
Lectin recognizing thermoresponsive double hydrophilic glycopolymer micelles by RAFT polymerization
Kan Suna,
S. W. Annie Blighb,
Hua-li Niea,
Jing Quan*a and
Li-min Zhu*a
aCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, 201620, P.R. China. E-mail: lzhu@dhu.edu.cn; Tel: +86 21 67792748
bDepartment of Life Sciences, Faculty of Science and Technology, University of Westminster, 115 New Cavendish Street, London W1W 6UW, UK
First published on 1st August 2014
Abstract
Thermoresponsive double hydrophilic block glycopolymer poly(N-isopropylacrylamide-co-6-O-vinyladipoyl-D-glucose)-b-poly(N-isopropylacrylamide) (P(NIPAm-co-OVAG)-b-PNIPAm) was prepared by a combination of enzymatic synthesis and reversible addition-fragment chain transfer (RAFT) polymerization protocols using P(NIPAm-co-OVAG) as a macro-RAFT agent. All the precisely synthesized glycopolymers were characterized by nuclear magnetic resonance spectroscopy and gel permeation chromatography. Detections using laser light scattering and transmission electron microscopy revealed that the block glycopolymer was able to self-assemble into micelles with various sizes and sphere morphologies in aqueous solutions. The glucose pendants in the glycopolymers had obvious interaction with lectin, concanavalin A (Con A), and their capacity of interaction with Con A was controlled by the number of glucose units in the glycopolymers. The block glycopolymer micelles have excellent biocompatibility with pig iliac endothelial cells using the MTT assay, however the glycopolymer–Con A micelles could be used to induce apoptosis in human hepatoma SMMC-7721 cells.
Introduction
Functional double hydrophilic block copolymers (DHBCs) containing at least one functional unit such as a temperature-responsive, light-sensitive, and pH-sensitive monomer have recently attracted significant interest for potential applications in drug delivery systems,1–4 switchable catalysts,5 and gene delivery vectors.6 Thermoresponsive DHBCs are one of the most important candidates of biomaterials in DHBCs and have capability of reversible micellization and dissociation responding to the change of temperature. When the temperature is above the lower critical solution temperature (LCST) of the thermoresponsive copolymer, the DHBCs self-assemble into micelles. Inversely, the formed micelles dissociate into aqueous solution when the temperature is below the LCST. Many studies have been focused on the connection of thermoresponsive DHBC with other functional monomer such as sugar-functional monomer, which are deemed to be promising for protein binding.7,8The glycopolymers that contain sugar pendant groups have attracted chemists to investigate for their biomedical applications and protein recognition.9–11 They have been used to mimic biological systems such as binding lectins through multipoint interactions.12,13 As a lectin, concanavalin A (Con A) shows high specificity for α-D-glucose and α-D-mannose, and can induce cancer cell death.14–17 The protein recognition is closely related to the sugar moiety of the glycopolymer, for example, a glycopolymer containing galactose can recognize peanut agglutinin (PNA).44
The architecture of glycopolymer plays a crucial role in its interaction with proteins.18,19 The precisely synthesized glycopolymers with great potential for use as novel biomaterials can be produced by controlled polymerization technique.18–20 RAFT polymerization as one of the most useful and amenable polymerization techniques can easily generate polymeric systems and control structures for a wide-variety of polymers with defined end and pendant functionalities, controlled molecular weights, narrow polydispersities and low toxicity using mild conditions.21–35 Miura et al. reported the advantage of RAFT polymerization was that it provided precise glycopolymer characteristics such as molecular weights with narrow polydispersities and random saccharide spacing.20
In this study we aimed to obtain a series of well-defined thermoresponsive double hydrophilic block glycopolymers which could be used to recognize protein in a controlled fashion and are not toxic. Thermoresponsive glycopolymers were fabricated through polymerization of a sugar-functional monomer 6-O-vinyladipoyl-D-glucose (OVAG) and a temperature sensitive monomer NIPAm via controlled RAFT polymerization using S-1-dodecyl-S′- (α,α′- dimethyl-α′′-acetic acid) trithiocarbonate (DDATC) as a chain transfer agent (CTA) and 2,2′-azo-bis-iso-butyronitrile (AIBN) as an initiator. The synthesis and characterization of well-defined thermoresponsive block glycopolymers poly(NIPAm-co-OVAG)-b-poly(NIPAm) by RAFT polymerization using poly(NIPAm-co-OVAG) as a macroagent are presented. The binding ability of these glycopolymers to Con A, their thermoresponsiveness, cell biocompatibility in free and bound lectin forms with pig idiliac endothelial (PIE) cells and human hepatoma SMMC-7721 cells are also reported.
Experimental
Materials
N-Isopropylacrylamide (NIPAm) (98%, Tokyo Chemical Co. Ltd.) was purified by recrystallization in n-hexane. Alkaline protease from Bacillus subtilis (EC 3.4.21.14, a crude preparation of alkaline serine protease, powder, 100 U per mg) was purchased from the Wuxi Xue Mei Technological Co. Ltd. (Wuxi, PR China). Dimethyl formamide (DMF) was purchased from the Sinopharm Chemical Reagent Co. (Shanghai, PR China). 2,2′-Azo-bis-iso-butyronitrile (AIBN; Sinopharm Chemical Reagent Co., Ltd., Shanghai, PR China, 97%) was purified by recrystallization from a 95% water solution. Adipic acid was purchased from the Sinopharm Chemical Reagent Co. (Shanghai, PR China). S-1-dodecyl-S′- (α, α′- dimethyl-α′′-acetic acid) trithiocarbonate (DDATC) was purchased from Sigma-Aldrich (Shanghai) Trading Co., Ltd. 2-[4-(2-Hydroxyethyl)-1-piperazinyl] ethanesulfonic acid (HEPES) was procured from the Nanjing Robiot Co. (Nanjing, China). Concanavalin A (Con A) was obtained from Sigma-Aldrich (Shanghai, China). Fluorescently-labeled Con A (FITC-Con A) was purchased from Vector Laboratories (Burlingame, CA, USA). Dimethyl sulfoxide (DMSO) was purchased from the China National Medicines Corporation Ltd. (Beijing, China). Dulbecco's modified Eagle medium (DMEM), penicillin–streptomycin and trypsin–EDTA solutions were obtained from Gibco (Tulsa, OK, USA). All solvents used in this work were of analytical grade and were dried by storing over activated 4 Å molecular sieves for 24 h prior to use. All other reagents were used as received. Water was distilled before use.Syntheses
Analytical Techniques
Lectin-binding assay
Cell viability study
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and incubated for 24 h. The glycopolymers in PBS solutions were added into the cells (final concentrations were 1, 5, 10, 25, 50, 100 mg L−1). After incubation for another 24 h, 20 μL of MTT solution (5 mg mL−1 in PBS buffer) was added to each well. Four hours later, the medium was removed and the samples in the wells were air dried. 200 μL of DMSO was added to dissolve the formed crystals. The optical density of the solution was measured at 570 nm using a microplate reader (Thermo Multiskan MK3, Thermo Scientific Company, Waltham, UK). The PIE cells without any treatment were used as the control.
000 cells per well and incubated for 24 h. The glycopolymers in PBS solutions were added into the cells (final concentrations were 1, 5, 10, 25, 50, 100 mg L−1). After incubation for another 24 h, 20 μL of MTT solution (5 mg mL−1 in PBS buffer) was added to each well. Four hours later, the medium was removed and the samples in the wells were air dried. 200 μL of DMSO was added to dissolve the formed crystals. The optical density of the solution was measured at 570 nm using a microplate reader (Thermo Multiskan MK3, Thermo Scientific Company, Waltham, UK). The PIE cells without any treatment were used as the control.Further experiments of the cytotoxicity of glycopolymers binding Con A were performed in which a series of solutions with 1 g L−1 of P(NIPAm-co-OVAG)-b-PNIPAm-1 and different concentration of Con A (0, 100, 300, 500, 1000 mg L−1) in HEPES buffer were initially incubated for 4 h. SMMC-7721 cells were seeded into 96-well plates at a density of 20000 cells per well and incubated for 24 h. The glycopolymers binding Con A in HEPES buffer were added to the cells (final concentrations of Con A were 0, 10, 30, 50, 100 mg L−1 and glycopolymer was 100 mg L−1 in every well). They were then assayed as above. The SMMC-7721 cells without any treatment were used as the control.
Results and discussion
Polymer synthesis
Thermoresponsive block glycopolymers P(NIPAm-co-OVAG)-b-PNIPAm with glucose was facilely synthesized by combining enzymatic synthesis with RAFT polymerization (Scheme 1) with good yields (Table 1). Diblock glycopolymers P(NIPAm-co-OVAG)-b-PNIPAm were prepared by RAFT polymerization of NIPAm in DMF at 70 °C using P(NIPAm-co-OVAG) as a marco-RAFT agent, which was prepared by RAFT polymerization of NIPAm and OVAG with different feed molar ratios (Table 1). In the polymerization reaction, S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (DDATC) was used as a RAFT agent because of its ability to control the reaction and its low toxicity property.40 The molar ratio of monomers to DDATC was fixed at 100:1. The conditions of the polymerization were listed in Table 1.
 | ||
| Scheme 1 (a) The synthesis of 6-O-vinyladipoyl-D-glucopyranose (OVAG) by controllable chemoenzymatic reaction; (b) the synthesis of P(NIPAm-co-OVAG)-b-PNIPAm by RAFT polymerization. | ||
| Experiment | Polymera | [NIPAm]0/[OVAG]0/[CTA]0/[AIBN]0b,c | Yieldd (%) |
|---|---|---|---|
| a PNO and PNO-b-PN were respectively shorted for P(NIPAm-co-OVAG) and P(NIPAm-co-OVAG)-b-PNIPAm.b [NIPAm]0, [OVAG]0, [AIBN]0 and [CTA]0 represent the initial concentrations of monomers, initiator and chain transfer agents (CTA), respectively.c In the synthesis of P(NIPAm-co-OVAG), CTA was S-1-dodecyl-S′-(α,α′- dimethyl-α′′-acetic acid)trithiocarbonate (DDATC), and P(NIPAm-co-OVAG) was macroCTA in the polymerization of P(NIPAm-co-OVAG)-b-PNIPAm.d Yield was determined by gravimetric analysis. | |||
| 1 | PNO-1 | 80/20/1/0.05 | 63 |
| 2 | PNO-2 | 90/10/1/0.05 | 67 |
| 3 | PNO-3 | 93/7/1/0.05 | 75 |
| 4 | PNO-b-PN-1 | 300/0/1/0.25 | 71 |
| 5 | PNO-b-PN-2 | 300/0/1/0.25 | 76 |
| 6 | PNO-b-PN-3 | 300/0/1/0.25 | 80 |
The1H NMR spectra of glycopolymers are shown in Fig. 1a and b, which reveal the presence of NIPAm and glucose moieties and the absence of vinyl groups. 1H NMR of NIPAm (D2O, 400 MHz) δ (ppm): 6.02 (qd, 2H, 1-H), 5.56 (dd, 1H, 2-H), 3.82 (m, 1H, 4-H), 1.00 (d, 6H, 5, 5′-H). 1H NMR of OVAG (D2O, 400 MHz) δ (ppm): 7.15 (dd, 1H, 13-H), 5.25 (d, 0.46H, 1-Hα), 4.85 (dd, 1H, 14-H trans), 4.65 (dd, 1H, 14-H cis), 4.55 (d, 0.54H, 1-Hβ), 4.25 (m, 2H, 6-H), 3.85 (m, 0.46H, 5-H), 3.60 (m, 3-Hα), 3.55 (m, 5-Hβ), 3.40 (m, 2-Hβ), 3.35 (m, 3-Hβ), 3.30 (m, 4-H), 3.15 (m, 0.52H, 2-Hα), 2.35 (m, 4H), 1.55(m, 4H). In the 1H NMR spectrum of P(NIPAm-co-OVAG)-b-PNIPAm-1, the signals at δ 7.15 (dd, 1H, 13-H) and 4.85 (dd, 1H, 14-H trans), 4.65 (dd, 1H, 14-H cis) of OVAG and 6.02 (qd, 2H), 5.56 (dd, 1H) of NIPAm have almost disappeared demonstrating polymerization has proceeded. In addition, the molar ratio of NIPAm and OVAG units in the resultant polymers was calculated based on the integral values of the signals at δ = 2.38–2.22 (2H of OVAG) and δ = 1.25–0.95 (6H of NIPAm) and the results are presented in Table 2. Meanwhile, when compared with P(NIPAm-co-OVAG)-1, the characteristic signals of NIPAm are enhanced because of the additional PNIPAm block. The calculated monomer ratio are basically consistent with the feed of the monomer ratio.
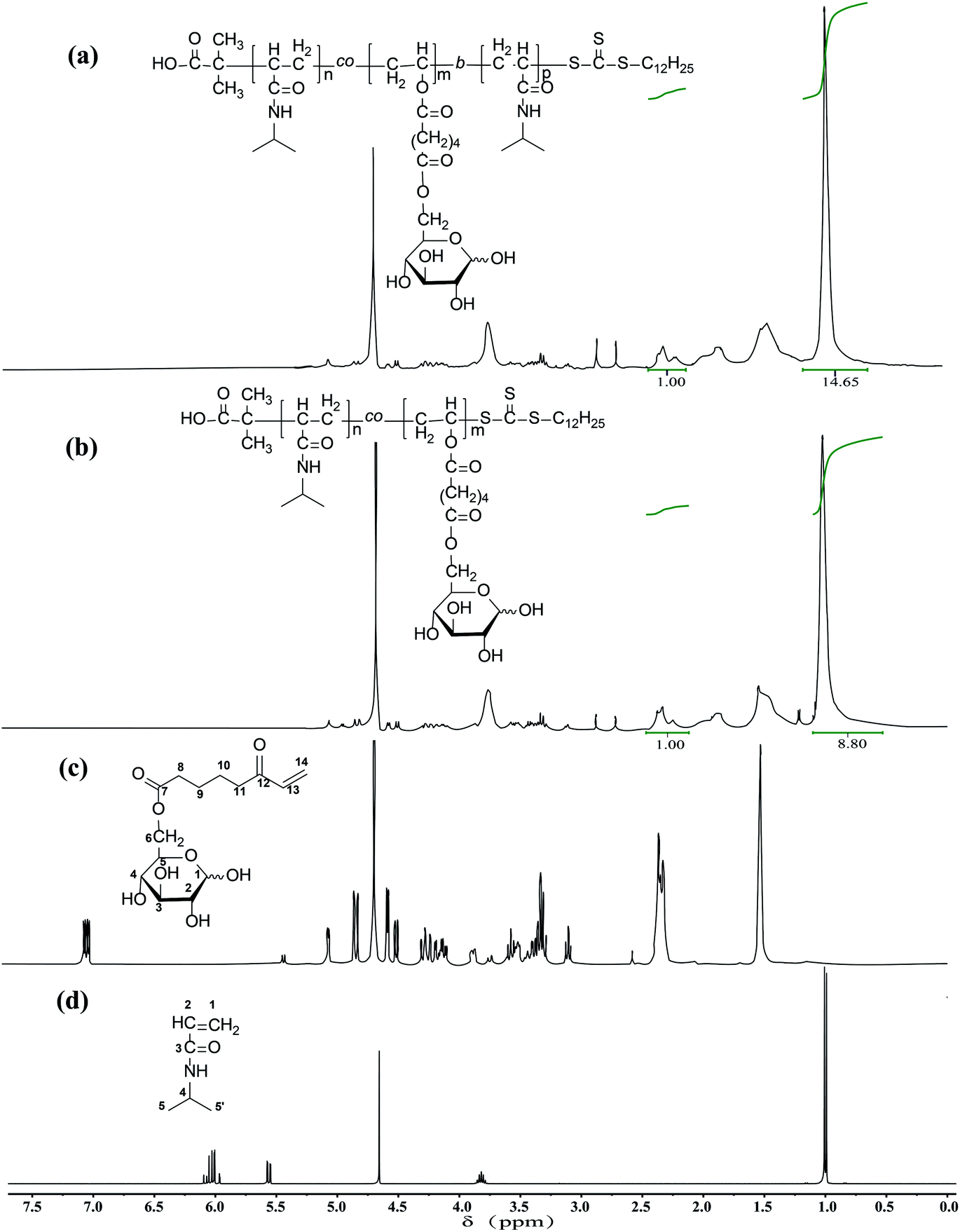 | ||
| Fig. 1 1H NMR spectra of (a) P(NIPAm-co-OVAG)-b-PNIPAm; (b) P(NIPAm-co-OVAG); (c) OVAG; (d) NIPAm in D2O. | ||
| Polymera | Mnb (× 104) | Mwb (× 104) | PDIb | [NIPAm]/[OVAG]c | LCSTd (°C) |
|---|---|---|---|---|---|
| a PNO and PNO-b-PN were respectively shorted for P(NIPAm-co-OVAG) and P(NIPAm-co-OVAG)-b-PNIPAm.b Mn, Mw and PDI were determined by GPC.c [NIPAm]/[OVAG] represented the molar ratio of NIPAm and OVAG in the resultant glycopolymers and was determined by 1H NMR.d LCST was determined by UV-vis spectroscopy. | |||||
| PNO-1 | 4.75 | 6.00 | 1.26 | 5.9 | 38.0 |
| PNO-2 | 4.55 | 5.49 | 1.20 | 9.1 | 37.0 |
| PNO-3 | 5.36 | 6.72 | 1.25 | 15.6 | 36.0 |
| PNO-b-PN-1 | 7.42 | 9.17 | 1.23 | 9.7 | 36.5 |
| PNO-b-PN-2 | 7.26 | 8.41 | 1.16 | 13.8 | 35.5 |
| PNO-b-PN-3 | 7.79 | 9.27 | 1.19 | 20.3 | 33.0 |
The molecular weights (Mn and Mw) and molecular weight distributions (Mw/Mn) of glycopolymers measured by GPC in THF are presented in Table 2. Compared to the macro-RAFT agents, all the diblock glycopolymers eluted at higher molecular weights and a clear shift can be observed. Fig. 2 shows the Mn and Mw of P(NIPAm-co-OVAG)-b-PNIPAm-1 is 7.42 × 104 and 9.17 × 104, Mw/Mn of P(NIPAm-co-OVAG)-b-PNIPAm-1 is 1.23. All the GPC traces of glycopolymers are symmetrical and unimodal, and the molecular weight distributions are relatively narrow. Because of the polydisperse of polymer, the values on Mn and Mw (and thus, also of Mw/Mn) are the values of samples with statistical means, which cannot be considered as the real ones. Compared with the results by GPC, the Mn,NMR (molecular weight determined by 1H NMR) is similar to the Mn,GPC, which is shown in Table 2. Meanwhile, in order to reduce the error caused by the structural difference between synthesized samples and standard sample, universal calibration curve was used. All data including Mn, Mw and Mw/Mn were the average numbers of three replicates.
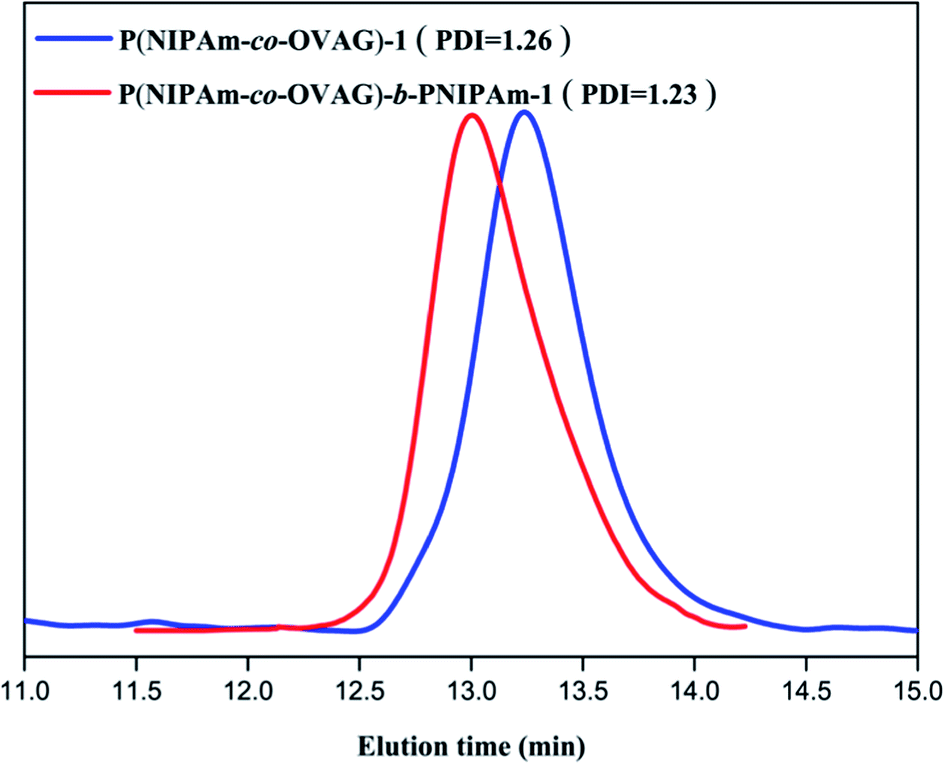 | ||
| Fig. 2 GPC traces of P(NIPAm-co-OVAG)-b-PNIPAm-1(red curve) and P(NIPAm-co-OVAG)-1 (blue curve) in THF. | ||
LCST determination of glycopolymers
It has been reported that the LCST value of PNIPAm-based copolymer can be precisely designed to be in the range 32–40 °C by adjusting the relative amount of monomers, and LCST value increases with the molar fraction of hydrophilic monomers in the copolymers.37 Fig. 3 shows the absorbance depends on the temperature of the glycopolymers micellar solution. The LCST of diblock glycopolymer P(NIPAm-co-OVAG)-b-PNIPAm-1 at 1.0 mg mL−1 was 36.5 °C (Fig. 3). Overall, the LCST values of glycopolymers increase with increasing molar fraction of OVAG in the glycopolymers (Table 2).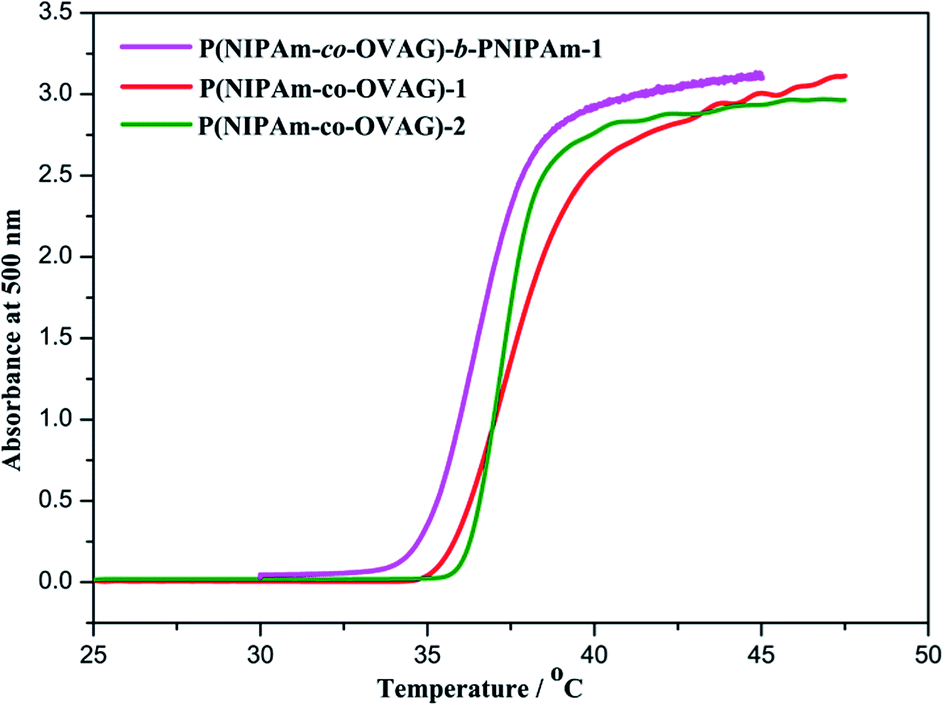 | ||
| Fig. 3 Turbidity curves recorded for 1.0 mg mL−1 solution of glycopolymers in water on UV-vis spectroscopy at 500 nm and the heating rate was 0.5 °C min−1. | ||
Meanwhile, it can be found that phase transition of P(NIPAm-co-OVAG)-1 lies in the range of 35–41 °C which is the widest in the glycopolymers studied. It is due to the increase of hydrogen bonds between hydroxyl groups of glucose and water which retard the process of phase transition with the increasing the molar ratio of OVAG. However, it also can be found that the range of phase transition of P(NIPAm-co-OVAG)-b-PNIPAm-1 is 35–38 °C and it is narrower than that of P(NIPAm-co-OVAG)-1. The reason for this change is the increasing proportion of PNIPAm and the formation of a block, which has also been observed in other similar studies.42,43
Self-assembled micellar behavior
From the plot of the relative intensity ratio (I3/I1) versus glycopolymer concentration (Fig. 4), an abrupt increase in the relative intensity ratio (I3/I1) was observed with increasing glycopolymer concentrations, indicating formation of micelles and the transfer of pyrene into the hydrophobic core of the micelles. The CMC values for P(NIPAm-co-OVAG)-b-PNIPAm-1 and P(NIPAm-co-OVAG)-1 were as low as 0.31 and 0.19 g L−1, respectively (Fig. 4), providing evidence for an apparent stability of micelles, which is consistent with the literature.39
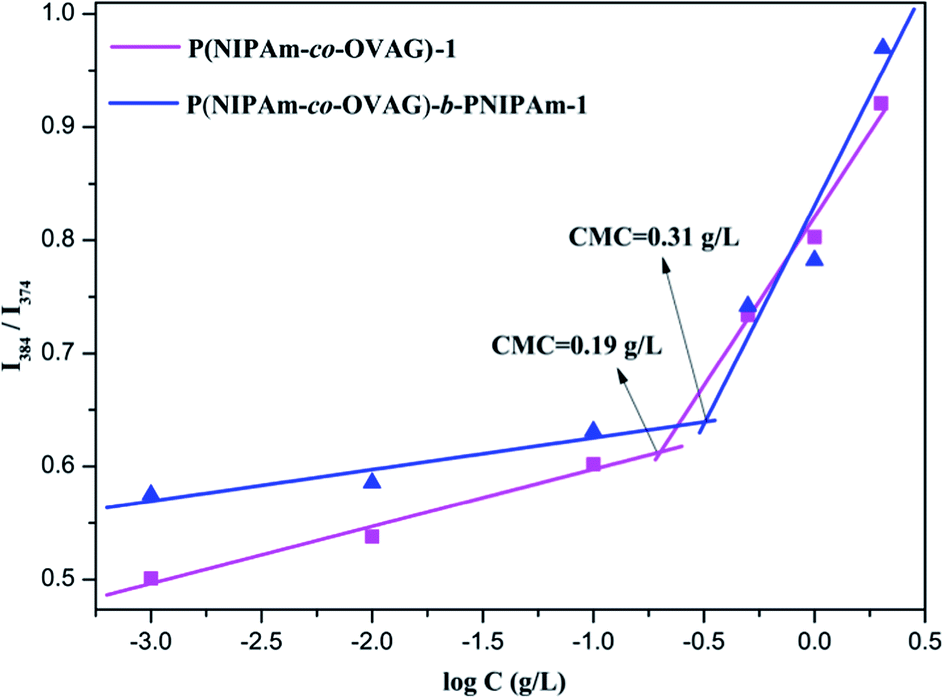 | ||
| Fig. 4 The critical micellar concentration (CMC) graphs of P(NIPAm-co-OVAG)-b-PNIPAm-1 (blue graph) and P(NIPAm-co-OVAG)-1 (purple graph). A series of concentrations are 2, 1, 0.5, 0.1, 0.01 and 0.001 mg mL−1. The CMC values of P(NIPAm-co-OVAG)-b-PNIPAm-1 and P(NIPAm-co-OVAG)-1 in aqueous media are about 0.19 g L−1 and 0.31 g L−1, respectively. | ||
Fig. 5(a) shows the temperature dependence of Rh and Rg for aqueous solution of P(NIPAm-co-OVAG)-b-PNIPAm-1. On heating, both Rh and Rg hardly changed between 25 °C and 31 °C. Obviously, for the glycopolymer, when the temperature is lower than the LCST, the glycopolymer is molecularly soluble, Rh is typically less than 35 nm and Rg is less than 65 nm. However, at elevated temperature to 33–35 °C, Rh and Rg increased dramatically. This suggests the occurrence of the coil-to-globule transition of the glycopolymer and aggregation of globose nanoparticles is due to the increasing specific surface area. At even higher temperatures, Rh and Rg maintain almost constant.
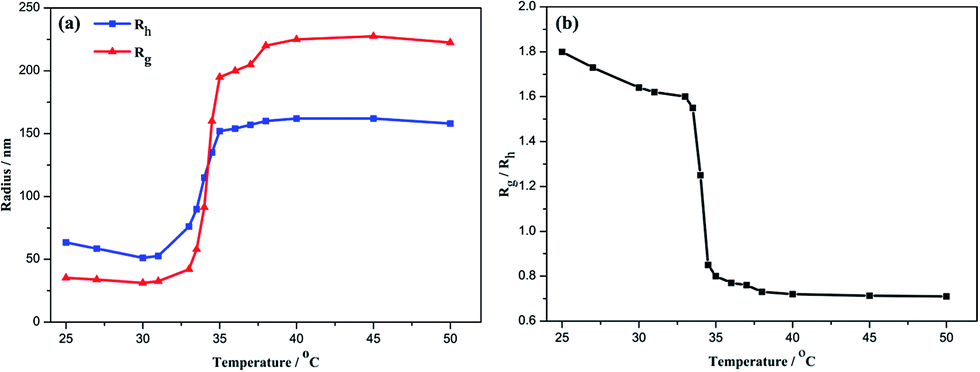 | ||
| Fig. 5 (a) The temperature dependences of average hydrodynamic radius (Rh, blue square), and average gyration radius (Rg, red circle) of P(NIPAm-co-OVAG)-b-PNIPAm-1 micelles in water at a concentration of 8 × 10−4 g mL−1. (b) The temperature dependence of Rg/Rh for P(NIPAm-co-OVAG)-b-PNIPAm-1 micelles in water. | ||
In order to understand in detail the process of the chain conformation changes, the temperature dependence of Rg/Rh were also studied (Fig. 5(b)). Rg/Rh is nearly a constant (∼1.6) in the heating process when temperature is lower than the LCST. Water is a good solvent for the block polymer P(NIPAm-co-OVAG)-b-PNIPAm-1, and glycopolymer is linear flexible chain coils in water. In the transition range of the heating process, Rg/Rh decreases sharply from 1.6 to 0.7 and remains ∼0.7 when the temperature is higher than 35 °C. This indicates that PNIPAm block and PNIPAm chain in the block polymer are induced from the extended coil conformation to the collapsed compact globule.
 | ||
| Fig. 6 TEM images of micelles self-assembled from P(NIPAm-co-OVAG)-2 (a and b) and P(NIPAm-co-OVAG)-b-PNIPAm-1 (c and d) at low (a and c) and high (b and d) magnification. | ||
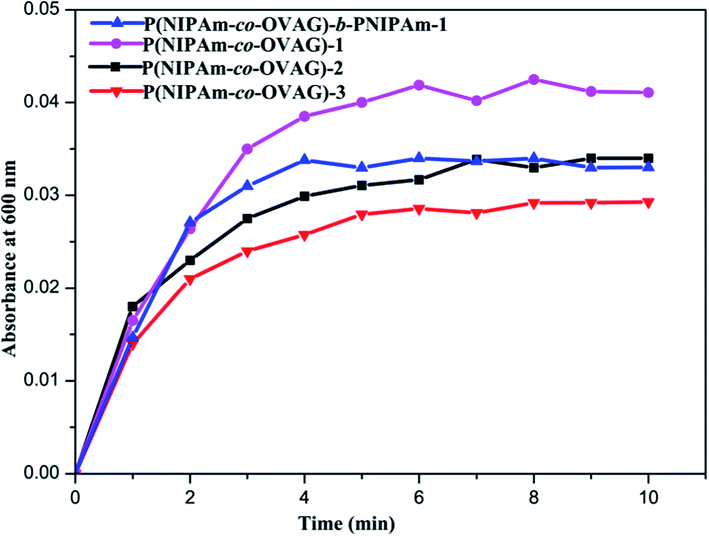 | ||
| Fig. 7 Turbidimetry assay experiments of P(NIPAm-co-OVAG)-1 (purple circle), P(NIPAm-co-OVAG)-2 (black square), P(NIPAm-co-OVAG)-3 (red down triangle), P(NIPAm-co-OVAG)-b-PNIPAm-1 (blue up triangle) micelles with Con A at 30 °C. | ||
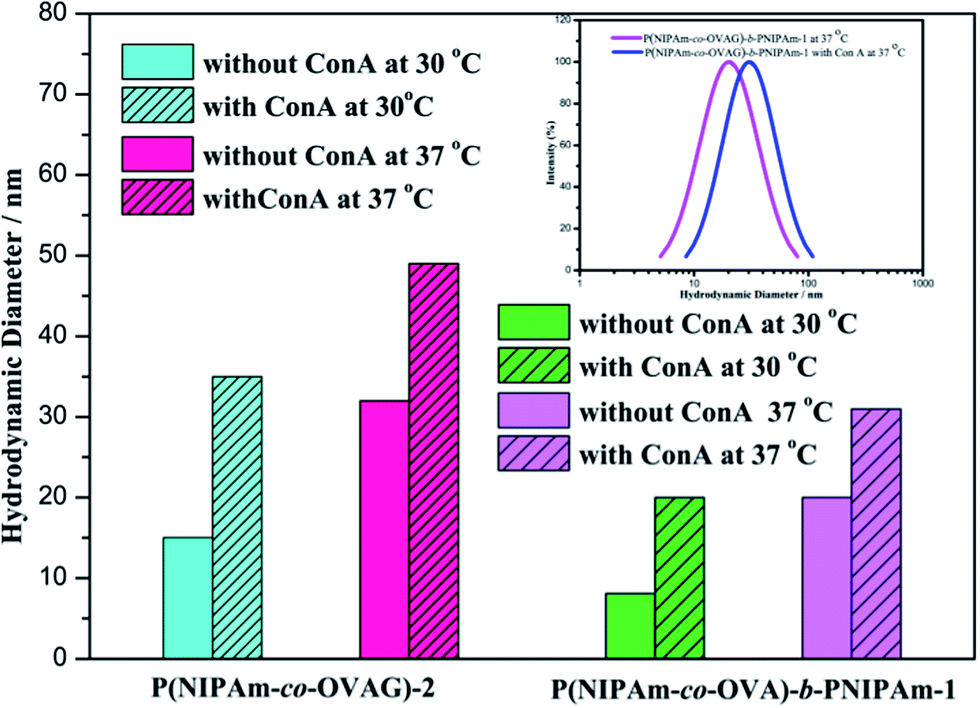 | ||
| Fig. 8 DLS experiments of the glycopolymers with Con A at different temperature. | ||
The reduction in size observed from the TEM study may be due to the collapse of the free segment of the hydrophilic chain of the polymer as well as the dehydration of the polymer chain.38
Lectin-binding assay
In Fig. 7, the turbidity increases with conjugation time to reach plateaus after 4 min and the maximum absorbance value increases with the portion of OVAG in P(NIPAm-co-OVAG). It proved that the more sugar moieties in glycopolymer and the greater recognizability to Con A. Multivalent arrays displayed by the glycopolymer allow high specific interactions between the glucose ligands on the polymers. Meanwhile, the maximum absorbance value of P(NIPAm-co-OVAG)-b-PNIPAm-1 is similar to P(NIPAm-co-OVAG)-2 because of analogical molar ratio of monomers. However, Fig. 7 also shows that glycopolymers contain the same portion of OVAG and have the different rate of increase of absorbance, P(NIPAm-co-OVAG)-b-PNIPAm-1 is a little faster than P(NIPAm-co-OVAG)-2. The reason of this phenomenon can be explained that the micellars of block glycopolymer may have more regular structure because of the existence of double block.
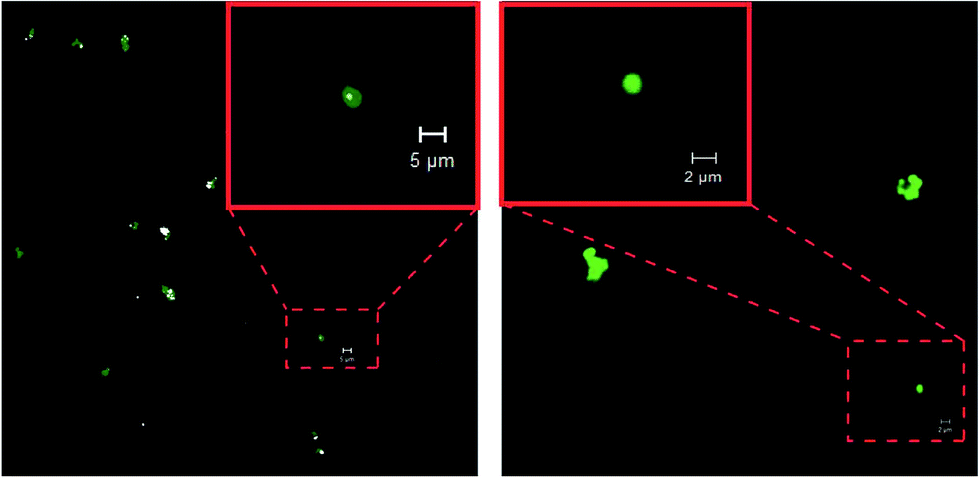 | ||
| Fig. 9 Confocal microscopy images of interaction of glycopolymer P(NIPAm-co-OVAG)-1 (a) and P(NIPAm-co-OVAG)-b-PNIPAm-1 (b) with FITC-Con A. | ||
Cell biocompatibility
The cell biocompatibility of the synthesized glycopolymers was explored using the MTT assay, the results are shown in Fig. 10. In Fig. 10(a), it is clear that the glycopolymer micelle solutions in different concentration have generally very good biocompatibility. The cell viability of glycopolymer-treated wells is the same or greater when compared to the untreated control group. It is consistent with the work of Dakrong Pissuwan group that polymers with trithiocarbonate end group have a low cytotoxity.35 To some degree, the glucose groups of glycopolymers can support the growth of cells. The data in Fig. 10(b) show the results of an analogous experiment but with Con A-loaded micelle solution. The cell viabilities after 24 h are reduced from the untreated control.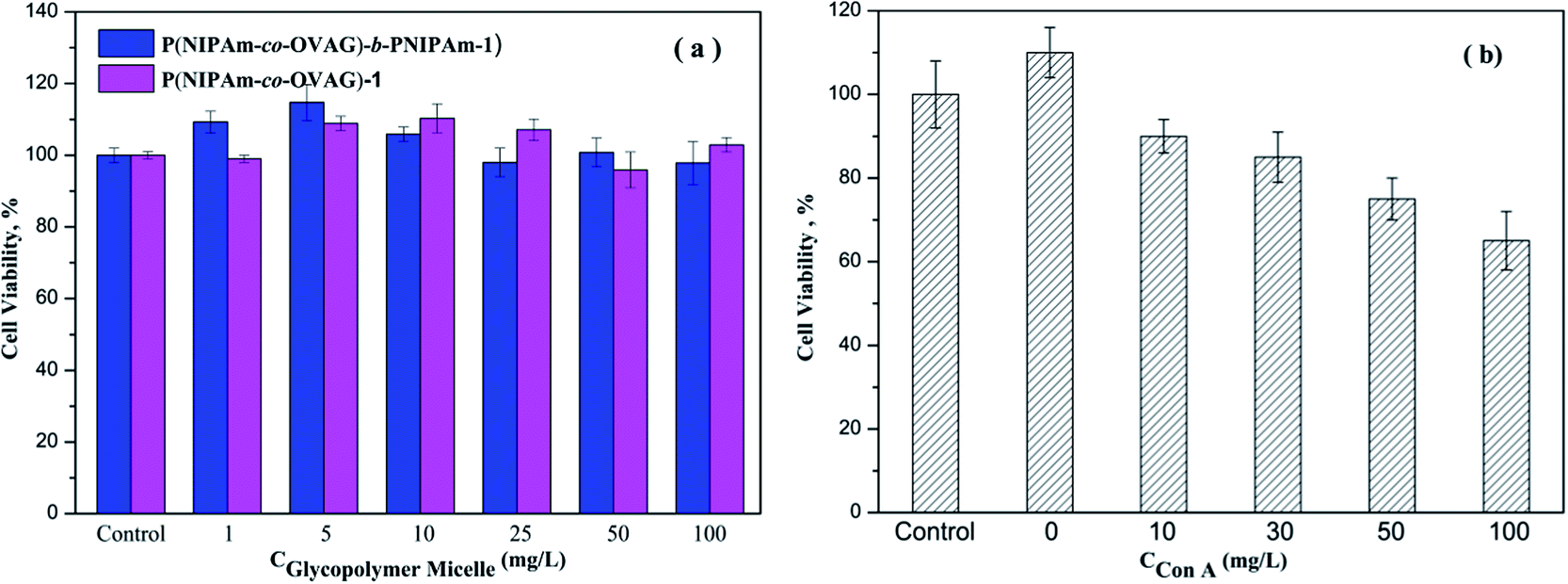 | ||
| Fig. 10 (a) Viability of PIECs after incubation with P(NIPAm-co-OVAG)-1 (purple bar) and P(NIPAm-co-OVAG)-b-PNIPAm-1 (blue bar) at various concentrations; (b) viability of SMMC-7721 cells after incubation with P(NIPAm-co-OVAG)-b-PNIPAm-1 binding different concentrations Con A. The viability was measured by a MTT assay. | ||
Conclusions
A series of multifunctional double hydrophilic block glycopolymers in the form of self-assembling micelles were successfully synthesized via controlled RAFT polymerization of a sugar-functional vinyl ester monomer and a thermoresponsive monomer. The conditions for preparing thermoresponsive P(NIPAm-co-OVAG)-b-PNIPAm diblock glycopolymers were effective. The LCST of the glycopolymer solution has been successfully controlled by changing the molar ratio of monomers in the reaction condition. In addition, the range of temperature transition can be decreased by adding the block of PNIPAm. The micelle structures formed by block glycopolymer were small and loose at low temperature, but large and compact spherical aggregations at temperature above the LCST. The protein recognition of the glucose moieties on the micelles were screened by using Con A. The ability of protein recognition not only related to the amount of sugar but also the structures of glycopolymers. The well-defined block glycopolymers had good cell biocompatibility and minimum cytotoxicity. When the glycopolymers were loaded with Con A, they were able to promote SMMC-7721 cell death.A wide range of block glycopolymers can be prepared by the developed method described in this work. Given their temperature sensitive and protein recognition properties, they have potential applications in targeting cancer cells scaffolds, as drug release carriers and for clinical diagnosis.
Acknowledgements
This investigation was supported by the Natural Science Foundation of China (no. 21303014), the UK-China Joint Laboratory for Therapeutic Textiles, the State Key Laboratory for the Modification of Chemical Fibers and Polymer Materials, the Key Laboratory of Science &Technology of Eco-Textile, Ministry of Education, and the Fundamental Research funds for the Central Universities, Langsha Group, Jofo (WeiFang) Nonwoven Co. Ltd.Notes and references
- C. Cheng, H. Wei, B.-X. Shi, H. Cheng, C. Li, Z.-W. Gu, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Biomaterials, 2008, 29, 497–505 Search PubMed.
- H. Wei, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS PubMed.
- J. E. Chung, M. Yokoyama, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 53, 119–130 CrossRef CAS.
- E. R. Gilies, T. B. Jonsson and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 11936–11943 Search PubMed.
- Z.-S. Ge, D. Xie, D.-Y. Chen, X.-Z. Jiang, Y.-F. Zhang, H.-W. Liu and S.-Y. Liu, Macromolecules, 2007, 40, 3538–3546 Search PubMed.
- B. R. Twaites, C. Heras Alarcón, D. Cunliffe, M. Lavigne, S. Pennadam, J. R. Smith, D. C. Gorecki and C. Alexander, J. Controlled Release, 2004, 97, 551–566 Search PubMed.
- X.-G. Jiang and B. Zhao, Macromolecules, 2008, 41, 9366–9375 CrossRef CAS.
- F. D. Jochum and P. Theato, Chem. Commun., 2010, 46, 6717–6719 RSC.
- P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638–2684 CrossRef.
- S. Muthukrishnan, G. Jutz, X. Andre, H. Mori and A. H. E. Mueller, Macromolecules, 2005, 38, 9–18 Search PubMed.
- Y. Luo, L. Liu, X.-B. Wang, H.-T. Shi, W.-H. Lv and J.-Y. Li, Soft Matter, 2012, 8, 1634–1642 Search PubMed.
- Y. Hoshino, M. Nakamoto and Y. Miura, J. Am. Chem. Soc., 2012, 134, 15209–15212 CrossRef CAS PubMed.
- R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013–2025 Search PubMed.
- L. Wang, G. R. Williams, H.-L. Nie, J. Quan and L.-M. Zhu, Polym. Chem., 2014, 5, 3009–3017 RSC.
- C.-P. Chang, M.-C. Yang, H.-S. Liu, Y.-S. Lin and H.-Y. Lei, Hepatology, 2007, 45, 286–296 Search PubMed.
- C.-Y. Li, H.-L. Xu, B. Liu and J.-K. Bao, Curr. Mol. Pharmacol., 2010, 3, 123–128 CrossRef CAS.
- Y. Kaneko, M. Harada, T. Kawano, M. Yamashita, Y. Shibata, F. Gejyo, T. Nakayama and M. Taniguchi, J. Exp. Med., 2000, 191, 105–114 CrossRef CAS.
- G. Yilmaz and C. R. Becer, Eur. Polym. J., 2013, 49, 3046–3051 CrossRef CAS PubMed.
- M. Ahmed, P. Wattanaarsakit and R. Narain, Eur. Polym. J., 2013, 49, 3010–3033 CrossRef CAS PubMed.
- Y. Miura, Polymer, 2012, 44, 679–689 CAS.
- Z.-C. Deng, M. Ahmed and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 614–627 CrossRef CAS PubMed.
- L. Albertin and N. R. Cameron, Macromolecules, 2007, 40, 6082–6093 CrossRef CAS.
- L. Albertin, M. H. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Macromolecules, 2005, 38, 9075–9084 CrossRef CAS.
- G.-J. Chen, L. Tao, G. Mantovani, J. Geng, D. Nystroem and D. M. Haddleton, Macromolecules, 2007, 40, 7513–7520 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 Search PubMed.
- S. R. S. Ting, A. M. Gregory and M. H. Stenzel, Biomacromolecules, 2009, 10, 342–352 Search PubMed.
- Y.-M. Chen and G. Wulff, Macromol. Chem. Phys., 2001, 202, 3273–3278 CrossRef CAS.
- T. Kakuchi, A. Narumi, T. Matsuda, Y. Miura, N. Sugimoto, T. Satoh and H. Kaga, Macromolecules, 2003, 36, 3914–3920 CrossRef CAS.
- M. Ejaz, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 2000, 33, 2870–2874 CrossRef CAS.
- S. Muthukrishnan, D. P. Erhard, H. Mori and A. H. E. Mueller, Macromolecules, 2006, 39, 2743–2750 CrossRef CAS.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS PubMed.
- L.-G. Yin, M. C. Dalsin, A. Sizovs, T. M. Reineke and M. A. Hillmyer, Macromolecules, 2012, 45, 4322–4332 CrossRef CAS.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344–1350 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS PubMed.
- D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis and V. Bulmus, Biomacromolecules, 2010, 11, 412–420 CrossRef CAS PubMed.
- L. Albertin, C. Kohlert, M. Stenzel, L. J. R. Foster and T. P. Davis, Biomacromolecules, 2004, 5, 255–260 CrossRef CAS PubMed.
- S.-F. Lou, H. Zhang, G. R. Williams, C. Branford-White, H.-L. Nie, J. Quan and L.-M. Zhu, Colloids Surf., B, 2013, 105, 180–186 Search PubMed.
- Y.-Y. Li, X.-Z. Zhang, H. Cheng, J.-L. Zhu, U.-N. Li, S.-X. Cheng and R.-X. Zhuo, Nanotechnology, 2007, 18, 505101 Search PubMed.
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850 CrossRef CAS PubMed.
- B. Trzebicka, D. Szweda, S. Rangelov, A. Kowalczuk, B. Mendrek, A. Utrata-Wesolek and A. Dworak, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 614–623 CrossRef CAS PubMed.
- A. Muñoz-Bonilla, O. León, V. Bordegé, M. Sanchez-Chaves and M. Fernandez-Garcia, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1337–1347 CrossRef PubMed.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS PubMed.
- M. E. Alf, T. A. Hatton and K. K. Gleason, Polymer, 2011, 52, 4429–4434 CrossRef CAS PubMed.
- S. R. S. Ting, E.-H. Min, P. Escalé, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |