
Effect of ligands on the characteristics of (CdSe)13 quantum dots†
Abstract
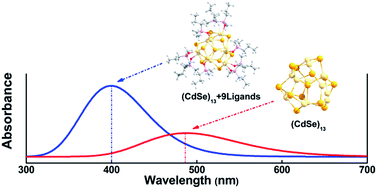
The widespread applications of quantum dots (QDs) have spurred an increasing interest in the study of their coating ligands, which can not only protect the electronic structures of the central QDs, but also control their permeability through biological membranes with both size and shape. In this work, we have used density functional theory (DFT) to systematically investigate the electronic structures of (CdSe)13 passivated by OPMe2(CH2)nMe ligands with different lengths and various numbers of branches (Me = methyl group, n = 0, 1–3) as well as different number of ligands ((CdSe)13 + [OPMe2(CH2)2Me]m (m = 0, 1, 9, 10)). Our results show that the absorption peak in the ultraviolet-visible (UV-vis) spectra displays a clear blue-shift, on the scale of ∼100 nm, upon the binding of ligands. Once the total number of ligands bound with (CdSe)13 reached a saturated number (9 or 10), no more blue-shift occurred in the absorption peak in the UV-vis spectra. On the other hand, the aliphatic chain length of ligands has a negligible effect on the optical properties of the QD core. Analyses of the bonding characteristics confirm that optical transitions are dominantly governed by the central QD core rather than the organic passivation. These findings might provide insights on the material design for the passivation of quantum dots for biomedical applications.
 Please wait while we load your content...
Please wait while we load your content...

