
Reshaping anisotropic gold nanoparticles through oxidative etching: the role of the surfactant and nanoparticle surface curvature†
Gang
Lu,  a
a
a
Abstract
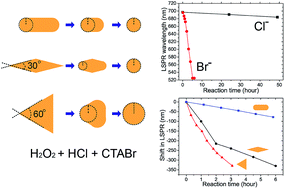
We report an investigation on the effect of stabilization agents and surface curvatures on oxidative etching of three classes of anisotropically shaped gold nanoparticles namely, rods, bipyramids and prisms. In particular, the dual role of the stabilizing agent CTAB in the etching process is explored, showing how it acts both as a source of bromine ions, accelerating etching and as a protection agent, resulting in anisotropic reshaping.
 Please wait while we load your content...
Please wait while we load your content...

