
Effect of surface chemistry on the double layer capacitance of polypyrrole-derived ordered mesoporous carbon
Abstract
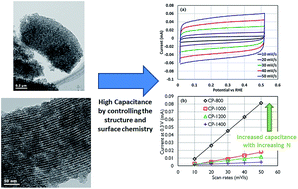
In this work, the effect of nitrogen on the double layer (DL) capacitance of nitrogen-doped ordered mesoporous carbon (NOMC) is studied. The nitrogen content of the NOMCs was controlled thermally. X-ray photoelectron spectroscopy shows decreasing nitrogen content with increasing heat treatment temperature. Despite the differing N content of the NOMCs, the BET surface area, pore size and distribution of the NOMCs did not change significantly with the heat treatment, though Raman spectroscopy and X-ray diffraction showed that the microcrystallinity was affected, exposing more of the basal planes with increased heat treatment temperature. The DL capacitance of the NOMCs was measured from cyclic voltammograms in a range that minimized the contribution of the space charge capacitance and pseudocapacitance, yielding information about the impact of oxygen and nitrogen functional groups on the pure Helmholtz behavior of NOMCs. It was found that nitrogen affects the DL capacitance by changing both the electronic properties and microstructure of the carbon. These enhancements resulted in NOMCs with very high areal Helmholtz capacitance of 25.1 μF cm−2.
- This article is part of the themed collection: Polymers for Electrochemical Energy Storage
 Please wait while we load your content...
Please wait while we load your content...

