled as N1 and N2 in Fig. 2) via a transition state. All three transition states TS(1H/2H-NAzTA-AZ), TS(1H/2H-AAzTA-AZ), and TS(1H/2H-AzTA-AZ) were characterized by a strongly elongated N1–N2 as compared with those in their corresponding 1H-AZ and 2H-AZ conformers. For example, N1–N2 in TS(1H/2H-NAzTA-AZ) is 1.45 Å and those in 1H-AZ and 2H-AZ are both around 1.35 Å for NAzTA. Each TS has a big imaginary frequency corresponding to the transfer of the hydrogen atom to give reactant or product. The transferred H atom is out of the ring plane in each TS. Table 4 presents the calculated activation energy (Ea) and the imaginary frequency (ν) of TSs at different levels. Fig. 3 shows the energy barrier diagram of H transfer processes. The Eas obtained at the B3LYP/6-31G* level are the highest and those obtained at the B3LYP/AUG-cc-PVDZ level are the lowest, suggesting the Eas are basis set dependent. In addition, the substituent groups play important roles in the 1H/2H tautomerization. Comparing the Eas of H transfer processes in NAzTA, AAzTA, and AzTA, it can be clearly observed that –NH2 facilitates the 1H/2H isomerization, while –NO2 gives an opposite effect. Conversion of the 1H-tautomer to 2H-tautomer requires the lowest energy barrier (194.26 kJ mol−1, B3LYP/6-311++G**) for AAzTA (with –N3 and –NH2 groups), while that of NAzTA (with –N3 and –NO2 groups) requires the highest energy barrier (219.24 kJ mol−1, B3LYP/6-311++G**).
Table 4 Activation energy (Ea, kJ mol−1) and imaginary frequency (ν, cm−1) of the transition states of 1H/2H tautomerisms
| |
B3LYP/6-31G* |
B3LYP/6-311++G** |
B3LYP/AUG-cc-PVDZ |
| Ea |
ν |
Ea |
ν |
Ea |
ν |
| TS(1H/2H-NAzTA-AZ) |
222.84 |
−1795.22 |
219.24 |
−1771.34 |
212.96 |
−1755.64 |
| TS(1H/2H-AAzTA-AZ) |
195.63 |
−1751.23 |
194.26 |
−1733.55 |
189.29 |
−1720.50 |
| TS(1H/2H-AzTA-AZ) |
209.69 |
−1757.01 |
207.28 |
−1736.23 |
201.35 |
−1719.12 |
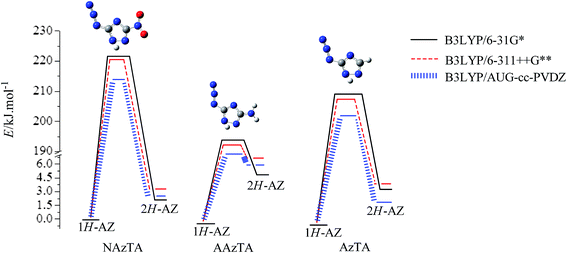 |
| | Fig. 3 Energy diagram for the tautomerization of the 1H- and 2H-tautomers. | |
3.2.2. AZ/TZ isomerization. AZ/TZ isomerizations of 1H- and 2H-tautomers of NAzTA, AAzTA and AzTA (i.e., 1H-NAzTA-AZ/TZ, 2H-NAzTA-AZ/TZ, 1H-AAzTA-AZ/TZ, 2H-AAzTA-AZ/TZ, 1H-AzTA-AZ/TZ, and 2H-AzTA-AZ/TZ) were investigated at three theoretical levels. Eas of the isomerization reactions and imaginary frequencies (νs) of TSs are listed in Table 5. The optimized structures of TSs at the B3LYP/6-311++G** level are presented in Fig. 4. Obviously, in TSs, the azido group bends and tends to form a tetrazole ring: The N3–N4–N5 angles (Fig. 4 for atomic numbering) all bend to be about 124°; The N3–N4 and N4–N5 bonds are all prolonged, for example, in 1H-NAzTA-AZ, the lengths of N3–N4 and N4–N5 are 1.246 and 1.124 Å, respectively, while in TS(1H-NAzTA-AZ/TZ), the corresponding data are 1.334 and 1.204 Å, respectively. Similar with the 1H/2H tautomerism, the predicted Eas of the AZ/TZ isomerizations are also basis set dependent. Values obtained at the B3LYP/6-31G* and B3LYP/AUG-cc-PVDZ levels are close to each other and lower than those obtained at the B3LYP/6-311++G** level. AZ/TZ isomerization is much easier to happen for 1H-AZs than for 2H-AZs, suggesting that the position of H affects the AZ/TZ isomerization. For example, the Eas of 1H-NAzTA-AZ/TZ and 2H-NAzTA-AZ/TZ at the B3LYP/6-311++G** level are 139.19 and 161.86 kJ mol−1, respectively. In addition, for whatever tautomers, 1H-AZ or 2H-AZ, the Ea increases in the order of AzTA < AAzTA < NAzTA, suggesting that the AZ/TZ isomerization is much easier to proceed in AzTA. To further observe how energetic groups affect the AZ/TZ isomerization, the same calculations were performed on 3,5-diazido-1,2,4-triazole (DAzTA). The structures of TSs, i.e., TS(1H-DAzTA-AZ/TZ) and TS(2H-DAzTA-AZ/TZ), and the corresponding Eas are also presented in Fig. 4 and Table 5, respectively. Compared with the Eas of the AZ/TZ isomerization of DAzTA, it can be found that, no matter at what calculation levels and no matter for which tautomer, the AZ/TZ isomerization of AzTA is the easiest to take place.
Table 5 Activation energy (Ea, kJ mol−1) and imaginary frequency (ν, cm−1) of the transition states of AZ/TZ isomerizations
| |
B3LYP/6-31G* |
B3LYP/6-311++G** |
B3LYP/AUG-cc-PVDZ |
| Ea |
ν |
Ea |
ν |
Ea |
ν |
| TS(1H-NAzTA-AZ/TZ) |
124.58 |
−163.49 |
136.19 |
−317.27 |
124.34 |
−298.50 |
| TS(2H-NAzTA-AZ/TZ) |
149.07 |
−369.25 |
161.86 |
−391.44 |
147.76 |
−371.27 |
| TS(1H-AAzTA-AZ/TZ) |
110.21 |
−270.99 |
121.33 |
−292.64 |
112.00 |
−281.26 |
| TS(2H-AAzTA-AZ/TZ) |
136.92 |
−323.41 |
148.87 |
−344.82 |
137.15 |
−329.92 |
| TS(1H-AzTA-AZ/TZ) |
109.66 |
−186.83 |
120.49 |
−307.93 |
110.64 |
−295.18 |
| TS(2H-AzTA-AZ/TZ) |
134.88 |
−346.06 |
146.76 |
−367.71 |
134.56 |
−349.82 |
| TS(1H-DAzTA-AZ/TZ) |
120.25 |
−285.95 |
131.71 |
−310.05 |
121.08 |
−294.08 |
| TS(2H-DAzTA-AZ/TZ) |
145.47 |
−345.77 |
158.08 |
−366.38 |
145.41 |
−350.52 |
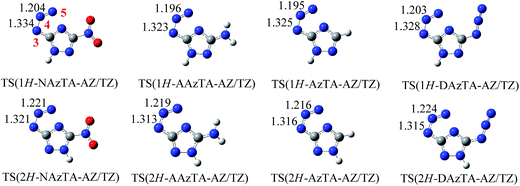 |
| | Fig. 4 Optimized transition states (bond lengths in Å) of the AZ/TZ isomerization for 1H-AZs (top) and 2H-AZs (bottom) at the B3LYP/6-311++G** level. | |
In addition, comparing the Eas of 1H/2H and AZ/TZ isomerization reactions, it seems that for 1,2,4-triazoles containing the –N3 group, the AZ/TZ isomerization is much easier to proceed than the 1H/2H tautomerization.
3.3. Aromaticity
Aromaticity of azide isomers (1H-AZs and 2H-AZs) and tetrazole isomers (1H-TZs and 2H-TZs) were evaluated by three aromaticity indexes, i.e., IHOMA, ISA, and ICSSZZ(1). IHOMA is one of the most popular indexes for measuring aromaticity. IHOMA = 1 stands for an entirely aromatic system, 0.50 < IHOMA < 1 for an aromatic system, 0 < IHOMA< 0.5 for a non-aromatic system, and IHOMA < 0 for an anti-aromatic system. The closer the IHOMA approaches to 1, the higher aromaticity a system has;34 ISA is an index about the electronic charge distribution between atoms in a given ring and has been applied by many studies.35,36 0.003 < ISA < 0.005 is generally the boundary of aromaticity and anti-aromaticity and the ring with a smaller ISA has a higher aromaticity;35 ICSSZZ(1) is a magnetic criterion to weigh the aromaticity. The positive and negative ICSSZZ(1)s suggest aromatic and anti-aromatic characters, respectively, and a bigger positive ICSSZZ(1) corresponds to a higher aromaticity. As a summary, larger IHOMA and ICSSZZ(1) together with smaller ISA imply a larger aromatic compound.
3.3.1. Aromaticity of azide isomers. Table 6 gives IHOMA, ISA, and ICSSZZ(1) of the triazole rings in the 1H-AZs and 2H-AZs. Clearly, for both 1H- and 2H-AZs, IHOMAs are higher than 0.5, ISAs are lower than 0.003, and ICSSZZ(1)s are all positive. In addition, 1H-AZs have higher IHOMA, higher ICSSZZ(1), and lower ISA than their corresponding 2H-AZs. These suggest that both 1H- and 2H-AZs have aromaticity and the order of aromaticity is 1H-AZs > 2H-AZs. That is to say, the 1H-AZs have higher stability than 2H-AZs, same as that obtained from energies. In other words, tautomerism from 1H- to 2H- conformer makes the stability decrease. Moreover, regardless of 1H-AZ or 2H-AZ tautomer, IHOMA, ISA, and ICSSZZ (1) of them all have the order of AzTA > NAzTA > AAzTA, that is, three indexes give the same order of aromaticity, i.e., AzTA > NAzTA > AAzTA.
Table 6 Predicted aromaticity indexes of 1H-AZ and 2H-AZ tautomers
| |
IHOMA |
ISA |
ICSSzz (1)/ppm |
| NAzTA |
1H-AZ |
0.92551 |
0.00098 |
25.22 |
| 2H-AZ |
0.89986 |
0.00124 |
24.88 |
| AAzTA |
1H-AZ |
0.86377 |
0.00118 |
21.34 |
| 2H-AZ |
0.85039 |
0.00137 |
21.10 |
| AzTA |
1H-AZ |
0.94099 |
0.00087 |
27.86 |
| 2H-AZ |
0.92864 |
0.00117 |
26.64 |
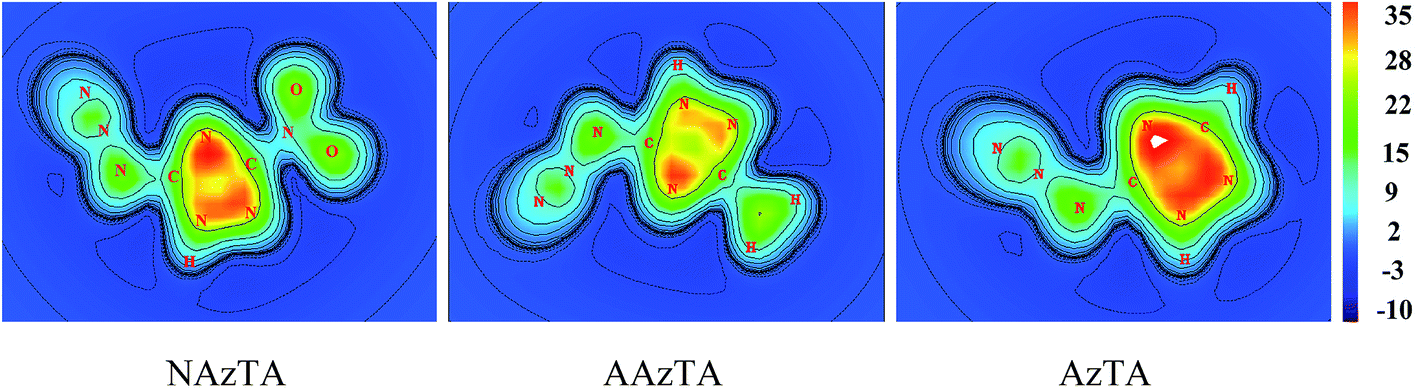
To more intuitively show the aromaticity of NAzTA, AAzTA, and AzTA, Fig. 5 plots the color-filled maps of ICSSZZ of 1H-AZs at 1 Å above the molecular plane. Obviously, the triazole ring center in 1H-tautomer of AzTA is surrounded by the darkest red color, representing the strongest shielding effect and thus the highest aromaticity in this area.
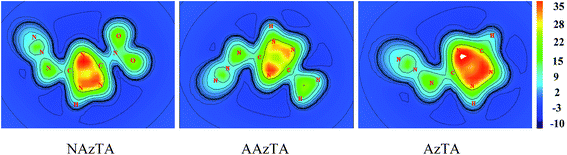 |
| | Fig. 5 Color-filled map of ICSSZZ at 1 Å above the molecular plane of 1H-azide isomers. | |
3.3.2. Aromaticity of tetrazole isomers. The AZ/TZ isomerization leads to the presence of tetrazole isomers. Table 7 lists the IHOMA, ISA, and ICSSZZ (1) of the triazole and tetrazole rings in 1H-TZs and 2H-TZs. As we can see from Table 7, for the triazole ring, all three indexes give the consistent results, i.e., the aromaticity of the triazole ring in 1H-TZs is lower than that in 2H-TZs. The order is contrary to that in azide tautomers, which indicates that the AZ/TZ isomerization largely affect the structural character of the triazole ring. While for the tetrazole ring, indexes do not give a consistent result. Taking the tetrazole ring of NAzTA for example, smaller IHOMA and ICSSZZ(1) correspond to a smaller ISA. That is, the order of aromaticity predicted by ISA is different from that by IHOMA and ICSSZZ(1). In addition, IHOMA suggests that triazole rings have higher aromaticity than the tetrazole rings, while ICSSZZ(1) give the contrary result. That is to say, IHOMA and ICSSZZ(1) can not always give a consistent order of aromaticity for tetrazole isomers, either. Which one is more reliable? Since the order of stability is generally triazole ring > tetrazole ring, the order of aromaticity should be triazole ring > tetrazole ring, so IHOMA is more reliable to weigh the aromaticity of various isomers. Overall, according to the data of IHOMA, no matter in 1H-TZ or 2H-TZ, the triazole ring and tetrazole ring both have good aromaticity and the aromaticity of the triazole ring is better. Comparing the azide isomer and tetrazole isomer, the latter has one more aromatic tetrazole ring, thus it has higher stability than the former. In other words, the isomerization from azide to tetrazole increases the stability of title compounds.
Table 7 Predicted aromaticity indexes of the triazole and tetrazole rings in the 1H- and 2H-tetrazole tautomers
| |
Triazole ring |
Tetrazole ring |
| IHOMA |
ISA |
ICSSZZ (1)/ppm |
IHOMA |
ISA |
ICSSZZ (1)/ppm |
| NAzTA |
1H-TZ |
0.86302 |
0.00331 |
24.59 |
0.77714 |
0.00447 |
31.82 |
| 2H-TZ |
0.92212 |
0.00270 |
26.98 |
0.86272 |
0.00455 |
33.77 |
| AAzTA |
1H-TZ |
0.77235 |
0.00247 |
18.97 |
0.76523 |
0.00394 |
28.62 |
| 2H-TZ |
0.80198 |
0.00115 |
25.65 |
0.79856 |
0.00587 |
29.52 |
| AzTA |
1H-TZ |
0.82162 |
0.00271 |
23.77 |
0.80434 |
0.00383 |
31.66 |
| 2H-TZ |
0.89614 |
0.00162 |
31.72 |
0.83887 |
0.00471 |
33.42 |
4. Conclusions
The structure, aromaticity, and isomerization of NAzTA, AAzTA, and AzTA were investigated using the DFT method. The main conclusions are as follows:
(1) In gas phase, 1H-tautomers have higher stability and populations than their corresponding 2H-tautomers, while the situation is contrary in solution and with the increasing dielectric constants of solvents, the stability of 2H-tautomers increases.
(2) The 1H/2H isomerization happens easiest in AAzTA and hardest in NAzTA. The AZ/TZ isomerization is easiest to proceed in AzTA, and then AAzTA and NAzTA in sequence. In addition, the AZ/TZ isomerization is much easier to take place than the 1H/2H tautomerization.
(3) For the azide isomers, the triazole ring in 1H-tautomer has higher aromaticity than that in 2H-one, which suggests the 1H/2H tautomerization makes the stability decrease. No matter it is 1H or 2H-tautomer, the aromaticity has the order of AzTA > NAzTA > AAzTA. For the tetrazole isomers, both the triazole ring and the tetrazole ring have high aromaticity and the aromaticity of the triazole ring is higher. One more aromatic ring in the tetrazole isomers demonstrates that the AZ/TZ isomerization increases the stability of title compounds.
Acknowledgements
We gratefully thank the National Natural Science Foundation of China (no. 21403110), the Research Fund for Natural Science Foundation of Jiangsu Province (no. BK20130755), and the “Excellent Plan and Zijin Star” Research Foundation of NUST for their support to this work. Yang would like to thank the Innovation Project for Postgraduates in Universities of Jiangsu Province (grant no. KYLX_0346) for financial support.
References
- I. Alkorta, F. Blanco, J. Elguero and R. M. Claramunt, Tetrahedron, 2010, 66, 2863–2868 CrossRef CAS PubMed.
- V. I. Minkin, A. D. Garnovskii, J. Elguero, A. R. Katritzky and O. V. Denisko, Adv. Heterocycl. Chem., 2000, 76, 157–323 CrossRef CAS.
- L. A. Burke, G. Leroy, M. T. Nguyen and M. Sana, J. Am. Chem. Soc., 1978, 100, 3668–3674 CrossRef CAS.
- W. M. Fabian, V. A. Bakulev and C. O. Kappe, J. Org. Chem., 1998, 63, 5801–5805 CrossRef CAS PubMed.
- M. H. V. Huynh, M. A. Hiskey, D. E. Chavez, D. L. Naud and R. D. Gilardi, J. Am. Chem. Soc., 2005, 127, 12537–12543 CrossRef CAS PubMed.
- A. Hammerl, T. M. Klapötke and R. Rocha, Eur. J. Inorg. Chem., 2006, 2006, 2210–2228 CrossRef.
- K.-Y. Lee, C. Storm, M. Hiskey and M. Coburn, J. Energ. Mater., 1991, 9, 415–428 CrossRef CAS.
- A. K. Sikder, M. Geetha, D. B. Sarwade and J. P. Agrawal, J. Hazard. Mater., 2001, A82, 1–12 Search PubMed.
- C. Heinemann, T. Müller, Y. Apeloig and H. Schwarz, J. Am. Chem. Soc., 1996, 118, 2023–2038 CrossRef CAS.
- H. Furuta, H. Maeda and A. Osuka, J. Org. Chem., 2001, 66, 8563–8572 CrossRef CAS PubMed.
- M. Mandado, N. Otero and R. A. Mosquera, Tetrahedron, 2006, 62, 12204–12210 CrossRef CAS PubMed.
- J. Yang, X. Gong and G. Wang, J. Mol. Model., 2014, 20, 2379–2388 CrossRef PubMed.
- V. G. Kiselev, P. B. Cheblakov and N. P. Gritsan, J. Phys. Chem. A, 2011, 115, 1743–1753 CrossRef CAS PubMed.
- D. C. Sorescu, C. M. Bennett and D. L. Thompson, J. Phys. Chem. A, 1998, 102, 10348–10357 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1992, 97, 9173–9177 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–655 CrossRef CAS PubMed.
- A. McLean and G. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS PubMed.
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS PubMed.
- Y. Takano and K. Houk, J. Chem. Theory Comput., 2005, 1, 70–77 CrossRef.
- T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS PubMed.
- G. Schreckenbach and T. Ziegler, J. Phys. Chem., 1995, 99, 606–611 CrossRef CAS.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- C. Corminboeuf, T. Heine, G. Seifert, P. von Ragué Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- T. Heine, P. V. R. Schleyer, C. Corminboeuf, G. Seifert, R. Reviakine and J. Weber, J. Phys. Chem. A, 2003, 107, 6470–6475 CrossRef CAS.
- Ed. T. Lu, http://multiwfn.codeplex.com/, 2013.
- P. von Ragué Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. van Eikema Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef PubMed.
- J. Q. Yang, F. Wang, J. Y. Zhang, G. X. Wang and X. D. Gong, J. Mol. Model., 2013, 19, 5367–5376 CrossRef CAS PubMed.
- J. Q. Yang, H. Yan, X. L. Zhang, G. X. Wang and X. D. Gong, Struct. Chem., 2014, 25, 931–940 CrossRef CAS PubMed.
- K. Lee, R. Gilardi, M. Hiskey and J. Stine, Mater. Res. Soc. Symp. Proc., 1996, 418, 43–48 CrossRef CAS PubMed.
- E. Garcia and K.-Y. Lee, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1682–1683 CrossRef.
- M. Kalinowska, E. Siemieniuk, A. Kostro and W. Lewandowski, J. Mol. Struct.: THEOCHEM, 2006, 761, 129–141 CrossRef CAS PubMed.
- S. Noorizadeh and E. Shakerzadeh, Phys. Chem. Chem. Phys., 2010, 12, 4742–4749 RSC.
- S. Noorizadeh and E. Shakerzadeh, Comput. Theor. Chem., 2011, 964, 141–147 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14560g |
|
| This journal is © The Royal Society of Chemistry 2015 |