
Growth mechanism of bioglass nanoparticles in polyacrylonitrile-based carbon nanofibers
Abstract
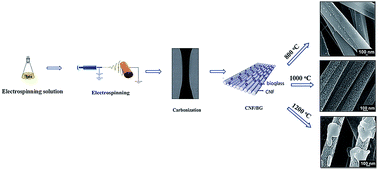
Polyacrylonitrile (PAN) electrospinning in combination with sol–gel method has been a common technique to produce inorganic nanoparticles containing composite carbon nanofibers (CNFs) for diverse applications. To investigate the morphology evolution and crystal transformation of inorganic components along with CNF formation, bioactive glass (BG) containing CNFs (CNF/BG) were prepared by sintering as-spun PAN/precursor composite nanofibers in a nitrogen atmosphere at temperatures of 800, 1000 and 1200 °C. Comprehensive characterizations were performed with TEM, SEM-EDXA and XRD. For samples sintered at 800 °C, numerous BG nanoparticles were observed inside the CNFs and mainly in an amorphous state. With the sintering temperature raised to 1000 °C, a number of spherical BG nanoparticles were detected on the surface of the resulting CNFs, with a crystal structure of wollastonite (β-CaSiO3) polycrystals. When the samples were sintered at 1200 °C, the BG nanoparticles on the surface of CNFs merged into forms with cuboid-like geometry, mainly consisting of pseudowollastonite (Ca3(Si3O9)) single crystals. Based on the geometry evolution and dynamic size distribution function analyses (Ostwald ripening and Smoluchowski equations), it was concluded that the growth of BG nanoparticles conformed to the ripening mechanism at 800 °C and migration–coalescence mechanism at 1200 °C, while the process involved both ripening and migration–coalescence mechanisms at 1000 °C.
 Please wait while we load your content...
Please wait while we load your content...

