
A green process for efficient lignin (biomass) degradation and hydrogen production via water splitting using nanostructured C, N, S-doped ZnO under solar light†
Abstract
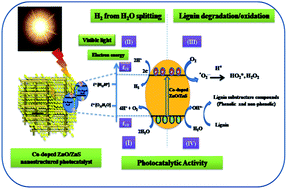
Herein, we have reported the simultaneous water splitting and lignin (biomass) degradation using C, N and S-doped ZnO nanostructured materials. The synthesis of C, N and S-doped ZnO was achieved via calcination of bis-thiourea zinc acetate (BTZA) complex. Calcination of the complex at 500 °C results in the formation of C, N, and S doping in a mixed phase of ZnO/ZnS, whereas calcination at 600 °C gives a single phase of ZnO with N and S-doping, which is confirmed by XRD, XPS and Raman spectroscopy. The band gap of the calcined samples was observed to be in the range of 2.83–3.08 eV. Simultaneous lignin (waste of paper and pulp mills) degradation and hydrogen (H2) production via water splitting under solar light has been investigated, which is hitherto unattempted. The highest degradation of lignin was observed with the sample calcined at 500 °C, i.e., C, N, S-doped ZnO/ZnS when compared to the sample calcined at 600 °C, i.e., N and S doped ZnO. The degradation of lignin confers the formation of a useful fine chemical as a by-product, i.e., 1-phenyl-3-buten-1-ol. However, excellent H2 production, i.e., 580, 584 and 643 μmol h−1 per 0.1 g, was obtained for the sample calcined at 500, 550 and 600 °C, respectively. The photocatalytic activity obtained is considerably higher as compared to earlier reported visible light active oxide and sulfide photocatalysts. The reusability study shows a good stability of the photocatalyst. The prima facie observations show that lignin degradation and water splitting is possible with the same multifunctional photocatalyst without any scarifying agent.
 Please wait while we load your content...
Please wait while we load your content...

