
PEG–lipid telechelics incorporating fatty acids from canola oil: synthesis, characterization and solution self-assembly†
Abstract
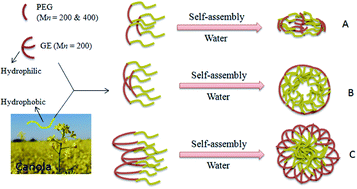
The synthesis of amphiphilic, ABA type, novel PEG–lipid telechelics and their solution self-assembly have been reported. Natural fatty acids were functionalized with a propargyl group. Poly(ethylene glycol) (PEG) and glycerol ethoxylate (GE) were functionalized with terminal azides. The functionalized PEG, GE and fatty acids were then conjugated using click chemistry. Characterization of these conjugates has been carried out with the help of 1HNMR, FTIR, and GPC and further evaluated for their solution self-assembly. The particle size and morphology of nanoparticles were observed with transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements. The results prove the high conjugation efficiency and self-assembly of telechelics into nanoparticles of different average sizes in solution. This study highlights that these novel telechelic nanoparticles may serve as a potential drug carriers. The strategy presented herein can potentially be extended to the preparation of natural fatty acid functionalized assemblies with other hydrophilic polymers for therapeutic applications.
 Please wait while we load your content...
Please wait while we load your content...

