
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriptycene based organosoluble polyamides: synthesis, characterization and study of the effect of chain flexibility on morphology†
Snehasish Mondal and
Neeladri Das*
Department of Chemistry, Indian Institute of Technology Patna, Patna 800 013, Bihar, India. E-mail: neeladri@iitp.ac.in; neeladri2002@yahoo.co.in; Tel: +91 6122552023
First published on 7th November 2014
Abstract
Synthesis of new triptycene-containing polyamides (TPAs) using 2,6-diaminotriptycene and various aromatic and aliphatic dicarboxylates (Yamazaki–Higashi phosphorylation polyamidation) is described. The effect of the polymer backbone flexibility on the surface morphology of the respective polymer is studied. Polyamides thus prepared are organosoluble and have relatively low solution viscosities (0.18–0.5 dL g−1). TGA indicated that triptycene polyamides containing aliphatic chains are thermally less stable than the wholly aromatic triptycene polyamides. These polyamides may be categorized as self-extinguishing materials. FTIR spectroscopy studies show that hydrogen bonding interactions are weaker in wholly aromatic polyamides (TPA1–TPA3) relative to those in semi-aromatic polyamides (TPA4–TPA6). FE-SEM images of TPAs show that replacement of aromatic dicarboxylates with aliphatic dicarboxylates results in a drastic change in the morphology of the polyamides.
Introduction
Polyamides are one of the most important and versatile classes of synthetic polymers known to date.1–7 Monomeric building blocks in polyamides can be either wholly aromatic (also known as aramides, e.g., Kevlar®, Nomex®),5–7 semi-aromatic (a combination of both aromatic and aliphatic monomers, e.g., Torgamide T®, Amodel®)8–10 and purely aliphatic (e.g., Nylon®).7,11,12 Polyamides have attracted research attention because of their high thermal stabilities, excellent mechanical properties and inherent chemical resistance.5,13–16 These properties are often attributed to inter-molecular hydrogen bonds (CO⋯HN) that strengthen inter-chain interactions.17 Applications of polyamides are manifold, which include material for products such as cords, ropes, fiber for garments (for daily use as well as for special use such as fire-fighting/military applications), hoses, and insulating jackets on electrical wires, sports equipment, and sheath for optical fibers. Aramides are also being used as “space age” materials for manufacturing goods that are employed for outer-space explorations.18This justifies importance of polyamides as synthetic polymers and their use as materials for use in advanced technologies. In this context, it must be mentioned that while aramides are thermally more stable than the purely aliphatic polyamides (Nylons), the former suffer from the disadvantage of poor solubility (due to intrinsic macromolecular inflexibility, stronger interchain interactions, or semicrystallinity).13,19–21 This restricts the processability and also limits applications of aramides to some extent. However, in the past few decades, there have been several approaches to improve the low solubility of wholly aromatic polyamides. This includes introduction of bulky groups (in the polymer backbone or as pendant groups in main chain)13,22–33 that disrupt packing and/or incorporation of flexible spacer (usually alkyl units) in the polymer backbone.34–39 These modifications in polymer design decrease inter-molecular hydrogen bonds (CO⋯HN), weaken inter-chain interactions and consequently improve solubility of the resulting polymer.
Triptycene, first synthesized by Bartlett and coworkers in 1942, is the simplest member of iptycene family.40 Triptycene has a rigid three dimensional (3D) structure with a paddle wheel configuration of three benzene rings that are connected together by a [2.2.2]bicyclic bridge. Triptycene based polymers were first reported by Klanderman and Faber in 1968.41 Over the past fifteen years, Swager, Chen and others have extensively explored triptycene chemistry.42–57 More recently, there is lot of excitement in polymer synthesis with the incorporation of triptycene in the polymer backbone or as a bulky pendent group in the main chain.58,59 Swager and coworkers have established that incorporation of triptycene building blocks in polymer chains enhances solubility and imparts high thermal stability. The rigid three dimensional paddle wheel structure of triptycene generates “internal free volume”, which in turn is responsible for the resulting interesting properties associated with polymers containing triptycene derivatives as monomeric units.47
However, to the best of our knowledge, there are only two literature reports of polyamides prepared from triptycene derived monomers. In the first example, Kasashima et al. utilized 1,5-disubstituted triptycene diamine as a monomer,60 while in the second report, Hsiao et al. have employed 1,4-disubstituted triptycene derivatives as polymer building blocks.58 On comparing the aforementioned two sets of triptycene polyamides, it is obvious that the orientation of the triptycene is different in the resulting polymer chains, since the relative position of the two functional groups on triptycene core is different.
It is known that on one hand strong interchain interactions (efficient packing of polymer chains) exist in polyamides, and on the other hand triptycene disrupts packing efficiency of polymer chains. It is reported that polyurethanes (PUs) containing triptycene motif have lesser extent of H-bonding relative to nontriptycene PUs.59 Considering the importance of polyamides in general and triptycene based polymers in particular, we were interested to design triptycene based polyamides. The objective is to study effect of flexibility of the polymer backbone on the morphological properties of the resultant polymer. Continuing our research efforts aimed at exploring newer applications of triptycene derivatives,61,62 herein we describe the syntheses of new series of alternating polyamides (wholly aromatic as well as semi-aromatic) using 2,6-diaminotriptycene as one of the monomeric building blocks, via Yamazaki–Higashi phosphorylation polyamidation reaction.63,64 Polymers were characterized by spectroscopic techniques (NMR and FTIR), gel permeation chromatography (GPC), thermal analysis (TGA and DSC), and powder X-ray diffraction (PXRD) techniques. Hydrogen bonding interactions in these polyamides were examined using FTIR spectroscopy. Field emission scanning electron microscope (FE-SEM) was used to study the changes in surface morphologies of the polyamides due to incorporation of aromatic or aliphatic dicarboxylate motifs in the polymer backbone.
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich or Alfa Aesar. Pyridine was dried in laboratory applying a common technique. Anhydrous NMP was purchased from Sigma-Aldrich.General methods and Instrumentation
NMR spectra were obtained using Bruker Avance II 400 Spectrometer. FTIR spectra were measured using PerkinElmer Spectrum-400 spectrometer. Molecular weight of each polymer was estimated using Agilent PL-GPC 50 integrated GPC spectrometer [using polystyrene standard and DMF (with 0.5% LiBr as additive) as an eluent at a flow rate 1.0 mL min−1 at 45 °C]. Viscosity of each polymer solution in DMF (0.5 g dL−1 concentration at 25 °C) was measured using BrookField DV-II + Pro Viscometer. UV−vis spectra were obtained using Shimadzu UV-2550 UV-vis spectrophotometer. TGA analyses were performed using SDT Q600 (TA Instruments) under nitrogen atmosphere at a scan rate 10 °C min−1. DSC analyses were carried out using DSC8000 (PerkinElmer) under nitrogen at a scan rate 10 °C min−1. PXRD data were obtained using Rigaku TTRAX III X-ray diffractometer. Melting points were measured using SRS EZ-Melt automated melting point apparatus. FE-SEM images were obtained using Hitachi S-4800 Field Emission Scanning Electron Microscope.General synthesis of polymer
In a two neck round bottom flask containing 2,6-diaminotriptycene (142 mg, 0.5 mmol), corresponding dicarboxylic acid (0.5 mmol), CaCl2 (55 mg, 0.5 mmol) and LiCl (22 mg, 0.5 mmol) were added. The round bottom flask was connected with a reflux condenser while maintaining inert atmosphere. Then NMP (1 mL), pyridine (0.25 mL) and triphenyl phosphite (TPP, 0.5 mL, 1.9 mmol) were added to the reaction mixture with a syringe. The reaction mixture was stirred at 110 °C. After 5 h, the viscous solution was cooled to room temperature and poured in methanol (100 mL). A fibrous precipitate was formed upon vigorous stirring. The precipitate was filtered off and washed with hot water and methanol. The residue was dried under vacuum at 100 °C to obtain the desired polyamide (TPA) as a white powder in excellent yield.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (amide I (H-bonded))), 1657 (m; νCO (amide I (non H-bonded))), 3285 (wb; νs (N–H)).O (amide I (H-bonded))), 1657 (vs; νCO (amide I (non H-bonded))), 3286 (wb; νs (N–H)).O (amide I (H-bonded))), 1652 (vs; νCO (amide I (non H-bonded))), 3280 (wb; νs (N–H)).O (amide I (H-bonded))), 1662 (vs; νCO (amide I (non H-bonded))), 2955 (w), 3292 (wb; νs (N–H)).O (amide I (H-bonded))), 1663 (vs; νCO (amide I (non H-bonded))), 2854 (w), 2926 (w), 3302 (wb; νs (N–H)).O (amide I (H-bonded))), 1662 (vs; νCO (amide I (non H-bonded))), 2851 (w), 2924 (w), 3292 (wb; νs (N–H)).
O (amide I (H-bonded))), 1657 (m; νCO (amide I (non H-bonded))), 3285 (wb; νs (N–H)).O (amide I (H-bonded))), 1657 (vs; νCO (amide I (non H-bonded))), 3286 (wb; νs (N–H)).O (amide I (H-bonded))), 1652 (vs; νCO (amide I (non H-bonded))), 3280 (wb; νs (N–H)).O (amide I (H-bonded))), 1662 (vs; νCO (amide I (non H-bonded))), 2955 (w), 3292 (wb; νs (N–H)).O (amide I (H-bonded))), 1663 (vs; νCO (amide I (non H-bonded))), 2854 (w), 2926 (w), 3302 (wb; νs (N–H)).O (amide I (H-bonded))), 1662 (vs; νCO (amide I (non H-bonded))), 2851 (w), 2924 (w), 3292 (wb; νs (N–H)).Results and discussion
Synthesis of monomer
2,6-Dinitrotriptycene (DNT) was prepared from triptycene based on synthetic protocol described by Swager and coworkers.65 Reduction of DNT thus obtained using hydrazine hydrate and Raney Nickel (in MeOH as solvent) resulted in the quantitative formation of the desired monomer 2,6-diaminotriptycene (DAT) (Scheme 1). The 1H NMR spectrum of the product obtained indicated the formation of pure 2,6-diaminotriptycene (ESI†). High purity DAT thus obtained was in turn utilized to prepare triptycene based polyamides described herein. Both DNT and DAT were characterized (NMR and melting point) and the data is in agreement with that reported in literature.65 | ||
| Scheme 1 Synthesis of 2,6-diaminotriptycene (DAT). | ||
Synthesis of polyamides
The Yamazaki–Higashi phosphorylation polyamidation reaction was utilized for synthesis of triptycene based polymers reported herein. This method has several advantages such as facile and efficient synthesis (high yield involving minimal number of steps) of polyamide and diversity in substrate scope. Employing 2,6-diaminotriptycene (DAT) and various aromatic and aliphatic dicarboxylic acids, two sets of polyamides have been synthesized [wholly aromatic polyamides (TPA1–TPA3) and semi-aromatic polyamides (TPA4–TPA6)] (Scheme 2). The reactions were carried out using 1-methyl-2-pyrrolidone (NMP) as solvent. The viscous solutions obtained by stirring each reaction mixture at 110 °C for 5 h were quenched in MeOH with vigorous stirring to yield fibrous precipitates in nearly quantitative yields. The molecular weight of each polyamide was determined by gel permeation chromatography (GPC) using polystyrene as a standard and DMF (containing 0.5% LiBr as additive) as an eluent. The number average molecular weight (Mn) obtained for TPA1–TPA6 was found to vary in the range 24 kDa to 33 kDa. In case of TPA4 and TPA5, the respective GPC traces indicate multimodal nature that may be due to phase separation during polymerization process leading to the formation of oligomeric species as well. The corresponding polydispersity index (PDI) (see ESI†) and degree of polymerization (Pn) varied from 1.19–1.35 and 54–71 respectively (Table 1). Comparison with PDI of previously reported triptycene based polyamides (see ESI†) suggests a relatively narrower molecular weight distribution in case of TPAs. The inherent viscosity of the polymer in DMF (at a concentration of 0.5 g dL−1) was found to vary from 0.18 to 0.51 dL g−1 (Table 1). Comparison of properties of the polyamides reported herein with those reported previously in literature has been summarized in ESI.† TPAs reported herein have relatively lower inherent viscosities than other triptycene based polyamides reported in literature. These low inherent viscosities may be explained in terms of reduction of interchain interaction due to presence of rigid three-dimensional triptycene unit in polymer backbone.65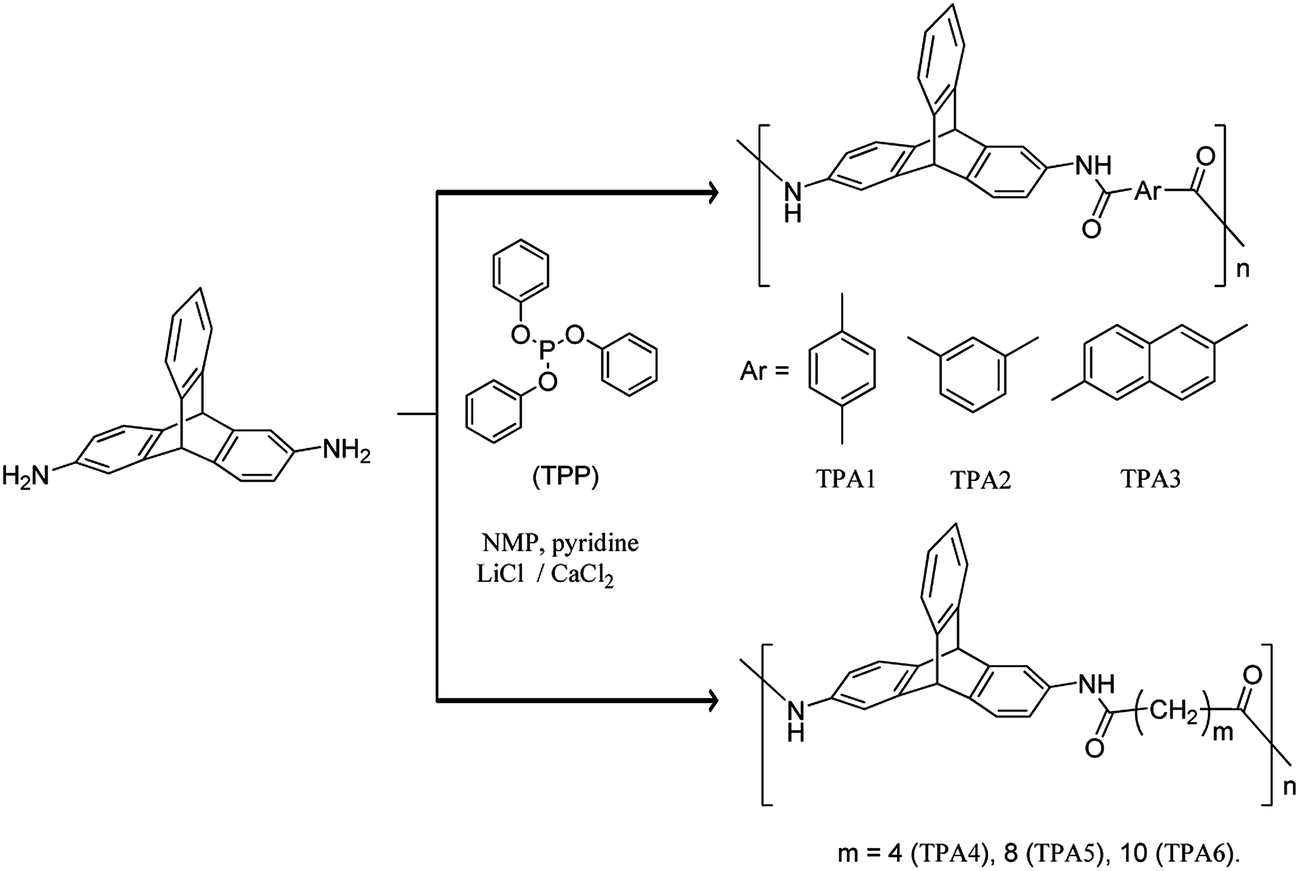 | ||
| Scheme 2 Synthesis of TPA1–TPA6. | ||
| Polymer | Yield (%) | Mna (Da) | PDIa | Pnb | ηinhc (dL g−1) | Tgd (°C) | Td at 10% weight losse (°C) | Char weightf (%) | LOI |
|---|---|---|---|---|---|---|---|---|---|
| a Number average molecular weight (Mn) and polydispersity index (PDI, Mw/Mn) were determined using polystyrene standard and DMF (with 0.5% LiBr as additive) as an eluent.b Degree of polymerization (Pn) determined from Mn.c Measured in DMF at a concentration of 0.5 g dL−1 at 25 °C.d Samples were heated from −50 °C to 300 °C at a scan rate 10 °C min−1 under N2 at a flow rate 20 mL min−1.e Samples were heated from 50 °C to 800 °C at a scan rate 10 °C min−1 under N2 at a flow rate 100 mL min−1, Td were measured at 10% weight loss.f Char weight of polyamides are calculated at 800 °C. | |||||||||
| TPA1 | 98 | 25.3k | 1.23 | 61 | 0.23 | >300 | 428 | 66.65 | 44.10 |
| TPA2 | 97 | 26.6k | 1.19 | 64 | 0.18 | >300 | 502 | 73.58 | 46.93 |
| TPA3 | 99 | 33k | 1.35 | 71 | 0.51 | >300 | 488 | 71.29 | 46.02 |
| TPA4 | 94 | 25.5k | 1.29 | 65 | 0.31 | >300 | 372 | 54.48 | 39.29 |
| TPA5 | 98 | 24k | 1.23 | 54 | 0.18 | >300 | 383 | 40.22 | 33.58 |
| TPA6 | 89 | 26.2k | 1.26 | 55 | 0.29 | >300 | 438 | 48.21 | 36.78 |
Spectroscopic characterization of polymer
The proton NMR spectrum in each case (Fig. 1 and ESI†) suggested polycondensation between 2,6-DAT and the respective dicarboxylic acid. Representative proton NMR spectrum of a wholly aromatic polyamide (TPA3) and a semi-aromatic polyamide (TPA6) are shown in Fig. 1. In the 1H NMR spectrum of TPA3, the singlet at 5.61 ppm is assigned to bridgehead proton of triptycene (Hf). The multiplet in the range δ = 10.30–10.50 ppm corresponds to the amide proton. The signal centered at δ = 7.02 ppm corresponds to the Ha proton of triptycene. Signals due to Hb, Hd and He proton of triptycene appear as multiplet in the range δ = 7.20–7.60 ppm. The peaks due to the protons of naphthalene moiety appear in the range δ = 7.90–8.30 ppm (Hi and Hj) and at δ = 8.60 (Hk). As expected, the integration ratio of all signals in the range δ = 6.90–8.70 ppm and that due to the bridgehead protons of triptycene (Hf) is approximately 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Similarly 1H NMR spectrum of the semi-aromatic polyamide TPA6 is consistent with the expected repeating unit structure. The singlet at δ = 5.40 ppm corresponds to bridge head proton of triptycene (Hf). Five signals in the aromatic region correspond to the five chemically inequivalent peaks of triptycene moiety. Incorporation of the bridging aliphatic dicarboxylate in the polyamide is obvious from the signals in the range δ = 1.20–2.30 ppm. The integration ratio of aromatic protons in the range δ = 6.95–7.79 ppm and aliphatic protons in the range δ = 1.20–2.30 ppm is equal to 1:2 as expected. Insets in Fig. 1 show detailed peak assignments for polyamide TPA6. In general, for all the triptycene polyamides being reported herein, the characteristic bridgehead protons of the triptycene unit appears in the region δ = 5.4–5.6 ppm while the amide protons appear in between δ = 9.70–10.35 (see ESI†) with integration ratio 1:1. Thus proton NMR spectral assignments in case of each polymer agree well the proposed polymer structures.
:1. Similarly 1H NMR spectrum of the semi-aromatic polyamide TPA6 is consistent with the expected repeating unit structure. The singlet at δ = 5.40 ppm corresponds to bridge head proton of triptycene (Hf). Five signals in the aromatic region correspond to the five chemically inequivalent peaks of triptycene moiety. Incorporation of the bridging aliphatic dicarboxylate in the polyamide is obvious from the signals in the range δ = 1.20–2.30 ppm. The integration ratio of aromatic protons in the range δ = 6.95–7.79 ppm and aliphatic protons in the range δ = 1.20–2.30 ppm is equal to 1:2 as expected. Insets in Fig. 1 show detailed peak assignments for polyamide TPA6. In general, for all the triptycene polyamides being reported herein, the characteristic bridgehead protons of the triptycene unit appears in the region δ = 5.4–5.6 ppm while the amide protons appear in between δ = 9.70–10.35 (see ESI†) with integration ratio 1:1. Thus proton NMR spectral assignments in case of each polymer agree well the proposed polymer structures.
 | ||
| Fig. 1 1H NMR spectra of TPA3 (Top) and TPA6 (bottom) recorded at ambient temperature in DMSO-d6 solvent. Insets: expansions of aromatic and aliphatic region of TPA6. | ||
FTIR spectroscopy was also used to confirm structural features of the polyamides. FTIR spectrum of TPA3 and TPA6 are shown as representative examples of a wholly aromatic polyamide and a semi-aromatic polyamide respectively in Fig. 2. FTIR spectrum of TPA3 has a broad band at 3280 cm−1 (νN—Hsym. str.) and a strong band at 1652 cm−1 [νCO stretching (amide I) of amide group]. Similarly in case of TPA6, the bands at 3292 cm−1 and 1662 cm−1 are assigned to νN—H symmetrical stretching and νCO stretching vibration of amide group respectively. In general, absence of symmetrical stretching band at 3400–3500 cm−1 (characteristics peak of primary amine) in the FTIR spectra of all polyamides reported herein confirm polycondensation reaction. FTIR spectra of other polyamides (TPA1, TPA2, TPA4 and TPA5) also exhibit strong peak for νCO stretching and broad band for νN—H symmetrical stretching in the range 1657–1663 cm−1 and 3285–3302 cm−1 respectively.
 | ||
| Fig. 2 FTIR spectra of TPA3 (left) and TPA6 (right) recorded at ambient temperature. | ||
Properties of polyamides
Previously, it has been shown that triptycene based polyamides are soluble in polar solvents (such as NMP, HMPA, DMAc, DMF, DMSO, m-cresol, THF, cyclohexanone or pyridine) at room temperature or upon heating. In the present study, polyamides (TPA1–TPA6) are no exception, since they have excellent solubilities in highly polar aprotic solvent such as NMP, DMAc, DMF, and DMSO at room temperature. However unlike the previously reported polyamides containing triptycene motifs, polymers (TPA1–TPA6) are also readily soluble in moderately polar solvents such as Pyridine at room temperature (see ESI†). The presence of bulky triptycene units in the polymer backbone inhibits efficient packing of the polymer chains and decreases interchain interactions, thereby contributing to improved solubilities.Thermal properties
Thermal properties of the polyamides were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA has been performed under a nitrogen atmosphere (heating rate: 10 °C min−1). Data obtained from these studies are tabulated in Table 1. TGA analysis curves and Td values clearly indicate that triptycene polyamides containing aliphatic chains are thermally less stable than the wholly aromatic triptycene polyamides (Fig. 3). This is also evident from the char yields of the polymers at 800 °C. In the case of wholly aromatic triptycene polyamides, the char yield is greater than 66%, while in case of the polyamides with flexible alkyl chains, the char yields are relatively low. This suggests that incorporation of flexible aliphatic chains in the polymer backbone results in deterioration in their thermal stability. Wholly aromatic TPAs reported herein have comparable thermal stability with other previously reported triptycene polyamides. | ||
| Fig. 3 TGA traces of TPA1–TPA6 under N2 at a scan rate 10 °C min−1. | ||
Based on the van Krevelen and Hoftyzer equation (eqn (1)),66,67 limiting oxygen index (LOI) of a given polymer can be calculated from its respective char yield.
| LOI = 17.5 + 0.4CR | (1) |
LOI values are often used to gauge flammability of polymers or their tendency to sustain flame. Materials with LOI value greater than 28 are classified as “self-extinguishing”.68 In case of the triptycene polyamides reported herein, LOI values are greater than 44 for wholly aromatic polymers (TPA1–3), where as for the semiaromatic ones (TPA4–6), LOI values are greater than 33. Thus, based on LOI values, it can be concluded that wholly aromatic polyamides are better fire-retardants than the semiaromatic analogues, albeit both class of triptycene polyamides may be categorized as “self-extinguishing” polymers.
The resulting polymers have high solubility (in common organic solvents) and high thermal stability. These properties are usually observed in case of cardo-polymers (i.e. polymer containing cardo69 groups such as xanthenes70). Although TPAs reported herein don't contain cardo-groups (lateral ring connected to the main macromolecular backbone by a quaternary carbon atom), these polymers show cardo specific properties such as have organosolubility and high thermal stability. These cardo-specific properties are attributed to the presence of rigid triptycene motifs.
From DSC analysis of TPAs, no glass transition temperature (Tg) was observed upto 300 °C (ESI†).
X-ray diffraction of the polymers
The wide angle X-ray diffraction (WAXD) patterns for polyamides (TPA1–TPA6) are shown in Fig. 4. Few sharp reflexes are observed in WAXD pattern only in case of TPA6 (% crystallinity = 2.78). In general, the broad spectra of TPAs suggest amorphous nature of these polyamides. This is due to the triptycene units in polymer backbone that reduce the extent of close packing and hence loss of crystallinity. | ||
| Fig. 4 WAXD pattern of the TPA1–TPA6 recorded at a scan rate 3° min−1 at ambient temperature. | ||
Optical properties
UV-vis spectra of the polyamides have been recorded in DMF (Fig. 5). TPAs containing aliphatic chain (TPA4–TPA6) show absorption band having maxima at 258 nm with a shoulder near 296 nm. The differences in length of the aliphatic chains do not have any marked change in the nature of the absorption spectra of these polyamides. The peaks are assigned to π–π* transitions in the aromatic rings of triptycene. In case of wholly aromatic TPAs (TPA1–TPA3), the highest intensity absorption bands have maxima beyond 280 nm. In case of TPA3, the value of λmax is highest among all triptycene polyamides reported herein, and this is attributed to the n–π* transitions in the carbonyl groups present in conjugation with the naphthalene ring present in the polymer backbone. | ||
| Fig. 5 Normalized UV-vis spectra of polyamides (TPA1–TPA6) in DMF at 298 K. | ||
Hydrogen bonding behavior of TPAs
Study of hydrogen bonding is important in case of polyamides as these interactions affect the thermal and mechanical properties of a given polymer. In general, strength of polyamides is often attributed to high degree of hydrogen bonding (interchain as well as intrachain). It is expected that presence of large number of bulky triptycene motifs in polymer backbone will restrict close packing of polymer chains and consequently interchain interactions via hydrogen bonding interactions will be minimized.FTIR spectroscopy can be conveniently used as a tool to study the extent of hydrogen bonding interactions in these triptycene based polyamides which arise due to interactions between the oxygen atom of the carbonyl (H-bond acceptor) and the N–H (H-bond donor) groups. It is expected that hydrogen bonded carbonyl will exhibit a CO stretching vibration band at a lower wavenumber than that due to the non-hydrogen bonded carbonyl group. This is due to the reduced electron density in the carbonyl group and consequential bond weakening upon its involvement in hydrogen bonding.59 In the FTIR spectra of the triptycene based polyamides reported herein, the stretching vibration bands of the carbonyl groups have split into two peaks. In case of each polyamide (TPA1–TPA6), the lower wavenumber band corresponds to hydrogen bonded carbonyl groups and the higher wavenumber band is attributed to non-hydrogen bonded carbonyl groups. This clearly indicates that in general, the hydrogen bonds are disrupted to a considerable extent by the presence of bulky triptycene unit. By comparing the relative intensity of the two aforementioned peaks, the strength of the hydrogen bonding interactions can be predicted.
As shown in Fig. 6, in case of wholly aromatic polyamides (TPA1–TPA3), wherein triptycene motifs are bridged by rigid aromatic spacers, the relative intensity of the hydrogen bonded carbonyl peak is less than that in case of the semi-aromatic polyamides (TPA4–TPA6), wherein triptycene motifs are bridged by aliphatic spacers. This suggests that in addition to the bulky triptycene units, the relatively rigid aromatic bridges also contribute to packing inefficiency, which is clearly reflected by the presence of a relatively less intense hydrogen bonded carbonyl peak.
 | ||
| Fig. 6 FTIR spectra in the carbonyl region of TPA1–TPA6 recorded at 298 K. | ||
The presence of more flexible aliphatic chains (in case of TPA4–TPA6) is a probable reason for a higher degree of hydrogen bonding interaction in semi-aromatic polymers in comparison to their wholly aromatic counterparts. The conclusion, that TPA4–TPA6 exhibit higher degree of hydrogen bonding interactions, is also supported by observing the nature of the N–H peak in these polymers. It is known that the narrower νN—H stretching vibration peak, correspond to a higher degree of hydrogen bonding.59 As shown in Fig. 7, triptycene based polyamides with bridging aromatic moieties show a relatively broader νN—H stretching vibration peak in comparison to the polyamides (TPA4–TPA6) with more flexible aliphatic hydrocarbon chains.
 | ||
| Fig. 7 FTIR spectra in the N–H region of TPA1–TPA6 recorded at 298 K. | ||
Surface morphology characterization
Morphology of powder polyamide samples were studied by field emission scanning electron microscopy (FE-SEM) analysis. In each case, a droplet of the suspended polyamide in hexane (0.1 wt%) was placed on a carbon tape and carefully dried under vacuum prior to FE-SEM analysis. The dried sample was then coated with platinum on an ion sputter coater and the images were taken at various levels of magnifications. Scanning electron micrographs depicting the general morphological features for TPAs are shown in Fig. 8. The Field emission SEM (FE-SEM) images, in case of wholly aromatic polyamides (TPA1–TPA3), indicate the presence of globular particles with diameter ranging from 50 to 550 nm. TPA1 and TPA2 have similar morphological features and this is due to the similar bridging aromatic group. In TPA3, the naphthyl group replaces the phenyl groups present in TPA1 and TPA2. This probably results in greater aggregation of the globular particles in TPA3. To calculate the particle size distribution of polyamides TPA1–TPA3, five different areas were randomly selected at higher magnification and dimension of each particle measured. The particle size distribution was subsequently plotted in the form of histograms. In each case, the particle size distribution (from SEM images) fitted well with a Gaussian curve as shown in Fig. 9. The average particle size was obtained from the fitted data.71,72 The average particle size thus determined in case of polyamides TPA1 and TPA2 are 185 nm and 166 nm respectively. In case of TPA3, the average particle size was relatively higher (213 nm), and this may be attribute to greater extent of aggregation in TPA3 when compared with polyamides TPA1 and TPA2. | ||
| Fig. 8 FE-SEM micrograph of TPA1–TPA6 at two different magnifications. | ||
 | ||
| Fig. 9 Particle size distribution histogram of TPA1–TPA3 (left to right), fitted with a Gaussian curve. | ||
Further from SEM images, it can be concluded that replacement of rigid aromatic bridging groups by relatively more flexible aliphatic chains results in considerable change in the surface morphologies. In case of semi-aromatic triptycene polyamides (TPA4–TPA6), SEM images revealed more aggregation as evident from plate-like morphologies.73,74 FTIR spectroscopy results clearly indicate that the degree of hydrogen bonding is greater in the semi-aromatic polyamides in comparison to the wholly aromatic triptycene polyamides. This greater extent of hydrogen bonding as well as the presence of favorable hydrophobic interactions between the aliphatic chains attribute to the observed change of surface morphologies in TPAs containing aliphatic bridges, thus leading to higher aggregation.
From FTIR and FE-SEM analyses, it is clearly observed that the interchain interactions of triptycene based polyamides vary extensively depending on the flexibility of polymer backbone as well as nature of the spacer. This parameter also significantly affects the surface morphology of the resulting polymer. In general, with increasing flexibility of the spacer, there is a considerable enhancement in the interchain interactions (as evident from FTIR) which leads to more aggregated plate like morphologies (as evident from FE-SEM). On the other hand for more rigid structure, there is an inhibition in interchain interactions which leads to the formation of less aggregated globular particles. This structure–property relationship may be employed to obtain different morphological characteristics as per requirement.
Conclusions
In conclusion, we report two series of triptycene based polyamides (TPAs) that were prepared utilizing DAT and various aromatic or aliphatic dicarboxylates via Yamazaki–Higashi phosphorylation polyamidation reaction in high yields (≥89%). In general, incorporation of rigid triptycene motifs in polyamide chains render the TPAs organosoluble (even in less polar solvents such as pyridine) and non-crystalline in nature. Irrespective of the bridging dicarboxylates used, solution viscosities studies of TPAs (0.5 g dL−1 polymer concentration in DMF) show extremely low viscosities (0.18–0.51 dL g−1). The polymers exhibit molecular weights (Mn) in the range between 25 and 33 kDa, as evident from GPC analysis. The wholly aromatic TPAs (TPA1–TPA3) exhibit higher Td (10% weight loss temperature under nitrogen) in comparison to the semi-aromatic TPAs (TPA4–TPA6). Wholly aromatic polyamides (TPA1–TPA3) exhibit significantly less hydrogen bonding than semi-aromatic polyamides (TPA4–TPA6) bearing flexible aliphatic segment. This is reflected in the morphological differences as observed in SEM images. The low inherent viscosities and organosoluble nature of the TPAs render facile solution processability of these thermally stable polymers. From an application viewpoint, based on the char yield and the corresponding LOI values, polyamides reported herein are “self-extinguishing” in nature and hence can be considered to as fire-retardant materials. In summary, TPAs reported herein can be considered as promising candidates for processable high temperature applications.Acknowledgements
N.D. thanks the CSIR, Govt. of India, New Delhi [CSIR No. 02(0126)/13/EMR-II] and the Indian Institute of Technology Patna, for financial support. S.M. thanks UGC, New Delhi for a Research Fellowship. The authors also acknowledge SAIF-Panjab University for providing analytical facilities.Notes and references
- M. Yang, K. Cao, L. Sui, Y. Qi, J. Zhu, A. Waas, E. M. Arruda, J. Kieffer, M. D. Thouless and N. A. Kotov, ACS Nano, 2011, 9, 6945–6954 CrossRef PubMed.
- K. Marchildon, Macromol. React. Eng., 2011, 5, 22–54 CrossRef CAS.
- M. Shadpour and D. J. Mohammad, J. Macromol. Sci., Part A: Pure Appl.Chem., 2011, 48, 644–679 CrossRef.
- S. Banerjee and S. Maji, in High Performance Polymers and Engineering Plastics, ed. Vikas M., Scrivener publishing, Salem, 2011, pp. 111–166 Search PubMed.
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef PubMed.
- M. Scholl, Z. Kadlecova and H.-A. Klok, Prog. Polym. Sci., 2009, 34, 24–61 CrossRef CAS PubMed.
- M. Trigo-López, P. Estévez, N. San-José, A. Gómez-Valdemoro, F. C. García, F. Serna, J. L. de la Peña and J. M. García, Recent Pat. Mater. Sci., 2009, 2, 190–208 CrossRef.
- S. Zulfiqar, M. Ishaq and M. I. Sarwar, Adv. Polym. Technol., 2010, 29, 300–308 CrossRef CAS.
- H. R. Kricheldorf, S. Böhme and G. Schwarz, Macromolecules, 2001, 34, 8879–8885 CrossRef CAS.
- K. Ishihara, S. Ohara and H. Yamamoto, Macromolecules, 2000, 33, 3511–3513 CrossRef CAS.
- V. Rajendran and M. J. Nanjan, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 829–838 CrossRef CAS.
- S. Chidambaram and M. J. Nanjan, Makromol. Chem., 1983, 184, 2225–2230 CrossRef CAS.
- D.-J. Liaw, F.-C. Chang, M.-K. Leung, M.-Y. Chou and K. Muellen, Macromolecules, 2005, 38, 4024–4029 CrossRef CAS.
- M. Arpin and C. Strazielle, Polymer, 1977, 18, 591–598 CrossRef CAS.
- P. E. Cassidy, in Thermally Stable Polymers, Marcel Dekker, New York, 1980, ch. 4 Search PubMed.
- H. H. Yang, in Aromatic High-Strength Fibres, John Wiley, New York, 1989, pp. 66–289 Search PubMed.
- S.-R. Sheng, X.-L. Pei, Z.-Z. Huang, X.-L. Liu and C.-S. Song, Eur. Polym. J., 2009, 45, 230–236 CrossRef CAS PubMed.
- E. Charles and J. Carraher, Introduction to Polymer Chemistry, Taylor & Francis, New York, 3rd edn, 2012 Search PubMed.
- D.-J. Liaw, B.-Y. Liaw, J.-R. Chen and C.-M. Yang, Macromolecules, 1999, 32, 6860–6863 CrossRef CAS.
- H. R. Kricheldorf and B. Schmidt, Macromolecules, 1992, 25, 5471–5476 CrossRef CAS.
- H. R. Kricheldorf and R. J. Burger, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 355–362 CrossRef CAS.
- D.-J. Liaw, in Macromolecular Nanostructured Materials, ed. Ueyama N. and Harada A., Kodansha & Springer, Tokyo, 2004, Ch. 2.2, pp. 80–100 Search PubMed.
- Y.-C. Huang, K.-L. Wang, C.-H. Chang, Y.-A. Liao, D.-J. Liaw, K.-R. Lee and J.-Y. Lai, Macromolecules, 2013, 46, 7443–7450 CrossRef CAS and reference therein.
- M.-D. Damaceanu, R.-D. Rusu, A. Nicolescu, M. Bruma and A. L. Rusanov, Polym. Int., 2011, 60, 1248–1258 CrossRef CAS.
- D.-J. Liaw, B.-Y. Liaw and C.-M. Yang, Macromolecules, 1999, 32, 7248–7250 CrossRef CAS.
- D.-J. Liaw and B.-Y. Liaw, Macromol. Chem. Phys., 1999, 200, 1326–1332 CrossRef CAS.
- D.-J. Liaw, B.-Y. Liaw and C. Y. Chung, Acta Polym., 1999, 50, 135–140 CrossRef CAS.
- D.-J. Liaw and B.-Y. Liaw, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1069–1074 CrossRef CAS.
- D.-J. Liaw and B.-Y. Liaw, Macromol. Symp., 1997, 122, 343–348 CrossRef CAS.
- I. K. Spiliopoulos, J. A. Mikroyannidis and G. M. Tsivgoulis, Macromolecules, 1998, 31, 522–529 CrossRef CAS.
- I. K. Spiliopoulos and J. A. Mikroyannidis, Macromolecules, 1998, 31, 1236–1245 CrossRef CAS.
- I. K. Spiliopoulos and J. A. Mikroyannidis, Macromolecules, 1996, 29, 5313–5319 CrossRef CAS.
- A. E. Lozano, J. de Abajo, J. G. de la Campa and J. Preston, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1987–1994 CrossRef CAS.
- D.-J. Liaw, B.-Y. Liaw, J.-R. Chen and C.-M. Yang, Macromolecules, 1999, 32, 6860–6863 CrossRef CAS.
- M. Yamashita, M. A. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1513–1518 CrossRef CAS.
- M. Lucas, P. Brock and J. L. Hedrich, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2179–2185 CrossRef CAS.
- M. R. Bellomo, G. D. Pasquale, A. L. Rosa, A. Pollicino and G. Siracusa, Polymer, 1996, 37, 2877–2881 CrossRef CAS.
- D.-J. Liaw, B.-Y. Liaw and M. Y. Tsai, Polym. J., 1997, 29, 474–477 CrossRef CAS.
- D.-J. Liaw and B.-Y. Liaw, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1075–1080 CrossRef CAS.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef CAS.
- B. H. Klanderman and J. W. H. Faber, J. Polym. Sci., Polym. Chem. Ed., 1968, 6, 2955–2965 CrossRef CAS.
- C.-F. Chen and Y.-X. Ma, Iptycene Chemistry: from Synthesis to Applications, Springer-Verlag, Berlin, 2013 Search PubMed.
- P.-F. Li and C.-F. Chen, J. Org. Chem., 2012, 77, 9250–9259 CrossRef CAS PubMed.
- Y. Jiang and C.-F. Chen, Eur. J. Org. Chem., 2011, 32, 6377–6403 CrossRef.
- C.-F. Chen, Chem. Commun., 2011, 47, 1674–1688 RSC.
- J. H. Chong and M. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301–3315 RSC.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef CAS PubMed.
- L. Zhao, Z. Li and T. Wirth, Chem. Lett., 2010, 39, 658–667 CrossRef CAS.
- S. A. Sydlik, P. A. Delgado, S. Inomata, B. VanVeller, Y. Yang, T. M. Swager and K. B. Wagener, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1695–1706 CrossRef CAS.
- B. VanVeller, D. Robinson and T. M. Swager, Angew. Chem., Int. Ed., 2012, 51, 1182–1186 CrossRef CAS PubMed.
- B. VanVeller, D. J. Schipper and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 7282–7285 CrossRef CAS PubMed.
- C. Simocko, Y. Yang, T. M. Swager and K. B. Wagener, ACS Macro Lett., 2013, 2, 1061–1064 CrossRef CAS.
- C. Zhang, Y. Liu, B. Li, B. Tan, C.-F. Chen, H.-B. Xu and X.-L. Yang, ACS Macro Lett., 2012, 1, 190–193 CrossRef CAS.
- Z. Chen and T. M. Swager, Macromolecules, 2008, 41, 6880–6885 CrossRef CAS.
- A. D. Regno, A. Gonciaruk, L. Leay, M. Carta, M. Croad, R. Malpass-Evans, N. B. McKeown and F. R. Siperstein, Ind. Eng. Chem. Res., 2013, 52, 16939–16950 CrossRef.
- T.-Y. Zhou, F. Lin, Z.-T. Li and X. Zhao, Macromolecules, 2013, 46, 7745–7752 CrossRef CAS.
- X. Zhu, C.-L. Do-Thanh, C. R. Murdock, K. M. Nelson, C. Tian, S. Brown, S. M. Mahurin, D. M. Jenkins, J. Hu, B. Zhao, H. Liu and S. Dai, ACS Macro Lett., 2013, 2, 660–663 CrossRef CAS.
- S.-H. Hsiao, H.-M. Wang, J.-S. Chou, W. Guo and T.-H. Tsai, J. Polym. Res., 2012, 19, 9902–9909 CrossRef.
- Z. Chang, M. Zhang, A. G. Hudson, E. B. Orler, R. B. Moore, G. L. Wilkes and S. R. Turner, Polymer, 2013, 54, 6910–6917 CrossRef CAS PubMed.
- Y. Kasashima, T. Kaneda, G. Saito, F. Akutsu, K. Naruchi and M. Miura, Macromol. Chem. Phys., 1994, 195, 2693–2697 CrossRef CAS.
- S. Chakraborty, S. Mondal, Q. Li and N. Das, Tetrahedron Lett., 2013, 54, 1681–1685 CrossRef CAS PubMed.
- S. Mondal, S. Chakraborty, S. Bhowmick and N. Das, Macromolecules, 2013, 46, 6824–6831 CrossRef CAS.
- J. Zhao, H. Xu, J. Fang and J. Yin, J. Appl. Polym. Sci., 2012, 126, 244–252 CrossRef CAS.
- N. Yamazaki, M. Matsumoto and F. Higashi, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1373–1380 CrossRef CAS.
- S. A. Sydlik, Z. Chen and T. M. Swager, Macromolecules, 2011, 44, 976–980 CrossRef CAS and references therein.
- D. W. Van Krevelen and P. J. Hoftyzer, Properties of polymers, Elsevier, Amsterdam, 3rd edn, 1976 Search PubMed.
- Z. Rafiee and L. Golriz, Polym. Eng. Sci., 2014, 54, 2252–2257 CAS.
- A. R. Horrocks, M. Tune and D. Price, Text. Prog., 1988, 18(1–3), 1–186 CrossRef.
- V. V. Korshak, S. V. Vinogradova and Y. S. Vygodski, J. Macromol. Sci., Rev. Macromol. Chem., 1974, 11, 45–142 CrossRef CAS.
- S.-R. Sheng, C.-X. Ma, J.-W. Jiang, Z.-Z. Huang and C.-S. Song, J. Appl. Polym. Sci., 2010, 116, 1650–1659 CAS.
- N. Suzuki, M. B. Zakaria, Y.-D. Chiang, K. C.-W. Wu and Y. Yamauchi, Phys. Chem. Chem. Phys., 2012, 14, 7427–7432 RSC.
- N. Baccile, D. Grosso and C. Sanchez, J. Mater. Chem., 2003, 13, 3011–3016 RSC.
- M. Ago, T. Endo and K. Okajima, Polym. J., 2007, 39, 435–441 CrossRef CAS.
- C. V. Rajaram, S. D. Hudson and L. C. Chien, Polymer, 1998, 39, 5315–5319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10476e |
| This journal is © The Royal Society of Chemistry 2014 |