
A comparative study on degradation characteristics of fluoropolymers irradiated by high energy heavy ions
Abstract
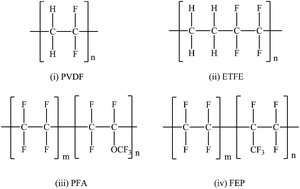
Energetic heavy ions passing through polymer foils cause modifications of properties, first of all structural changes. In order to evaluate ion beam modification of fluoropolymers, a comparison of polyvinylidene fluoride (PVDF), ethylene-tetrafluoroethylene (ETFE), tetrafluoroethylene-per-fluoromethoxyethylene (PFA), and tetrafluoroethylene-hexa-fluoropropylene (FEP) was undertaken. Foils were irradiated with heavy ions (Sm, Au) of a kinetic energy of a few MeV u−1 and fluences up to 6 × 1012 ions cm−2 at room temperature in vacuum. The ion induced changes in the molecular structure were studied with Fourier transform infrared spectroscopy (FT-IR) and on-line mass spectrometry (Residual Gas Analysis, RGA). The results in terms of macromolecular changes and formation of fragments were compared and related to the individual polymer structures. In general, the polymers lose various small molecular fragments by bond scission of polymeric backbone and side chains, typically fluorine and hydrogen fluoride, as well as trifluoromethyl, and form new bonds, mainly CC double bonds. From the results, the main degradation reactions are derived. The findings are of technical interest for space applications when devices are hit by highly energetic heavy ions from galactic cosmic rays on long-term missions.
 Please wait while we load your content...
Please wait while we load your content...

