 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of the diffusion-layer thickness during electrodeposition on the synthesis of nano core/shell Sn–O–C composite as an anode of lithium secondary batteries†
Moongook Jeonga,
Tokihiko Yokoshimab,
Hiroki Narab,
Toshiyuki Mommaab and
Tetsuya Osaka*ab
aGraduate School of Advanced Science and Engineering, Waseda University 3-4-1, Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: osakatets@waseda.jp; Fax: +81-3-3205-2074; Tel: +81-3-5286-3202
bFaculty of Science and Engineering, Waseda University 3-4-1, Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 20th May 2014
Abstract
Electrodeposition was conducted from an organic carbonate solvent via the potentiostatic technique through three consecutive steps in order to synthesise Sn–O–C composite, which delivered a discharge capacity of 596 mA h g of Sn−1 after 50 cycles. However, the composite anode suffered from a significantly low initial discharge capacity, delivering a discharge capacity of 79 mA h g of Sn−1 until the 5th cycle. It was deduced that the improbably low initial capacity was induced by the deposition of Li-rich compounds, which were formed by electrolyte decomposition accompanied by the reduction product of supporting electrolyte salts during the electrodeposition process, on the surface layer. In order to improve the poor initial capacity, we modified the chemical composition of the surface layer by means of implementing the agitation of the electrolyte during the deposition process. This gave rise to varying the diffusion-layer thickness during the deposition process due to the enhancement of convection by movement of the electrolyte itself. As a result, we achieved improvement of the initial discharge capacity, delivering 572 mA h g of Sn−1 at the 1st cycle and 586 mA h g of Sn−1 at the 50th cycle. It was revealed that the surface layer was composed of a decomposition product of the organic carbonate solvent. Furthermore, a smaller particle size of the Sn–O–C composite was obtained via electrolyte agitation, giving rise to homogeneous shell formation on the Sn compound core. Herein, we thoroughly examined the influence of varying diffusion-layer thickness during the deposition process on the properties of the Sn–O–C composites from an electrochemical standpoint.
Introduction
Recently, research on tin-based materials for an Li-alloying anode of lithium ion batteries (LIBs) has been accelerated due to its higher theoretical capacity (Li4.4Sn; 993 mA h g−1) than that of a commercial graphite anode (LiC6; 372 mA h g−1), which has been considered as one of solutions for the development of energy resources for electric vehicles.1 In spite of the high energy density of tin-based materials for an anode of LIBs, the material has a significant shortcoming to attain a stable cycle life due to its drastic volume change (250–400%) during the charge/discharge cycle. Once the volume has been changed, it creates a mechanical crack and pulverization in the active material with repeated cycles. Consequently, it suffers from electrical isolation and disintegration, causing rapid capacity failure within the initial cycles.2,3 Therefore, withstanding the volume change by controlling the material properties is a challenging task for achieving a stable cycle life. To solve the abovementioned problems, diverse strategies have been attempted, such as through a Sn nanostructure,4–6 an embedded matrix buffer,7–13 or a mesoporous structure.14–17 The Sn nanostructure can reduce mechanical stress induced during the electrochemical cycles by minimizing Sn particles. A suitably embedded buffer matrix, such as a carbon composite or non-active inter-metallic compound, can endure the massive volume changes by acting as a buffer matrix and absorbing the drastic volume changes. The porous space of mesoporous Sn anodes can avoid aggregation and pulverization during cycling.Meanwhile, the passivating layer between an electrode and electrolyte after cycling has been understood as an important phenomenon in the chemistry of LIBs, which is the so-called solid electrolyte interphase (SEI).18 The importance of SEI has been well recognized in a number of reports.18–22 Decomposition of organic electrolytes occurs on both the anode and cathode mostly during the several initial cycles causing SEI formation. The decomposed solvent and Li salt generate a passivating layer, behaving as an electronic insulator while also acting as a conductor for Li+ ions to pass through. The electronic insulation behaviour is a significant characteristic of SEI since it suppresses further decomposition of electrolytes during the following cycles, causing SEI thickening, which increases internal resistance, self-discharge and low Faradaic efficiency.23 Furthermore, high Li+ ion conductivity is essential for the SEI layer to avoid an increase in overpotential during cycling. Thus, the SEI layer determines the performance of LIBs, such as cycle life, durability and even safety.24
In our previous work, we accomplished the electrodeposition of a Sn–O–C composite anode from an organic carbonate solvent showing a discharge capacity of 465 mA h g of Sn−1 after 100 cycles, which was synthesized by the galvanostatic deposition technique.25 It was clarified that the Sn deposition and electrolyte decomposition were simultaneously carried out during the electrodeposition process, giving rise to an inorganic/organic composite with a structure of polycrystalline Sn metal surrounded by SEI formation. The stable cycle life was derived from the inorganic/organic composite acting as a buffer matrix against stress for volume change. We also thoroughly examined the electrodeposition mechanism showing three different deposition steps that emulated a trace of the potential transient profile during the galvanostatic deposition, as revealed in our previous report. The results revealed that the deposition mechanism was composed of multi-stages including 2.3, 1.5 and 0.3 V vs. Li/Li+ for Sn deposition, catalytic decomposition of the organic carbonate solvent, and a reaction between Li+ ions and deposited Sn, respectively.26 The cycle performance of the Sn–O–C composite as an anode material was strongly influenced by the method of controlling the duration of the specific step at which different electrochemical reactions occur.
In the present work, we attempted to deposit the Sn–O–C composite from an organic carbonate solvent via the potentiostatic technique with three consecutive steps including 2.3, 1.5 and 0.3 V vs. Li/Li+ as the deposition mechanism, which was suggested in our previous work. Consequently, the Sn–O–C composite with a nano core/shell structure was synthesized. We confirmed that the chemical composition of the shell layer strongly influenced the cycle performance, such as the poor initial capacity and Coulombic efficiency of the Sn–O–C composite. To improve the unacceptable initial performance, we implemented electrolyte agitation through simple magnetic stirring. Herein, we comprehensively examined the effects of electrolyte agitation during the deposition process with electrochemical standpoints by comparing them with the characteristics of the deposited composites without electrolyte agitation. Furthermore, the influence of agitation on the morphological characteristics and chemical compositions of the deposited Sn–O–C composites accompanied by their cycle performances were also closely investigated as candidates for anode materials for lithium secondary batteries. Hereafter, the Sn–O–C composites electrodeposited with electrolyte agitation and without electrolyte agitation are simply referred to as “a composite deposited with bath agitation” and “a composite deposited without bath agitation”, respectively.
Results and discussion
The influence of electrolyte agitation through simple magnetic stirring was electrochemically analysed and the results are shown in Fig. 1 as curves of the current transient on potentiostatic deposition with three steps accompanied by the corresponding applied potentials. These results indicated a rapid decrease in the current density during the initial few seconds, which was obtained with each current transient of the three potentiostatic steps for both composites deposited with and without bath agitation. The sharp decrease in initial current density was followed by a fairly constant value, implying that nucleation mostly occurred during the initial process at each deposition step. In Fig. 1a, constant values of the current transient of the composite deposited with bath agitation obtained unstable transient curves of ca. −0.23, −0.25 and −0.26 mA cm−2 from the three potentiostatic steps at 2.3, 1.5 and 0.3 V vs. Li/Li+, respectively. However, the composite deposited without bath agitation attained a remarkably lower and more stable current density value of ca. −0.046, −0.048 and −0.052 mA cm−2 obtained from the three potentiostatic steps at 2.3, 1.5 and 0.3 V vs. Li/Li+, respectively (Fig. 1b). These are typical effects of electrolyte agitation during the deposition process, since the movement of the bulk solution by electrolyte agitation generally results in an enhancement of the convection, giving rise to the vigorous delivery of metal ions to the cathode surface.27 According to the electrochemical standpoint, it is thought that there are three basic mechanisms of mass transport: diffusion, migration and convection. The contribution of migration to the delivery of metal ions is insiginificant.27,28 Therefore, it is supposed that the main driving forces contributing to electrodeposition are derived from a concentration gradient (diffusion) and movement of the reacting species (convection). Scheme 1a and b are schematic illustrations accounting for variations of diffusion-layer thickness dependent on deposition time. To begin with a consideration of the deposition without bath agitation (Scheme 1b), in general, the metal ion concentration on the cathode surface is reduced after a certain deposition time of t1. Afterwards, the decrease in the metal ion concentration continuously rises with a further deposition time of t2. Consequently, the diffusion layer thickens with an increase in deposition time. Meanwhile, the metal ion concentration for the deposition with bath agitation (Scheme 1a) also decreases after a certain deposition time of t1. However, it is assumed that the continuous decrease in the metal ion concentration does not occur with a further deposition time of t2 due to the enhanced mass transport behaviour from the contribution of the convection caused by bath agitation. As a result, a thinner diffusion layer can be generated, which can be electrochemically explained with limiting current density (iL) expressed as
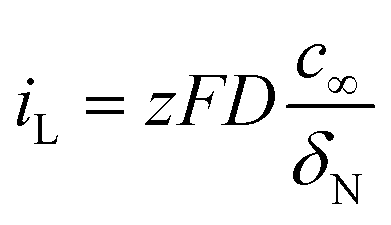 | (1) |
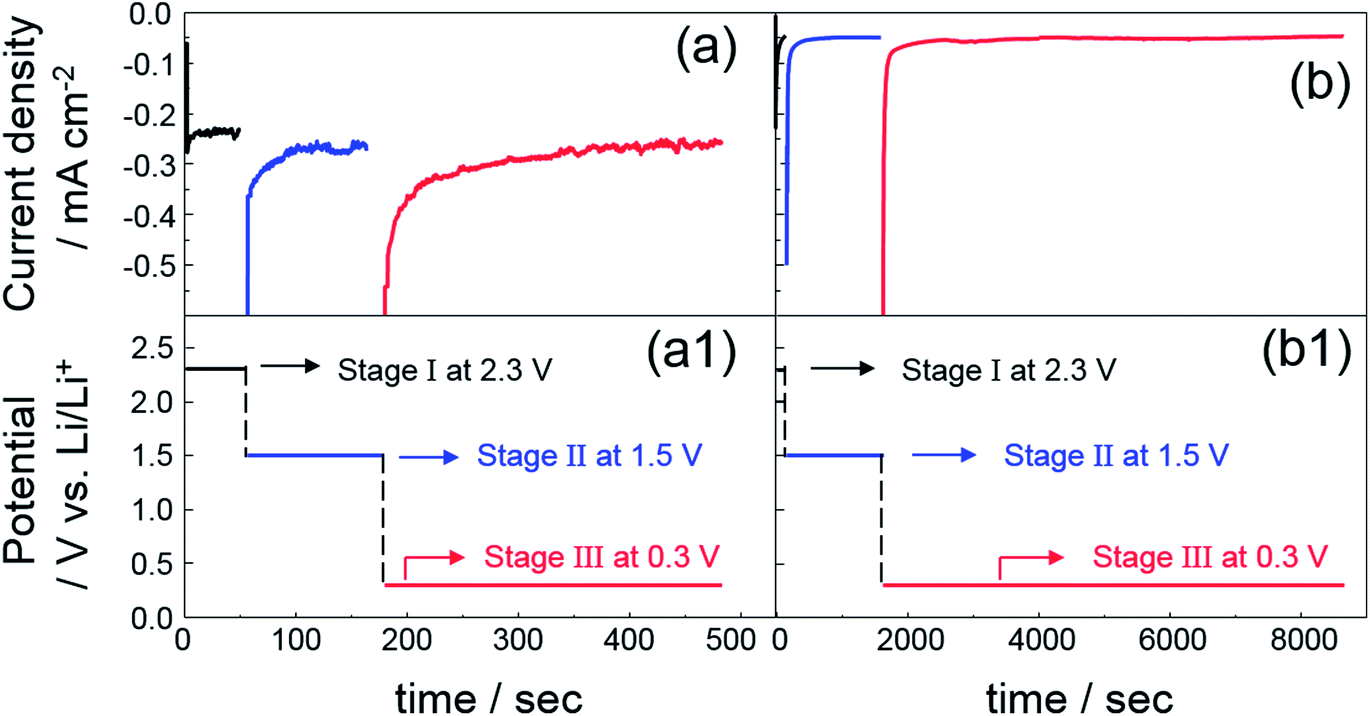 | ||
| Fig. 1 Current transient curves (a and b) on potentiostatic deposition with three steps and their applied potentials (a1 and b1) at each step. (a and a1) deposition with bath agitation by simple magnetic stirring and (b and b1) without bath agitation. | ||
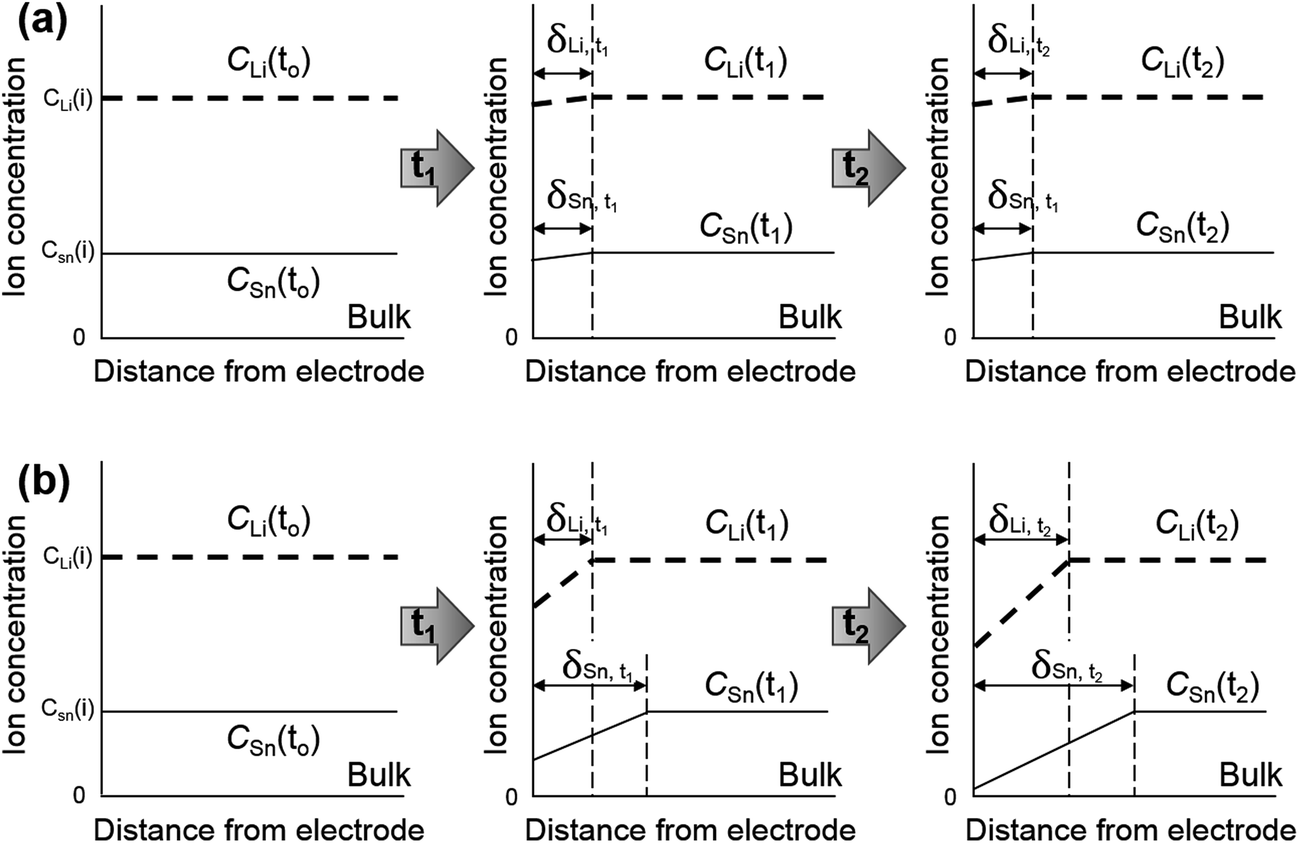 | ||
| Scheme 1 Schematic diagrams illustrating variation of diffusion-layer thickness dependent on deposition time, (a) deposition with bath agitation by simple magnetic stirring and (b) without bath agitation. | ||
Morphologies of the as-prepared Sn–O–C composites deposited with and without bath agitation are shown in Fig. 2 with low and high magnification FE-SEM images. Both Sn–O–C composites showed small particles on a nano-sized scale. It was also observed that inter-particle void spaces were formed by randomly overlapped particles in both composites deposited with and without bath agitation. In Fig 2a, the composite deposited with bath agitation indicated many aggregated structures composed of small particles with a size of 50–90 nm, causing the formation of clustered particles with a size of 400–500 nm. The inter-particle void space was produced by aggregation of the particle clusters, not of individual small particles. However, the morphologies of the composite deposited without bath agitation indicated relatively large particles with a size of 400–500 nm. From the electrochemical standpoint, the particle size was defined by the relationship between the rate of nucleation and the growth of crystals.29 From morphological observation, it was thought that the composite deposited with bath agitation had a high rate of nucleation and slow growth, resulting in the formation of a small particle structure. On the other hand, the composite deposited without bath agitation showed a comparably larger particle size, which was derived from the growth of nuclei during the deposition process. In general, the particle size can be explained as the critical radius of the nucleus, rc, which is a function of overpotential, η;30
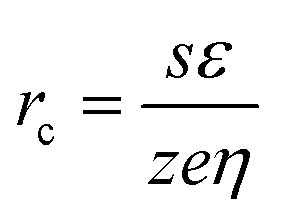 | (2) |
| ηtot = ηact + ηconc | (3) |
 | ||
| Fig. 2 Low and high magnification FE-SEM images of the as-prepared Sn–O–C composite deposited with (a) and without (b) bath agitation. | ||
Herein, the electrodeposition was conducted by a potentiostatic deposition technique. In other words, the ηtot was identical and constant for both deposition processes at the individual stages. However, the ηconc of the deposition with bath agitation was smaller than that of the deposition without bath agitation due to movement of the reacting species themselves by enhancement of convection caused by magnetic stirring. Thus, the ηact of the deposition with bath agitation became larger than that of the deposition without bath agitation, resulting in the relationship between the rate of nucleation and the growth of crystals, as mentioned before.
To investigate the structure of the as-prepared composites, FE-STEM images with EDS element mapping were observed. The FE-STEM images of the composite deposited with bath agitation (Fig. 3a) indicated that a particle consisted of a dark part covered by a thin amorphous layer with a thickness of ca. 6 nm showing a core/shell structure. The core showed lattice fringes of the (200) planes, which corresponded to the d-spacing value of 2.9 Å of the tetragonal tin. Another particle also verified the lattice fringes of the (101) planes corresponding to a d-spacing value of 2.7 Å (ESI, Fig. S1a†). Furthermore, the selected area electron diffraction (SAED) pattern (ESI, Fig. S1b†) showed point and ring patterns, indicating the presence of single and polycrystalline planes, which were assigned to (101), (200), (211) and (400) planes of the tetragonal Sn. These results imply that the core of the particle consisted of a single crystal of tin and the particles were aggregated with other particles, causing a polycrystalline structure with various ring patterns corresponding to different planes of the tetragonal Sn. EDS element mapping of tin and oxygen were respectively indicated as having a red and green colour with a particle size of less than 100 nm. The overlapped mapping image of the two elements revealed that oxygen homogeneously covered tin particles, implying that oxygen was one of the elements composing the amorphous shell layer. The chemical state of oxygen and its bonding with other elements will be discussed in detail later with the XPS results. In Fig. 3b, the composite deposited without bath agitation also showed a tin core covered by an amorphous layer with a thickness of ca. 14 nm and an irregular particle size of 100–500 nm. The d-spacing value of its core tin was not clearly shown, probably since the particles were too large for an electron beam to transmit throughout. An overlapped EDS mapping image of tin and oxygen indicated that the oxygen shell layer inhomogeneously covered the tin core. This inhomogeneous coating layer was also shown in another HR-TEM image (ESI, Fig. S1c†). It is believed that the large particle size hindered the homogeneous formation of the oxygen shell layer during the electrodeposition process without bath agitation.
 | ||
| Fig. 3 FE-STEM images with EDS element mapping of the as-prepared Sn–O–C composite deposited with (a) and without (b) bath agitation. | ||
The crystalline structure of the tin core in the as-prepared composites was also analysed by GIXRD patterns (ESI, Fig. S2†). Strong peaks at 2θ = 30.6, 32.0, 43.8 and 44.9° that were assigned to the 200, 101, 220 and 211 crystal planes and weak peaks at 2θ = 55.3, 62.5, 63.7, 64.5, 72.4, 73.1, 79.4 and 89.4° that were assigned to the 301, 112, 400, 321, 420, 411, 312 and 501 crystal planes, respectively, of the tetragonal phase tin were observed (JCPDS # 04-0673). The results validated that the cores of both composite particles were composed of β-tin. Meanwhile, peaks at 2θ = 23.1 and 35.5° appeared in Fig. S2b† although they were not able to be assigned to stable phases of Sn.
In Fig. 4, XPS was analysed to examine the surface chemical state and the composition of the shell layer of as-prepared composites. O 1s spectra of the composites deposited with and without bath agitation showed deconvoluted curves with two and three main peaks, respectively (Fig. 4a and b). The identical peaks corresponding to oxygen atoms in carbonate and organic species at 531.7 eV were obtained from both composites.22,31,32 The peak at 532.2 eV was attributed to the oxygen atom in O–C.31 However, the deconvoluted peaks assigned to 528.2 and 533.2 eV indicating Li2O and lithium alkyl carbonate (ROCO2Li) and/or lithium alkoxide (RCH2OLi) were obtained with only the composite deposited without bath agitation.22,33,34 Similar results were indicated by the C 1s spectra of both composites (Fig. 4c and d). The spectra were deconvoluted into three peaks. Among them, two peaks assigned to identical positions at 284.8 and 286.2 eV, indicating carbon bonding in CH2–CH2 and C–O, respectively, were obtained from both composites. However, a carbon atom that bonded to three oxygen atoms in the carbonate species that was assigned to 289.3 eV was indicated by only the composite deposited with bath agitation.31,32 The carbon atom in lithium alkyl carbonate (ROCO2Li) and/or lithium alkoxide (RCH2OLi) assigned at 286.6 eV was also shown by only the composite deposited without bath agitation.31–33 Fig. 4e and f present the XPS spectra of Cl 2p core peaks. A negligible peak with low intensity was shown in the result of the composite deposited with bath agitation. On the other hand, the composite deposited without bath agitation obviously showed two strong deconvoluted curves assigned to 199.4 and 201.1 eV indicating LiCl of Cl 2p3/2 and Cl 2p1/2, respectively.35 From these results, it is suggested that the shell layer of the Sn–O–C composite was produced by electrolyte decomposition during the deposition process, causing SEI formation. In particular, the SEI formation consisting of decomposition species of an organic carbonate solvent was obtained from the composite deposited with bath agitation, herein EC and PC. Conversely, the shell formation of the composite deposited without bath agitation was composed of not only decomposition species of an organic carbonate solvent but also inorganic compounds derived from reduction products of LiClO4, used as a supporting electrolyte, such as Li2O, lithium alkyl carbonate (ROCO2Li), lithium alkoxide (RCH2OLi) and LiCl. As discussed with morphological and structural characteristics, it was assumed that the chemical compositions of the deposits were also strongly affected by the bath agitation, which enhanced mass transfer of the metal ion.
 | ||
| Fig. 4 XPS spectra of the as-prepared Sn–O–C composite deposited with (a), (c), (e) and without (b), (d), (f) bath agitation. | ||
To summarize the structures of the composites, the core/shell structure of both Sn–O–C composites consisted of a Sn compound core with SEI formation of a shell layer that was derived from the electrolyte decomposition process. The chemical composition of the shell layer differed with the deposition process, either with or without bath agitation (i.e., decomposition compounds of an organic carbonate solvent for the composite deposited with bath agitation as opposed to decomposition compounds of an organic carbonate solvent accompanied by a reduction product of LiClO4), forming Li-rich compounds for the composite deposited without bath agitation.
For further investigation of the chemical structure and composition on the surface of the as-prepared composites, XPS spectra of Sn 3d5 and Li 1s were examined with Ar ion etching for 30 s with 3 s intervals. As shown in Fig. 5a, the composite deposited with bath agitation showed a strong peak of Sn 3d5 assigned to 484.8 eV, indicating metallic Sn(0) after the first Ar ion etching. Small satellite peaks at 486.6 eV corresponding to Sn(IV) were also shown during Ar ion etching for 30 s.31,36,37 However, a Li 1s peak was not obtained during Ar ion etching for 30 s. On the other hand, the composite deposited without bath agitation indicated a Sn 3d5 peak assigned to 484.8 eV showing a low peak intensity during the initial Ar ion etching time followed by a gradual increase with a further Ar ion etching time. A similar tendency was also obtained with a small satellite peak assigned to 486.6 eV. Furthermore, XPS spectra of Li 1s showed a strong peak during initial Ar ion etching, assigned to 56.2 eV indicating a Li atom in Li2O.38 The intensity of this peak gradually decreased with the Ar ion etching time and finally disappeared after 30 s of Ar ion etching. This result suggested that the Li-rich compounds existed on the surface of the Sn–O–C composite deposited without bath agitation.
 | ||
| Fig. 5 XPS spectra of Sn 3d5 and Li 1s of the as-prepared Sn–O–C composite deposited with (a) and without (b) bath agitation. Ar ion etching was conducted for 30 s with 3 s interval. | ||
To discuss the different chemical compositions of the as-prepared Sn–O–C composites, we need to recall the variations in ion concentrations and diffusion-layer thicknesses during the deposition process as discussed in Scheme 1. In our previous work, we reported that both Sn and Li ions were involved in the electrodeposition process at 0.3 V vs. Li/Li+.26 Thus, it is required to take the diffusion-layer thickness variations of a Li ion into account along with those of the Sn ion. In the case of the deposition process without bath agitation, the diffusion-layer would gradually thicken with deposition time, as discussed in Scheme 1b. The Sn ion concentration would decrease faster than that of the Li ion due to its low concentration in the deposition bath and higher standard reduction potential. Consequently, the thicker diffusion-layer probably formed with the Sn ion rather than the Li ion. Thus, the Li ion could easily take part in the electrochemical reaction, resulting in a shell layer with SEI formation including reduction products of LiClO4 forming Li-rich compounds, such as Li2O, lithium alkyl carbonate (ROCO2Li), lithium alkoxide (RCH2OLi) and LiCl as discussed with the XPS results. On the other hand, in the case of the deposition process with bath agitation, a constant diffusion-layer thickness of both metal ions could be achieved due to an enhanced convection by simple magnetic stirring, as discussed in Scheme 1a. Thus, a sufficient amount of Sn ion can be constantly supplied from the bulk electrolyte, giving rise to a more preferable electrochemical reduction of the Sn ion than that of the Li ion, which attained a chemical composition of the SEI layer mainly containing a decomposed organic solvent without Li-rich compounds derived from reduction products of LiClO4.
Both composites were electrochemically tested to investigate the influence of the distinct core/shell structure on the anode performance for lithium secondary batteries. Fig. 6a and b indicated the cycle performances and Coulombic efficiencies of both composites. In Fig. 6a, a high initial discharge capacity of 572 mA h g of Sn−1 at the 1st cycle was obtained from the composite deposited with bath agitation. A constant capacity value was prolonged with the highest discharge capacity of 646 mA h g of Sn−1 at the 18th cycle and 586 mA h g of Sn−1 at the 50th cycle. On the other hand, cycle performances of the composite deposited without bath agitation delivered a poor initial capacity. The variation of the discharge capacity against the cycle number seems to be divided in to three stages: (i) a significantly low initial discharge capacity of 67 mA h g of Sn−1 from the 1st cycle until the 5th cycle, delivering a discharge capacity of 79 mA h g of Sn−1; (ii) a gradual increase in the first stage to a discharge capacity of 560 mA h g of Sn−1 at the 30th cycle; (iii) and a stable stage up to the 50th cycle, delivering discharge capacity of 596 mA h g of Sn−1. Low Coulombic efficiencies of 45.9% and 12.4% were obtained at the 1st cycle from the composites deposited with and without bath agitation, and they increased up to 90.8% and 63.2% at the 2nd cycle, respectively. The considerably low Coulombic efficiency at the 1st cycle was probably due to SEI formation and further reduction of the remaining oxide states of the active material.25,26,39–43 The composite deposited with bath agitation indicated a Coulombic efficiency of higher than 90% after the 2nd cycle and finally reached 96.8% at the 50th cycle. Conversely, a comparably low Coulombic efficiency was obtained from the composite deposited without bath agitation showing a value higher than 90% after the 35th cycle followed by a gradual increase up to 93% at the 50th cycle. It is assumed that the Li-rich compounds in the SEI shell layer, such as Li2O, lithium alkyl carbonate (ROCO2Li), lithium alkoxide (RCH2OLi) and LiCl may be related to the low initial capacity along with retarded Coulombic efficiency. It has been reported that a Li ion can pass through media under two driving forces, such as an externally applied electric field and a concentration gradient of Li ion.44 During lithiation, the positively charged ions move via migration and diffusion through the electrolyte towards the active material where they react and lithiate into a solid phase of active material particles. Then, the solid-state diffusion of Li ions is conducted with a driving force of a Li concentration gradient in the active material particles.45 On the basis of this assumption, it is suggested that the Li-rich components in the shell layer of the composite deposited without bath agitation caused difficulty in the diffusion of the Li ion in the solid-state media, resulting in a low initial capacity. Thus, it is believed that the remarkably enhanced initial capacity and Coulombic efficiency of the composite deposited with bath agitation was achieved due to the compositions in its shell layer without Li-rich compounds, which allows Li ions to easily diffuse within the solid-state phase by a high Li concentration gradient.46
 | ||
| Fig. 6 Plots of (a) discharge capacity against the number of cycles, (b) their Coulombic efficiency, (c) potential profile and (d) differential capacity plot at the 1st cycle. | ||
Fig. 6c shows the potential profiles of the composites deposited with (dot line) and without (bold solid line) bath agitation. It is easily recognized that the composite deposited with bath agitation showed distinctive plateaus, indicating multi-electrochemical reactions during the charge/discharge cycle. However, the unclear plateaus were shown in the potential profile of the composite deposited without bath agitation. Furthermore, these results also indicated a high initiating potential of the delithiation process obtained at ca. 0.4 V, which assumed that the Li ion had difficulty completely diffusing into the solid-state of the active material, as aforementioned. To discuss the detailed investigation on the effects of distinctive composition in the shell layer on the electrochemical reaction between Li+ ions and deposited Sn during cycling, differential capacity plots (DCPs) were analysed with the Sn–O–C composite deposited with and without bath agitation. The results are indicated in Fig. 6d along with the 1st charge/discharge cycle. The composite deposited with bath agitation distinguishably showed a small cathodic peak at 0.6–0.7 V and a strong, sharp peak at 0.35 V derived from the phase transition of the LixSn alloying reaction. Four broad and sharp anodic peaks were also obviously shown at 0.5, 0.6, 0.7 and 0.8 V, implying the multistep nature of this phase change.47,48 However, the peaks corresponding to the phase transition of LixSn were scarcely recognizable with the composite deposited without bath agitation, which reveals that the electrochemical reaction during the 1st cycling was significantly suppressed. The 1st cycle of DCPs was enlarged by the selected y axis range [ESI, Fig. S3†], indicating several peaks of the composite deposited without bath agitation, which are hardly recognizable in Fig. 6d. Small and broad cathodic peaks at 0.6 and 0.3–0.4 V were obtained with four anodic peaks at 0.5–0.8 V, respectively, which implies that the phase transition reaction was carried out in spite of a significantly weak peak intensity compared to that of the composite deposited with bath agitation.
For further examination of the electrochemical reaction during cycling, the DCPs were plotted with the 2, 5, 10, 15, 20, 30, 40 and 50th charge/discharge cycles (Fig. 7). As discussed previously, distinctive cathodic and anodic peaks of DCPs for charge/discharge cycles were shown at the 2nd cycle and continued up to the 50th cycle from the composite deposited with bath agitation. The obvious peaks at the selected cycle indicated that the electrochemical reaction could be kinetically carried out during initial cycles with the composite deposited with bath agitation. Small peaks at 0.65 V for the charge process and 0.7 and 0.8 V for the discharge process began to form a broad and indistinct peak, which was presumably attributed to the partially remaining alloy state of LixSn without complete delithiation after several tens of cycles.25,47,48 Meanwhile, the DCPs of the composite deposited without bath agitation showed significantly indistinct cathodic and anodic peaks during the 2nd and 5th charge/discharge cycles. Afterward, the DCPs indicated a gradual increase in peak intensity attained by the 10th, 15th and 20th cycles followed by a constant value at the 30th, 40th and 50th cycles. These results affirm improvement of the kinetics of the electrochemical reaction of the composite deposited without bath agitation with further cycling. These results also suggest that its electrochemical reaction during the 1st stage was probably suppressed by an inorganic passivating compound and Li-rich compounds in the shell layer. Furthermore, the larger particle size would lengthen the Li ion diffusion length. Nevertheless, the volume change of the active material may have undergone repeated charge/discharge cycles, causing the formation of cracks on the surface layer. Consequently, the electrolyte was able to permeate into the inner layer of the active material, allowing the inside fresh Sn phases to react with the Li ions. Finally, a stable cycle performance was achieved after several tens of cycles (see Fig. S4,† showing an increase in cracks with further cycles).
 | ||
| Fig. 7 DCPs of the 2, 5, 10, 15, 20, 30, 40 and 50th charge/discharge cycles of the Sn–O–C composite deposited with (a) and without (b) bath agitation. | ||
Conclusions
We electrodeposited the Sn–O–C composite with a core/shell structure from an organic carbonate solvent via the potentiostatic technique with three consecutive steps including 2.3, 1.5 and 0.3 V vs. Li/Li+. The Sn–O–C composite delivered a low initial discharge capacity of 67 mA h g of Sn−1 at the 1st cycle, which continued until the 5th cycle, delivering a discharge capacity of 79 mA h g of Sn−1 with 12.4% and 73.4% of Coulombic efficiencies, respectively. After the 50th cycle, a discharge capacity of 596 mA h g of Sn−1 with 93% Coulombic efficiency was obtained. The significantly poor initial capacity was derived from the deposition of Li-rich compounds such as Li2O, lithium alkyl carbonate (ROCO2Li), lithium alkoxide (RCH2OLi) and LiCl at the shell layer. In order to improve the low initial capacity and poor Coulombic efficiency, modification of the chemical composition of the shell layer was conducted by implementing the agitation of the electrolyte with simple magnetic stirring during the electrodeposition process. It was suggested that a constant diffusion-layer thickness during the deposition process was obtained from the enhancement of the convection by movement of the electrolyte itself. As a result, the shell layer consisting of a decomposition product from an organic carbonate solvent and a smaller particle size of the Sn–O–C composite was obtained, delivering a discharge capacity of 572 mA h g of Sn−1 and 586 mA h g of Sn−1 at the 1st cycle and 50th cycle, respectively, with improved Coulombic efficiency.Experimental
All the chemical reagents were purchased at an analytical grade and used as supplied. The electrodeposition bath was prepared by dissolving 2.5 mmol L−1 SnCl2 (Sigma-Aldrich) into 1.0 mol L−1 of LiClO4 in ethylene carbonate (EC)–propylene carbonate (PC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v), Kishida, H2O content < 20 ppm).
:1 (v/v), Kishida, H2O content < 20 ppm).
A Ni plate with a dimension of 10 × 10 mm was used as a substrate for the electrodeposition of the Sn–O–C composite after ultra-sonication in trichloroethylene and ethanol followed by acid treatment with 10% HCl. A piece of tin was used as a counter electrode. A lithium metal and EC–PC solution containing 1.0 mol L−1 LiClO4 were sealed in a glass tube by a glass flit to serve as a reference electrode.
All processes for the electrodeposition were performed in an argon-filled glove box at a dew point lower than −100 °C. The Sn–O–C composites were synthesized via a potentiostatic deposition technique with three consecutive steps at 2.3, 1.5 and 0.3 V vs. Li/Li+ by passing a charge quantity of 0.01, 0.04 and 0.21 C cm−2, respectively, using a potentiostat (Hokuto Denko, HZ-5000). The electrodeposition process was carried out either with or without electrolyte agitation using simple magnetic stirring at a rate of 700 rev. min−1. All the characteristics of the Sn–O–C composites electrodeposited either with or without electrolyte agitation were closely compared with each other.
The morphological observation and the crystalline structure analysis of the electrodeposited Sn–O–C composites were conducted by field emission scanning electron microscopy (FE-SEM, Hitachi, S-4500), grazing incidence X-ray diffraction (GIXRD, Rigaku, Rint-Ultima III) and field emission scanning transmittance electron microscopy (FE-STEM, JEOL, JEM-2100F) with energy dispersive X-ray spectroscopy (EDS, JEOL, JED-2300T) for element mapping. The surface chemical state was analysed by X-ray photoelectron spectroscopy (XPS, Physical Electronics, PHI 5000 VersaProbe) with Ar ion etching for 30 s performed at 3 s intervals using 5 kV ion beam voltage. The C 1s hydrocarbon peak was set at 284.8 eV to correct binding energy. For the XPS measurement, the sample was taken in a transfer vessel in the glove box; afterward, it was moved and placed directly into the preparation chamber under vacuum conditions without contacting any atmosphere. The galvanostatic charge/discharge test of the Sn–O–C composite anodes deposited with or without bath agitation were conducted with a loading current of C/10, between 0.01 and 1.1 V vs. Li/Li+ using a charge/discharge unit (Hokuto Denko, HJ-1010M). A three-electrode system containing 1.0 mol L−1 LiClO4 in EC–PC (1:1 v/v) (Kishida, H2O content < 20 ppm) as an electrolyte and lithium foils as counter and reference electrodes was used.
Acknowledgements
This work was partially supported by Advanced Low Carbon Technology Research and Development Program (JST-ALCA) Special Priority Research Area “Next-generation Rechargeable Battery”.Notes and references
- K. T. Lee, Y. S. Jung and S. M. Oh, J. Am. Chem. Soc., 2003, 125, 5652 CrossRef CAS PubMed.
- Y. Yu, L. Gu, X. Lang, C. Zhu, T. Fujita, M. Chen and J. Maier, Adv. Mater., 2011, 23, 2443 CrossRef CAS PubMed.
- X. Li and C. Wang, J. Mater. Chem. A, 2013, 1, 165 CAS.
- D. D. Vaughn II, O. D. Hentz, S. Chen, D. Wang and R. E. Schaak, Chem. Commun., 2012, 48, 5608 RSC.
- X. Zhao, M. Cao and C. Hu, RSC Adv., 2012, 2, 11737 RSC.
- J. S. Chen, M. F. Ng, H. B. Wu, L. Zhang and X. W. Lou, CrystEngComm, 2012, 14, 5133 RSC.
- H. Mukaibo, T. Sumi, T. Yokoshima, T. Momma and T. Osaka, Electrochem. Solid-State Lett., 2003, 6, A218 CrossRef CAS PubMed.
- H. Mukaibo, T. Momma, M. Mohamedi and T. Osaka, J. Electrochem. Soc., 2005, 152, A560 CrossRef CAS PubMed.
- J. Chen, S.-J. Bull, S. Roy, H. Mukaibo, H. Nara, T. Momma, T. Osaka and Y. Shacham-Diamand, J. Phys. D: Appl. Phys., 2008, 41, 13 CrossRef.
- S.-H. Ng, S.-Y. Chew, J.-Z. Wang, J. Chen, S.-X. Doua and H.-K. Liua, Dalton Trans., 2009, 723 RSC.
- H. Zhang, H. Song, X. Chen, J. Zhou and H. Zhang, Electrochim. Acta, 2012, 59, 160 CrossRef CAS PubMed.
- W. Yue, S. Yang, Y. Ren and X. Yang, Electrochim. Acta, 2013, 92, 412 CrossRef CAS PubMed.
- X. Li, Y. Zhong, M. Cai, M. P. Balogh, D. Wang, Y. Zhang, R. Li and X. Sun, Electrochim. Acta, 2013, 89, 387 CrossRef CAS PubMed.
- Y. Xu, J. Guo and C. Wang, J. Mater. Chem., 2012, 22, 9562 RSC.
- H. Nara, Y. Fukuhara, A. Takai, M. Komatsu, H. Mukaibo, Y. Yamauchi, T. Momma, K. Kuroda and T. Osaka, Chem. Lett., 2008, 37(2), 142 CrossRef CAS.
- X. Li, X. Meng, J. Liu, D. Geng, Y. Zhang, M. N. Banis, Y. Li, J. Yang, R. Li, X. Sun, M. Cai and M. W. Verbrugge, Adv. Funct. Mater., 2012, 22, 1647 CrossRef CAS.
- X. Li, A. Dhanabalan, L. Gu and C. Wang, Adv. Energy Mater., 2012, 2, 238 CrossRef CAS.
- E. Paled, J. Electrochem. Soc., 1979, 126(12), 2047 CrossRef PubMed.
- K. Kanamura, S. Shiraishi and Z.-I. Takehara, J. Electrochem. Soc., 1994, 141(9), L108 CrossRef CAS PubMed.
- D. Aurbach, I. Weissman, A. Schechter and H. Cohen, Langmuir, 1996, 12, 3991 CrossRef CAS.
- K. Okita, K.-I. Ikeda, H. Sano, Y. Iriyama and H. Sakaebe, J. Power Sources, 2011, 196, 2135 CrossRef CAS PubMed.
- B. Philippe, R. Dedryvére, J. Allouche, F. Lindgren, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2012, 24, 1107 CrossRef CAS.
- P. B. Balbuena and Y. Wang, Lithium ion secondary batteries solid-electrolyte interphase, Imperial College Press, 2004 Search PubMed.
- A. Funabiki, M. Inaba, T. Abe and Z. Ogumi, J. Electrochem. Soc., 1999, 146(7), 2443 CrossRef CAS PubMed.
- T. Momma, M. Jeong, T. Yokoshima, H. Nara, A. Toyoda and T. Osaka, J. Power Sources, 2013, 242, 527 CrossRef CAS PubMed.
- M. Jeong, T. Yokoshima, H. Nara, T. Momma and T. Osaka, J. Electrochem. Soc., 2014, 161, D3025 CrossRef PubMed.
- W. Chen, Y. He and W. Gao, J. Electrochem. Soc., 2010, 157(8), E122 CrossRef CAS PubMed.
- N. Kanani, Electroplating: Basic Principles, Processes and Practice, Elsevier Advanced Technology, Oxford, 2004 Search PubMed.
- Y. D. Gamburg and G. Zangari, Theory and Practice of Metal Electrodeposition, Springer, 2011 Search PubMed.
- M. Paunovic and M. Schlesinger, Fundamentals of Electrochemical Deposition, Wiley-Interscience, 2006 Search PubMed.
- J.-T. Li, J. Światowska, V. Maurice, A. Seyeux, L. Huang, S.-G. Sun and P. Marcus, J. Phys. Chem. C, 2011, 115, 7012 CAS.
- J. Światowska, V. Maurice, S. Zanna, L. Klein and P. Marcus, Electrochim. Acta, 2007, 52, 5644 CrossRef PubMed.
- S. Malmgrena, K. Cioseka, M. Hahlina, T. Gustafssona, M. Gorgoic, H. Rensmob and K. Edström, Electrochim. Acta, 2013, 97, 23 CrossRef PubMed.
- L. Martina, H. Martinez, D. Poinotb, B. Pecquenardc and F. L. Cras, J. Phys. Chem. C, 2013, 117, 4421 Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, ULVAC-PHI, Inc., 1995 Search PubMed.
- B. Zhang, Y. Yu, Z. Huang, Y.-B. He, D. Jang, W.-S. Yoon, Y.-W. Mai, F. Kangd and J.-K. Kim, Energy Environ. Sci., 2012, 5, 9895 CAS.
- H. Zhang, H. Song, X. Chen and J. Zhou, J. Phys. Chem. C, 2012, 116, 22774 CAS.
- R. Younesi, S. Urbonaite, K. Edström and M. Hahlin, J. Phys. Chem. C, 2012, 116, 20673 CAS.
- D. Aurbach, A. Nimberger, B. Markovsky, E. Levi, E. Sominski and A. Gedanken, Chem. Mater., 2002, 14, 4155 CrossRef CAS.
- J.-S. Bridel, S. Grugeon, S. Laruelle, J. Hassoun, P. Reale, B. Scrosati and J.-M. Tarascon, J. Power Sources, 2010, 195, 2036 CrossRef CAS PubMed.
- T. Momma, S. Aoki, H. Nara, T. Yokoshima and T. Osaka, Electrochem. Commun., 2011, 13, 969 CrossRef CAS PubMed.
- H. Nara, T. Yokoshima, T. Momma and T. Osaka, Energy Environ. Sci., 2012, 5, 6500 CAS.
- H. Nara, T. Yokoshima, M. Otaki, T. Momma and T. Osaka, Electrochim. Acta, 2013, 110, 403 CrossRef CAS PubMed.
- M. Parka, X. Zhanga, M. Chunga, G. B. Lessa and A. M. Sastry, J. Power Sources, 2010, 195, 7904 CrossRef PubMed.
- K. Smith and C.-Y. Wang, J. Power Sources, 2006, 161, 628 CrossRef CAS PubMed.
- J.-I. Lee, J.-H. Park, S.-Y. Lee and S. Park, Phys. Chem. Chem. Phys., 2013, 15, 7045 RSC.
- M. Noh, Y. Kwon, H. Lee, J. Cho, Y. Kim and M. G. Kim, Chem. Mater., 2005, 17, 1926 CrossRef CAS.
- S.-L. Chou, X.-W. Gao, J.-Z. Wang, D. Wexler, Z.-X. Wang, L.-Q. Chend and H.-K. Liu, Dalton Trans., 2011, 12801 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information of HR-TEM, SAED pattern, GIXRD, DCPs. See DOI: 10.1039/c4ra03435j |
| This journal is © The Royal Society of Chemistry 2014 |