Recent advances in Mn-based oxides as anode materials for lithium ion batteries
Yuanfu Deng*ab,
Lina Wana,
Ye Xiea,
Xusong Qin*b and
Guohua Chenbc
aThe Key Laboratory of Fuel Cell Technology of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou, China. E-mail: chyfdeng@scut.edu.cn
bCenter for Green Products and Processing Technologies, Guangzhou HKUST Fok Ying Tung Research Institute, Guangzhou 511458, China. E-mail: qinxusong@ust.hk
cDepartment of Chemical and Biomolecular Engineering, The Hong Kong University of Science and Technology, Hong Kong, China
First published on 14th May 2014
Abstract
The development of new electrode materials for lithium-ion batteries (LIBs) is of great interest because available electrode materials may not meet the high-energy demands for electronic devices, especially the demands for good cyclic and rate performance. Mn-based oxides have received substantial attention as promising anode materials for LIBs due to their high theoretical specific capacities, low charge potential vs. Li/Li+, environmental benignity and natural abundance. Herein, the preparation of Mn-based oxide nanomaterials with various nanostructures and chemical compositions along with their applications as negative electrodes for LIBs are reviewed. The review covers MnO, Mn3O4, Mn2O3, MnO2, CoMn2O4, ZnMn2O4 and their carbonaceous composite/oxide supports with different morphologies and compositions. The aim of this review is to provide an in-depth and rational understanding of the relationships among the chemical compositions, morphologies and electrochemical properties of Mn-based anode materials and, to understand how electrochemical performance can be improved using materials engineering strategies. Special attention has been paid to the discussion of challenges in the practical applications of Mn-based oxides in LIB full cells.
1. Introduction
Since the beginning of the 21st century, the call for more energy and a cleaner environment has forced many countries to invest large amounts of money in developing environmentally-friendly energy storage systems.1 Compared to lead–acid, nickel–cadmium and nickel–metal hydride batteries, rechargeable lithium-ion batteries (LIBs) have the advantages of high energy density, long cycle life and memory-free effect, making them widely applicable in the portable electronic and electric tool market.2–4 In the near future, they are expected to play even more important roles in electric vehicles (EVs) and plug-in hybrid electric vehicles (PHEVs).4 However, they currently still cannot meet the demands dictated by the powering of EVs and PHEVs or by the large-scale energy storage systems needed for renewable energies. Therefore, a significant improvement in the lithium storage capacity of LIBs is urgently needed to enhance their feasibility for the aforementioned large-scale applications.The charge–discharge mechanism of LIBs is based on the rocking-chair concept. A typical commercial LIB consists of a cathode (e.g., LiCoO2) and an anode (e.g., graphite) together with an electrolyte-filled separator that allows lithium (Li) ion transfer but prevents direct contact between the electrodes. When the battery is charging, Li ions deintercalate from the cathode and intercalate into the anode. Conversely, the Li ions intercalate into the cathode via the electrolyte during discharge. During charge–discharge, Li ions flow between the anode and the cathode, enabling the conversion of chemical energy into electrical energy and the storage of electrochemical energy within the battery. The reactions in a typical battery are LiCoO2 + 6C ↔ Li1−xCoO2 + LixC6 (0 < x < 1).5–8
LIB energy density depends on the capacities and operating potentials of the respective electrode materials. The identification of electrode materials that can yield higher capacity and energy density, e.g., cathode materials with higher operating voltages (≥4.0 V vs. Li/Li+)9–12 and anode materials with lower operating voltages (≤1.0 V vs. Li/Li+), while having low environmental cost is essential.8 Research on anode materials for LIBs has been directed towards new materials that involve three different mechanisms: (1) intercalation–deintercalation mechanism, e.g., compounds with a two-dimensional (2D) layer structure13 or a three-dimensional (3D) network structure14 that can reversibly intercalate/deintercalate Li+ into/out of the lattice; (2) elements (M) that can form alloys with Li metal (LixM), e.g., Si,15–17 Sn/SnO2,18–23 Ge,24 Sb,25 Zn,26 In, Bi, and Cd;27 and (3) materials that can act as an anode in the so-called redox or “conversion” reaction with Li.28 These materials are mainly oxides, fluorides, oxyfluorides, sulfides, nitrides and phosphides.29 As a consequence, a large number and wide variety of metal-containing oxides including binary and complex oxides have been explored for Li cycling.8,30–32 In addition, modern nanotechnology has been employed to prepare many metal oxides as anode materials for LIBs; these materials contain various morphologies and structures for Li cycling.7 As a result, research on metal oxides as anodes for LIBs has made significant progress in recent years.8
Among the metal oxides used as anode materials for LIBs, Mn-based oxides have obvious advantages because of their high specific capacities and low toxicity, cost, and operating voltage (average discharge and charge voltages of 0.5 V and 1.2 V vs. Li/Li+, respectively) compared to the iron,33–35 cobalt36–38 and Ni-based oxides.39,40 Table 1 lists the experimental values of the plateau potentials and theoretical specific capacities associated with conversion reactions in Mn-based oxides.41–45 In a Li/MnxO half-cell, the electrochemical processes can be summarized by the following equation:
| MnOx + 2xLi+ + 2xe− ↔ Mn + xLi2O |
This reaction for Li storage in Mn-based oxides based on the “conversion mechanism” has four major obstacles for practical application:29 (1) the strong structural reorganization that must take place to accommodate the chemical changes induces large changes in volume, resulting in severe particle agglomeration and unsatisfactory cycling performance; (2) the low rate performance arising from their low conductivity; (3) an unacceptably large voltage hysteresis in terms of round-trip energy density loss, which is observed between the discharge and charge steps; and (4) a virtually ubiquitous large coulombic inefficiency observed in the first cycle.
Different strategies have been proposed to resolve the aforementioned problems of Mn-based anodes, and four of them are widely practiced. The first strategy is to fabricate nanosized particles to shorten the diffusion length for electrons and lithium ions.46 The second strategy is to synthesize Mn-based oxides with porous structures containing porous 1D nanorods/nanowires47 and hollow nanotubes,48 2D nanoflakes,49 and 3D hollow or hierarchical porous nanostructures.50 The vacant space provided by the hollow or porous structures can accommodate the structural strain to facilitate fast Li+ insertion/extraction kinetics by enabling electrolyte uptake through the nanoparticles; this leads to improved rate and cycling performance. The third strategy is to introduce a considerable proportion of carbon into the Mn-based oxides to form homogenous oxides/carbon nanocomposites.46 It has been suggested that the carbon-based materials can provide a cushioning effect against the volume strain because of their elastic character. Meanwhile, carbon-based materials could also increase the electrical conductivity of the nanocomposites, which is essential to the rate performance and cyclability. Carbon is also active at the low voltages at which some of the conversion reactions occur, partially compensating for the decrease in capacity associated with the dilution of the active phase. The fourth strategy is to optimize the electrodes by adjusting the ratios of active materials, binders and conductive agents or by choosing novel binders/conductive agents.51 The proper selection of the ratios of these materials has been demonstrated to generate significant improvement in the rate and cyclic performance of metal oxides-based52 and other anode materials.51 Because of the potential applications of nano-scale Mn-based oxides as anode materials for LIBs, it is of great importance to have a more detailed discussion on these kinds of materials along with their various nanostructures, chemical compositions and electrode fabrication processes.
Cabana et al.29 gave an excellent review of the different conversion reactions of metal oxides, sulfides, nitrides, phosphides, and fluorides in 2010; the development of Mn-based oxides as anode materials for LIBs before 2010 were extensively reviewed in this work. Wu et al.30 gave another good review on the topic of metal oxides as anode materials for LIBs in 2012. Reddy et al.8 reviewed the detailed development of metal oxides and oxysalts as anode materials for LIBs. In addition to highlighting some of the recent advances in Mn-based oxides as anode materials for LIBs, these three excellent review papers demonstrated that the field of metal oxides as anode materials is a very active and important LIB research area. Although some recent advances on this topic have been reviewed in the aforementioned papers, continuous updates are still required to reflect the rapid developments of strategies for improving the electrochemical performance of Mn-based oxides. In order to better understand the status of research on this topic, this review mainly discusses the recent developments, current status and major strategies related to the performance enhancement of Mn-based oxides with various structures and chemical compositions. The oxides reviewed in this work include MnO, Mn3O4, Mn2O3, MnO2, CoMn2O4 and ZnMn2O4 as well as their carbon/metal oxides-based composites. The key challenges and opportunities for their future developments and applications are also discussed. Given the broad nature and rapid development of the field of Mn-based oxides, the authors apologize for the inevitable oversights of some important contributions.
2. Binary Mn-based oxides as anode materials
Mn-based binary oxides have been widely investigated as anode materials for LIBs in the past decades after the works of Tarascon et al. in 2000.28 Many binary Mn-based oxides including MnO, Mn3O4, Mn2O3, MnO2 and their carbon-based nanocomposites with different nano-structures have found useful applications in LIBs.82.1 MnO and its nanocomposites
Although MnO has a high theoretical capacity of 755.6 mA h g−1, which is twice as the capacity of graphite, and a lower electromotive force (1.032 V vs. Li/Li+) than other transition metal oxides (TMO) anodes such as Fe2O3, Co3O4, NiO, CuO, and so forth, few examples of the Li cyclability of pure MnO anode materials have been reported in recent years.46,49,53,54 This may be attributed to the fact that manganese is one of the first row transition metals, which are very difficult to be reduced into the metallic state.29 Zhong et al.46 investigated commercial MnO powders as anode-active materials for LIBs. According to the results of ex situ XRD, TEM and a galvanostatic intermittent titration technique, they suggested that lithium is stored reversibly in MnO through the conversion reaction and an interfacial charging mechanism. In addition, they found that a layer of the solid electrolyte interphase with a thickness of 20–60 nm is covered on MnO particles after full insertion. The cyclic performance of MnO is significantly improved by decreasing particle size. Following this work, MnO anode materials have been fabricated with microsphere,53 nanoflake49 and thin-film54 forms to improve their cycling and rate performance. In 2011, Zhong et al.53 synthesized porous MnO microspheres by decomposing MnCO3 under argon atmosphere at 600 °C for 1 h. The as-prepared MnO material shows a reversible capacity of 800 mA h g−1, and an initial coulombic efficiency of 71%. It can deliver 600 mA h g−1 at a rate of 400 mA g−1. They further confirmed the conversion reaction mechanism by Mn K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopies, indicating that pristine MnO is reduced to Mn0 after discharging to 0 V, and that part of the reduced Mn0 is not oxidized to Mn2+ after charging to 3 V. Li et al.49 developed a novel method for the preparation of porous MnO nanoflakes on nickel foam (Fig. 1a–d) by the reduction of a hydrothermally-synthesized MnO2 precursor under hydrogen. The porous MnO nanoflakes showed good capacity retention capability and rate performance (Fig. 1e), with a capacity of 568.7 and 708.4 mA h g−1 at the 2nd and 200th charge–discharge cycles, respectively, at a current density of 246 mA g−1; the capacity was 376.4 mA h g−1 at a current density as high as 2460 mA g−1. Thin films of MnO were successfully deposited on Cu foils by radio-frequency (RF) sputtering under an Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 (95:5 vol%) reduction atmosphere at 500 °C.54 These films have approximately 0.5 mm thicknesses and exhibit an initial coulombic efficiency of 75%; after 100 cycles, their reversible specific capacity is 700 mA h g−1 and 428 mA h g−1 at 0.05 C and 20 C, respectively. These values demonstrate that the sputter-grown MnO films exhibit excellent cyclability and rate performance in comparison with MnO powders. This study suggests that pure phases with low oxidation states and certain porosities might be favourable, accounting for the improved electrochemical performance.
:H2 (95:5 vol%) reduction atmosphere at 500 °C.54 These films have approximately 0.5 mm thicknesses and exhibit an initial coulombic efficiency of 75%; after 100 cycles, their reversible specific capacity is 700 mA h g−1 and 428 mA h g−1 at 0.05 C and 20 C, respectively. These values demonstrate that the sputter-grown MnO films exhibit excellent cyclability and rate performance in comparison with MnO powders. This study suggests that pure phases with low oxidation states and certain porosities might be favourable, accounting for the improved electrochemical performance.
 | ||
| Fig. 1 (a and b) Low-magnification SEM images of the samples before and after reduction; (c and d) high-magnification SEM images of the samples before and after reduction; and (e) the discharge capacity of MnO nanoflakes at various current densities. Reproduced with permission.49 Copyright 2012, RSC. | ||
In addition to phase-pure MnO, MnO-based nanocomposites have also been intensively studied as negative electrodes due to their improved electrochemical properties (Table 2).46,55–74 The group of Li46 synthesized carbon-coated commercial MnO (MnO-L) by ball milling with sugar followed by pyrolysis at 600 °C in Ar atmosphere (C/MnO-L). These materials have a reversible capacity of ∼650 mA h g−1 at 50 mA g−1 (0.08 C), and are almost stable up to 150 cycles in the voltage range of 0.01–3.0 V. A good rate performance (400 mA h g−1 at a rate of 400 mA g−1) was also found. Additionally, they demonstrated that the C/MnO-L nanocomposites have a lower voltage hysteresis (<0.7 V with a charging voltage of 1.2 V) during the discharge–charge cycles compared to other metal oxides.
| Materials | Preparation method | 1st discharge–charge capacity (mA h g−1) | Capacity retention | Rate performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| MnO powder | Commercial + ball milling | ∼1266/766 at 50 mA g−1 | 550 (10 cycles) | 400 at 400 mA g−1 | 46 |
| MnO microspheres | Precipitation + annealing | 1126/800 at 50 mA g−1 | 704 (50 cycles) | 600 at 400 mA g−1 | 53 |
| Porous MnO nanoflakes | Hydrothermal method | 781/570 at 246 mA g−1 | 648 (100 cycles) | 376 at 2460 mA g−1 | 49 |
| Crystalline MnO films | Radio-frequency (RF) sputtering + annealing | 550/413 at 0.05 C | 350 (100 cycles) | 150 at 20 C | 54 |
| Amorphous MnOx/C | Aerosol spray pyrolysis | 1083/650 at 200 mA g−1 | 601 (130 cycles) | 500 at 800 mA g−1 | 55 |
| MnOx/OMC nanocomposite | Wet-impregnation + annealing | 1390/860 at 100 mA g−1 | 950 (50 cycles) | 500 at 2000 mA g−1 | 56 |
| Mesoporous MnO–C | Microwave-polyol process + annealing | 1456/1000 at 200 mA g−1 | 1224 (200 cycles) | 731 at 1500 mA g−1 | 57 |
| Monodisperse MnO–C hollow microspheres | A facile biotemplating technique | 1021.9/755.6 mA g−1 | 702.2 (50 cycles) | 230 at 3000 mA g−1 | 58 |
| (MnO/MWNTs) | Hydrothermal + annealing | 1050/686 at 14.4 mA g−1 | 484 (200 cycles) at 144.1 mA g−1 | 455 at 310 mA g−1 | 59 |
| MnO-attached graphene | Impregnation + annealing | 685/635 at 0.2 C | 490 (30 cycles) at 1C | 410 at 5 C | 60 |
| MnO nanoparticles/graph-ene composite | In situ carbothermal reduction | 1080/700 at 100 mA g−1 | 782 (60 cycles) | 402 at 1000 mA g−1 | 61 |
| Nitrogen-doped MnO/graphene nanosheets | Hydrothermal method + ammonia annealing | 1193/829 at 50 mA g−1 | 772 (90 cycles) at 100 mA g−1 | 202 at 5000 mA g−1 | 62 |
| (MnO/RGOS) | Liquid phase deposition + annealing | 1001/648 at 100 mA g−1 | 666 (50 cycles) | 454 at 400 mA g−1 | 63 |
| MnO/graphene | Liquid phase deposition + annealing | 900/891 at 200 mA g−1 | 2014 (150 cycles) | 843 at 2000 mA g−1 | 64 |
| Coaxial MnO–C nanotubes | Self-templates method | 1014/678 at 75.5 mA g−1 | 600 (10 cycles) | 500 at 188.9 mA g−1 | 65 |
| MnO–C core–shell nanorods | In situ reduction method | 1090/780 at 200 mA g−1 | 600 (40 cycles) | 66 | |
| MnO@carbon core–shell nanowires | Solvothermal + annealing | 1196/840 at 100 mA g−1 | 801 (200 cycles) at 500 mA g−1 | 462 at 2000 mA g−1 | 66 |
| MnO@1-D carbon composites | Annealing of C4H4MnO6 | 1249/738 at 500 mA g−1 | 660 (1000 cycles) | 250 at 6 Ag−1 | 67 |
| Porous MnO–C nanotubes | Hydrothermal method + annealing | 1129/810 at 100 mA g−1 | 763 (100 cycles) | 303 at 3200 mA g−1 | 68 |
| MnO–C nanorods | Hydrothermal method + annealing | 1408/707 at 200 mA g−1 | 481 (50 cycles) | 252 at 1000 mA g−1 | 69 |
| Porous carbon-modified MnO disks | Microwave-polyol process + annealing | 1387/997 at 100 mA g−1 | 1044 (140 cycles) | 535 at 1000 mA g−1 | 70 |
| Nano-MnO–C composites | An alcoholysis process + annealing | 1650/900 at 75.5 mA g−1 | 939 (30 cycles) | 589 at 7550 mA g−1 | 71 |
| MnO@C core–shell nanoplates | Hydrothermal method + annealing | 1365/770 at 200 mA g−1 | 563 (30 cycles) | 550 at 300 mA g−1 | 72 |
| MnOx/C nanocomposite | Sol–gel method + annealing | 1055/700 at 200 mA g−1 | 500 (100 cycles) | 320–345 at 2000 mA g−1 | 73 |
| Hierarchical micro/nanostructured MnO | Hydrothermal method | 1051/750 at 98 mA g−1 | 783 (200 cycles) | 350 at 1572 mA g−1 | 74 |
Inspired by the positive effects of carbon-coating, the MnO–C nanocomposites as anode materials for LIBs have received considerable interest.55–59 For example, unique amorphous MnOx–C nanocomposite particles with interspersed carbon have been synthesized using aerosol spray pyrolysis.55 The amorphous MnOx–C nanoparticles showed a high reversible capacity of approximately 650 mA h g−1 under a current density of 200 mA g−1, with an exceptional capacity retention of 93% after 130 cycles. The coulombic efficiency of the amorphous MnOx–C remained stable during cycling after the first few cycles. They suggested that the observed 100% coulombic efficiency is attributed to the absence of new SEI formation during cycling, where carbon blocks liquid electrolyte penetration into MnOx–C particles, reducing the contact area between MnOx and the electrolytes. Chae et al.56 obtained an MnOx/OMC (OMC = ordered mesoporous carbon) nanocomposite by a simple wet-impregnation of Mn(NO3)2 aqueous solution onto OMC nanorods followed by thermal treatment at 450 °C in an Ar flow (Fig. 2a). The MnOx/OMC composite exhibited a high reversible capacity (>950 mA h g−1) after 50 deep charge–discharge cycles; it also exhibited excellent cycling stability (Fig. 2b), acceptable coulombic efficiency (97%) and good rate capability (750 mA h g−1 at 1000 mA g−1) (Fig. 2b and c). The incorporation of insulating and high density MnOx into OMC nanorods resulted in synergistic benefits including higher volumetric and specific capacities, and a smaller redox voltage hysteresis compared to OMC nanorods.
 | ||
| Fig. 2 (a) Schematic diagram for the preparation of MnOx-loaded OMC nanorods; (b) specific capacities of OMC nanorods and MnOx/OMC with cycle number; and (c) rate performances of MnOx/OMC, OMC nanorods and graphite cycled at different current densities. Reproduced with permission.56 Copyright 2012, RSC. | ||
Very recently, Luo et al.57 developed an efficient microwave-polyol process with subsequent thermal treatment for the synthesis of MnO nanoparticles encapsulated uniformly in 3D mesoporous interconnected carbon networks (MnO-MICN). Sodium citrate plays an important role in the morphology of the Mn-based precursors. Interestingly, the specific capacities (∼30% of the overall capacity) of the nanocomposite anode materials gradually increases after 100 cycles. The authors suggested that a new electrochemical reaction might be occurring; for instance, Mn2+ may be oxidized to a higher oxidation state. After 200 cycles at 200 mA g−1, the specific discharge capacity reaches 1224 mA h g−1 with a coulombic efficiency of ∼99%. Moreover, a specific capacity of 731 mA h g−1 is retained after 200 cycles at 1500 mA g−1, indicating good rate performance of the nanocomposites. Inspired by natural microalgae that have desirable characteristics (easy availability, biological activity, and a carbon source), Xia et al.58 developed a green and facile biotemplating method to fabricate monodisperse MnO–C microspheres. Due to the unique hollow porous structure where MnO nanoparticles were tightly embedded in a porous carbon matrix, forming a penetrative shell, the MnO–C microspheres exhibited a high reversible specific capacity of 700 mA h g−1 at 0.1 A g−1, excellent cycling stability with 94% capacity retention after 50 cycles, and good rate performance of 230 mA h g−1 at 3 A g−1. More recently, manganese monoxide nanocomposites and multi-walled carbon nanotubes (MnO/MWNTs) have been hydrothermally prepared by MWNTs and KMnO4 followed by a sintering process.59 These nanocomposites also show improved electrochemical performances as anode materials for LIBs, with a reversible capacity as high as 770.6 mA h g−1 at the current density of 7.21 mA g−1 and good capacity retention (over 92.4% after 200 cycles at 216.14 mA g−1 between 0.01 and 3.0 V).
The strategy of preparing MnO/graphene composites has been demonstrated to be an effective route to obtain enhanced lithium-storage properties because graphene has superior conductivity, large surface area, structural flexibility, and chemical stability. In 2011, a composite of graphene nanosheets (GNs) supported by MnO nanocrystals was fabricated through a chemical-wet impregnation followed by thermal reduction.60 The MnO-attached GN anode delivers a reversible capacity of 635 mA h g−1 at 0.2 C at voltages between 0.01 and 3.5 V, a high coulombic efficiency (92.7%) during the 1st cycle and good rate capability [capacity retention (5/0.2 C: >70%)]. They also investigated the effect of Mn/C atomic ratio on coulombic efficiency and found that the reversibility of Li+ intercalation/deintercalation gradually increases with the Mn/C atomic ratio. This result may be due to two major reasons: (i) Mn ions tend to terminate dangling bonds located at the edges of GNs, forming MnO-terminated edges of graphene fragments, and (ii) the appropriate amount of MnO spacers tend to stabilize the GN network, offering more active sites for reversible Li-storage. After this work, MnO/graphene nanocomposites with different MnO structures have been synthesized by different routes and used as anode materials for LIBs. Qiu et al.61 prepared a MnO nanoparticle/graphene composite (the diameters of the MnO nanoparticles range from 20 to 250 nm) via in situ carbothermal reduction of Mn3O4 on the surface of graphene nanosheets. The detachment and agglomeration of MnO nanoparticles were effectively prevented by the tight interface between the MnO nanoparticles and graphene. The MnO nanoparticle/graphene composites show a high specific capacity of approximately 700 mA h g−1 at 100 mA g−1, excellent cyclic stability, and good rate capability as an anode material for LIBs. Zhang et al.62 synthesized a nitrogen-doped MnO/graphene nanosheet (N-MnO/GNS) hybrid material by a hydrothermal method followed by ammonia annealing (Fig. 3a). Because of its unique N-doped nanostructure and efficiently-mixed conducting network, this hybrid nanostructure exhibits a reversible electrochemical lithium storage capacity as high as 772 mA h g−1 at 100 mA g−1 after 90 cycles (Fig. 3b), and an excellent rate capability of 202 mA h g−1 at a high current density of 5 A g−1 (Fig. 3c).
 | ||
| Fig. 3 (a) Schematic illustration of the preparation of N-MnO/GNS hybrid materials; (b) comparison of cycling performance of N-MnO, N-GNS, and N-MnO/GNS hybrids at a current density of 100 mA g−1; and (c) rate capability of N-MnO and N-MnO/GNS at various current densities. Reproduced with permission.62 Copyright 2012, ACS. | ||
Mai et al.63 synthesized a MnO/reduced graphene oxide sheet (MnO/RGOS) hybrid by a two-step electrode design consisting of a liquid phase deposition of MnCO3 nanoparticles on the surface of graphene oxide sheets followed by heat treatment in nitrogen flow. The MnO/RGOS hybrid electrode shows a reversible capacity of 665.5 mA h g−1 after 50 cycles at a current density of 100 mA g−1 and delivers 454.2 mA h g−1 at a rate of 400 mA g−1. The MnO/RGOS hybrid electrode exhibits an enhanced reversible capacity with respect to that of the MnO electrode, which is attributed to the re-oxidation of the Mn/Li2O nanocomposite to MnO nanoparticles. In addition, the authors suggested that the incorporation of RGOS is not very effective in decreasing the hysteresis voltage of the MnO electrode by the galvanostatic intermittent titration technique (GITT) as well as by the electrochemical impedance spectroscopy (EIS). This remarkable hysteresis has both thermodynamic and kinetic origins. In 2013, Sun et al.64 reported a facile solution-based method combined with a subsequent reduction process for the large-scale fabrication of a MnO/graphene nanocomposites. The prepared MnO/graphene hybrid anode exhibits an initial reversible capacity of around 890.7 mA h g−1 at a current density of 200 mA g−1 and a reversible capacity as high as 2014.1 mA h g−1 after 150 discharge–charge cycles (Fig. 4). In addition, the composite shows excellent rate capability (625.8 mA h g−1 at 3000 mA g−1) and superior cyclability (843.3 mA h g−1 even after 400 cycles at 2000 mA g−1 with only 0.01% capacity loss per cycle). Its specific capacity is much higher than the theoretical capacities of both MnO (∼756 mA h g−1) and graphene (∼744 mA h g−1). The lithiation and delithiation behaviours suggest that the further oxidation of Mn(II) to Mn(IV), similar to the recent report of Luo et al.,57 and the interfacial lithium storage upon cycling contribute significantly to the enhancement of specific capacity.
 | ||
| Fig. 4 (a) Discharge and charge voltage profiles of the synthesized MnO/graphene anode at a current density of 200 mA g−1; (b) cycling performance of the prepared MnO/graphene hybrid electrode cycled at 200 mA g−1 and (c) at various current densities of 200, 400, 800, 1200, 1600, 2000, and 3000 mA g−1; and (d) discharge and charge curves at a current density of 2000 mA g−1. All were carried out in the voltage range 3–0.01 V vs. Li. Reproduced with permission.64 Copyright 2013, Wiley. | ||
One dimensional (1D) nanostructures have attracted considerable attention as anode materials for LIBs since they allow better accommodation of volume changes during repetitive charge–discharge cycles. 1D nanostructures also possess direct 1D electronic pathways for efficient charge transport. In 2011, 1D coaxial MnO–C nanotubes with an average diameter of approximately 450 nm, a wall thickness of approximately 150 nm, a length of 1–5 μm and a 10 nm thick carbon layer were first reported by Ding et al.65 using β-MnO2 nanotubes as self-templates in acetylene at 600 °C. This sample exhibits a reversible capacity of about 500 mA h g−1 at a current density of 188.9 mA g−1 and a capacity retention of 83.9%. Although the as-prepared sample shows better lithium storage properties compared to bare MnO nanotubes (58.2%) and MnO nanoparticles (25.8%), its electrochemical performance is still not satisfactory due to a quick fading in capacity. The enhanced reversible capacity and cycling stability for coaxial MnO–C-NTs are attributed to the conductive carbon layer on the MnO nanotubes, which can provide improved electronic transport to the MnO nanotube core and can be considered as a buffering layer to alleviate the volume changes caused by repetitive cycling. In recent years, the reduction of MnOx (x > 1) with special nanostructural features has been more widely practiced. The group of Wang66 synthesized MnO–C core–shell nanorods by an in situ reduction method using MnO2 nanowires as a precursor and block copolymer F127 as a carbon source in a gas flow of 5 vol% H2 in Ar. As anode materials for lithium ion batteries, the as-prepared MnO–C core–shell nanorods exhibit a higher specific capacity than MnO microparticles; they have a specific discharge capacity of approximately 700 mA h g−1 after 40 cycles under a current density of 200 mA g−1. Li et al.67 reported a facile synthetic method for the preparation of MnO@carbon core–shell nanowires with a jointed appearance based on the reduction of Mn2O3 precursors followed by graphitization of C2H2. The MnO@carbon core–shell nanostructures could deliver reversible capacities as high as 801 mA h g−1 at a high current density of 500 mA g−1, with excellent electrochemical stability over 200 cycles. The remarkable electrochemical performance is mainly attributed to the highly-uniform carbon layer around the MnO nanowires, which is not only effective in buffering the structural strain and volume variations of anodes during repeated electrochemical reactions, but also greatly enhances the conductivity of the electrode material. They also suggested that their strategy is simple but very effective, and appears to be sufficiently versatile, with the ability to be extended to other high-capacity electrode materials with large volume variations and low electrical conductivities. More recently, Zhu's group68 developed a novel route to synthesize MnO@1D carbon composites through a pyrolysis process using C4H4MnO6 as the single-molecule precursor. The MnO nanoparticles are uniformly dispersed inside or adhered to the surface of the 1D carbon nanotubes, which overlap each other to form carbon scaffolds. This special structural feature not only improves the electronic conductivity, but also provides a support for loading MnO nanoparticles, resulting in a high specific capacity (1482 mA h g−1 at 200 mA g−1), good rate performance (420 mA h g−1 at 2.0 A g−1) and excellent capacity retention capability (810 mA h g−1 at 1460 mA g−1 after 1000 cycles).
The solvo/hydrothermal approach has been demonstrated as a suitable route for the preparation of 1D MnO–C nanocomposites. For examples, Sun's group48 developed and employed a facile hydrothermal method followed by thermal annealing to synthesize porous MnO–C nanotubes (Fig. 5). Electrochemical results demonstrate that the porous MnO–C nanotubes can deliver a reversible capacity as high as 763.3 mA h g−1 after 100 cycles at a charge–discharge current density of 100 mA g−1 (0.13 C, 1 C = 755.6 mA g−1); the capacity is 618.3 mA h g−1 after 200 cycles at a rate of 0.66 C (Fig. 6). The authors suggested that the superior cyclability and rate capability are attributed to the hollow interior, porous structure, the 1D structure and the uniformly-dispersed carbon in the porous MnO–C nanotubes. Su et al.69 reported a two-step hydrothermal treatment and subsequent sintering at 600 °C to prepare hierarchical MnO@C nanorods using KMnO4 and citric acid as raw materials. The ultra-small MnO nanocrystals (<5 nm) were homogeneously dispersed in a carbon matrix and further coated with a well-proportioned carbon shell. This sample exhibits a reversible capacity of 481 mA h g−1 after 50 cycles at a current density of 200 mA g−1, which is higher than that of carbon-coated MnO nanoparticles.
 | ||
| Fig. 5 (a) Low and (b) high magnification SEM images; (c) TEM image and (d) XRD pattern of the porous MnO–C nanotubes. Reproduced with permission.48 Copyright 2012, RSC. | ||
 | ||
| Fig. 6 (a) Charge–discharge curves and (b) cycle performances of commercial MnO particles (short dash dot) and the porous MnO–C nanotubes (solid) at a charge–discharge rate of 0.13 C; (c) cycle performance of the porous MnO–C nanotubes at a charge–discharge rate of 0.66 C; (d) rate performance of the porous MnO–C nanotubes. Reproduced with permission.48 Copyright 2012, RSC. | ||
The 2D nanostructures of MnO-based nanocomposites are not as well-researched as their 1D counterparts, likely due to the difficulty in fabricating well-defined MnO-based nanocomposites. Sun et al.70 prepared porous carbon-modified MnO disks with approximately 50 nm average thicknesses and up to 3 mm diameters by a microwave-polyol process followed by thermal annealing of the disk-like Mn-complex precursor. In their work, they demonstrated that each C–MnO disk has a single-crystal-like nature and is built up by the assembly of ∼12 nm carbon-modified MnO nanocrystals having the same crystallographic orientations. Because the unique assembled nano-architecture involves three-dimensionally interconnected nanopores and carbon modification as well as small MnO nanocrystal particle sizes, the as-synthesized C–MnO nanocomposites exhibit high capacities (an initial capacity of 1387.2 mA h g−1 with a coulombic efficiency of 71.9% at a current density of 100 mA g−1 in the first cycle) and excellent cycling stability (a reversible capacity of 1044.2 mA h g−1 after 140 cycles at 100 mA g−1). It is also worthy to note that the capacity drops firstly and then increases upon cycling, which has been observed in the case of MnO–C,53 MnO/reduced graphene oxide63 and MnOx/C.55 The authors suggested that the mechanism behind the U-shaped capacity retention curve is not clear, but may be attributed to the formation of high oxidation state products49 or the mixed effects of Mn cluster aggregation and reversibility improvement of the conversion reaction in manganese oxide due to the formation of defects and deformations.55 Although the carbon materials (amorphous carbon, carbon nanotube and graphene) have apparent beneficial effects on the electrochemical cycling and rate performance of MnO-based composites, they have very low electrochemical activity and limited lithium storage capacity. Therefore, the carbon content in the composites needs to be carefully adjusted in order to find a balance between electrode capacity and cycle life. Chen's group71 synthesized carbon-coated nano-MnO powders (MnO–C) with different carbon contents by the reduction of Mn3O4 with a particle size of about 20 nm, using glucose as both a carbon source and a reducing agent. The electrode fabricated using 10.7 wt% carbon-coated MnO–C shows the best cycling stability and rate performance, with a high reversible capacity of 939.3 mA h g−1 after 30 cycles at 0.1 C. By controlling the treatment temperature and reaction time, Zhang et al.72 synthesized MnO@C core–shell nanoplates with controllable carbon shell thicknesses via the thermal treatment deposition of acetylene on Mn(OH)2 nanoplate precursors (Fig. 7a). By controlling the temperature of carbon deposition and the reaction time, the thickness of the carbon shell can be tuned from 3.1 to 13.7 nm. The electrochemical experimental results show that the MnO@C nanoplates with a carbon shell thickness of 8.1 nm display a higher reversible capacity (770 mA h g−1 at a current density of 200 mA g−1) and better cyclability than those with carbon shell thicknesses of 3.1, 4.0, 4.2, 10.9 and 13.7 nm (Fig. 7b).
 | ||
| Fig. 7 (a) Schematic illustrating the formation process of MnO@C; (b) discharge capacity vs. cycle number for MnO@C products synthesized at different conditions at a current density of 200 mA g−1; and (c) the rate performance of the MnO@C products synthesized at 550 °C for 10 h. Reproduced with permission.72 Copyright 2012, RSC. | ||
In summary, results from this series of studies demonstrate the high capacities of MnO-base composites and the improved cyclic and rate performances generated by many strategies; however, these data were obtained using Li/MnO half-cells. At present, few papers73,74 have reported full cells using MnO-based oxides as anodes and LiMn2O4 or LiNi0.5Mn1.5O4 as cathodes. A lithium ion battery using LiMn2O4 as the cathode and MnOx/mesoporous carbon as the anode was reported by Lee et al.73 The capacity of the MnOx/C anode decayed gradually from 700 mA h g−1 to 500 mA h g−1 after 100 cycles, and the MnOx/mesoporous carbon/LiMn2O4 full cell operates at an average working voltage of 3.3 V with a capacity of 105 mA h g−1 (Fig. 8).
 | ||
| Fig. 8 (a) Voltage profile; (b) cycling performance of LMO–MnOx/C (A) cell at 0.2 C for 100 cycles and then at 0.1 C for up to 188 cycles; (c) cycling performance of LMO–MnOx/C (B) cell at 0.2 C for up to 195 cycle; and (d) rate performance of LMO–MnOx/C (B) cell. Voltage cut-off range is between 2.0 and 4.1 V. Reproduced with permission.73 Copyright 2013, Elsevier. | ||
In a recent paper, Sun's group74 successfully developed a facile route to synthesize a hierarchical micro/nanostructured MnO anode and a spinel LiNi0.5Mn1.5O4−δ cathode using olive-shaped hierarchical micro/nanostructured MnCO3 as a precursor. Both the MnO and LiNi0.5Mn1.5O4−δ materials possess excellent lithium storage properties due to their unique hierarchical micro/nanostructures. The MnO in a MnO/Li half-cell can deliver a reversible capacity of 782.8 mA h g−1 after 200 cycles at a rate of 0.13 C along with a stable reversible capacity of 350 mA h g−1 at a high rate of 2.08 C. A full lithium ion battery using MnO as anode and LiNi0.5Mn1.5O4–δ as cathode demonstrates a high discharge specific energy ca. 350 W h kg−1 after 30 cycles at 0.1 C, an average working voltage at 3.5 V and long cycle stability. The MnO/LiNi0.5Mn1.5O4−δ full cell can maintain a discharge specific energy of 227 W h kg−1 after 300 cycles at a higher rate of 0.5 C (Fig. 9). Considering these two successful examples, full cells assembled with MnO-based nanocomposite anodes may present a promising potential application for next-generation LIBs.
 | ||
| Fig. 9 (a) Charge–discharge profiles and (b) cycling performance of the MnO/LNMO full cell at a rate of 0.1 C; (c) cycling performance of the MnO/LNMO full cell at a rate of 0.5 C; and (d) the discharge specific energies of the MnO/LNMO full cell at different rates (0.05 C charge). Reproduced with permission.74 Copyright 2013, ACS. | ||
2.2 Mn3O4 and its nanocomposites
In 1983, the group of Goodenough75 reported the electrochemical insertion of Li into Mn3O4. However, few studies75–78 have reported this material as an anode for LIBs, partially due to its extremely low electrical conductivity (∼10−7 to 10−8 S cm−1), limiting its capacity to ∼400 mA h g−1, even with Co doping. The optimization of synthetic conditions is therefore necessary to obtain single-phase Mn3O4 materials. In 2010, Li's group79 reported a facile solvothermal process for the controllable synthesis of Mn3O4 nanocrystals with different sizes and shapes including dots, rods, and wires in the presence of the surfactants dodecanol and oleylamine. The as-prepared monodisperse nanocrystals, acting as ideal building blocks, can be rationally assembled into 3D Mn3O4 colloidal spheres using a facile ultrasonication strategy. This paper gave a good example of the preparation of pure Mn3O4 nanomaterials; subsequently, this mixed-valent oxide (Mn3O4) and its nanocomposites have been extensively investigated as anode materials by the “conversion reaction” of lithium, and they continue to be studied (Table 3).80–89 Shen et al.80 synthesized Mn3O4 nanorods with diameters of 50–150 nm and lengths of approximately 1–2 μm via the heat-treatment of MnCO3 nanorods, which were obtained from a facile hydrothermal method, in nitrogen atmosphere. When applied as anode materials for LIBs, the Mn3O4 nanorods exhibited a reversible lithium storage capacity of 1050 mA h g−1 in the first few cycles. However, the pure Mn3O4 nanorods show a rapid capacity loss with increasing cycle number; the capacity decreased to 108 mA h g−1 at the 100th cycle. Engineering the morphology of Mn3O4 nanomaterials has been an effective strategy to enhance their electrochemical performance. Therefore, other pure Mn3O4 materials with different morphologies and microstructures including octahedral Mn3O4 crystals with exposed high-energy facets,81 sponge like Mn3O4 nanostructures,43 mesoporous stacked Mn3O4 nanosheets,82 foam-like porous Mn3O4,83 mesoporous Mn3O4 nanotubes,84 order-aligned Mn3O4 nanostructures85 and Mn3O4 octahedrons with typical diameters around 300–400 nm (ref. 86) have been synthesized and intensively investigated as anode materials for LIBs. In these Mn3O4 nanomaterials, the mesoporous Mn3O4 nanotubes84 and Mn3O4 octahedra86 show high specific capacities and excellent cycling performances. Specifically, the mesoporous Mn3O4 nanotubes84 with a high surface area (42.18 m2 g−1) and an average pore size of 3.72 nm obtained from the hydrogen reduction of β-MnO2 nanotubes under a H2/Ar atmosphere at 280 °C for 3 h exhibit a high specific capacity of 641 mA h g−1 after 100 cycles at a high current density of 500 mA g−1 (Fig. 10). The Mn3O4 octahedra,86 which were straightforwardly prepared at room temperature using a one-step de-alloying method (Fig. 11a), have ultra-long cycle lifetimes with capacity retentions of 81.3% and 77.8% after 500 cycles at 100 and 300 mA g−1, respectively (Fig. 11b and c).| Materials | Preparation method | 1st discharge–charge capacity (mA h g−1) | Capacity retention | Rate performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| SpongelikeNanosized Mn3O4 | Precipitation method | 1327/869 at 40 mA g−1 | 800 (40 cycles) | 500 at 10C | 43 |
| Mn3O4 nanorods | Hydrothermal + annealing method | 1050/555 at 0.1 C | 108 (100 cycles) | 80 | |
| Mn3O4 octahedral | Hydrothermal method | 1009/700 at 50 mA g−1 | 404 (20 cycles) | 269 at 500 mA g−1 | 81 |
| Mesoporous stacked Mn3O4 nanosheets | Chemical bath deposition (CBD) route | 824/300 at 0.1 C | 400 (60 cycles) | 298 at 1.5 C | 82 |
| Porous MnxCo3−xO4 nanocubes | A New facile strategy | discharege1395 at 200 mA g−1 | 733 (30 cycles) | 473 at 1600 mA g−1 | 83 |
| Mesoporous Mn3O4 nanotubes | Hydrothermal + hydrogen reduction | 1065/641.5 at 100 mA g−1 | 641 (100 cycles) at 500 mA g−1 | 525 at 1000 mA g−1 | 84 |
| Order-aligned Mn3O4 nanostructures | Electrochemically depositing method | 919/622 at 936 mA g−1 | 882 (85 cycles) | 500 at 40 Ag−1 | 85 |
| Mn3O4 octahedrons | Dealloying of MnAl alloy | 918.3/536.8 at 100 mA g−1 | 746 (500 cycles) | 240 at 1500 mA g−1 | 96 |
| Fe-doped MnxOy | Nanocasting technique + controlled calcination | 1358/800 at 200 mA g−1 | 620 (100 cycles) | 409 at 1600 mA g−1 | 87 |
| Fe2O3–Mn3O4 nanocomposite | Electrostatically-derived selfassembly method | 1050/919.8 at 90 mA g−1 | 230 (25 cycles) | 88 | |
| Mn3O4–graphene hybrid | Two-step solution-phase reactions | 1350/950 at 40 mA g−1 | 730 (40 cycles) | 390 at 1600 mA g−1 | 91 |
| Mn3O4–graphene composite | Ultra-sonication + annealing | 1375/750 at 75 mA g−1 | 720 (100 cycles) | 92 | |
| Mn3O4–graphene nanocomposite | Microwave hydrothermal technique | 1354/901 at 40 mA g−1 | 900 (50 cycles) | 400 at 1000 mA g−1 | 93 |
| Mn3O4–graphite nanosheet | Solvothermal route | 1079/762 at 50 mA g−1 | ∼600 (50 cycles) | ∼250 at 600 mA g−1 | 94 |
| Mn3O4/ordered mesoporous carbons | Precipitation + annealing method | 1843/773 at 100 mA g−1 | 802 (50 cycles) | 95 | |
| Mn3O4/(MWCNTs) nanocomposite | Solvothermal route | 1380/750 at 100 mA g−1 | 592 (50 cycles) | 387 at 1000 mA g−1 | 96 |
| Mn3O4/C nanorod | Solvothermal route | 1246/723 at 40 mA g−1 | 473 (50 cycles) | 97 | |
| Mn3O4 nanoparticles/graphene | An in situ transformation method | 1600/700 at 60 mA g−1 | 500 (40 cycles) | 200 at 1500 mA g−1 | 98 |
| Mn3O4 nanocrystals/(RGO) | Ultra-sonication-assisted method | 1630/1050 at 40 mA g−1 | 900 (20 cycles) | 200 at 1600 mA g−1 | 99 |
| Mn3O4/SACNT | Thermal decomposition | ≈1900/1108.4 at 1 C | 390 (100 cycles) | 342 at 10 C | 100 |
| Mesoporous Mn3O4 nanosheet/graphene | Precipitation | 1553/973 at 400 mA g−1 | 910 (230 cycles) | 360 at 8 A g−1 | 101 |
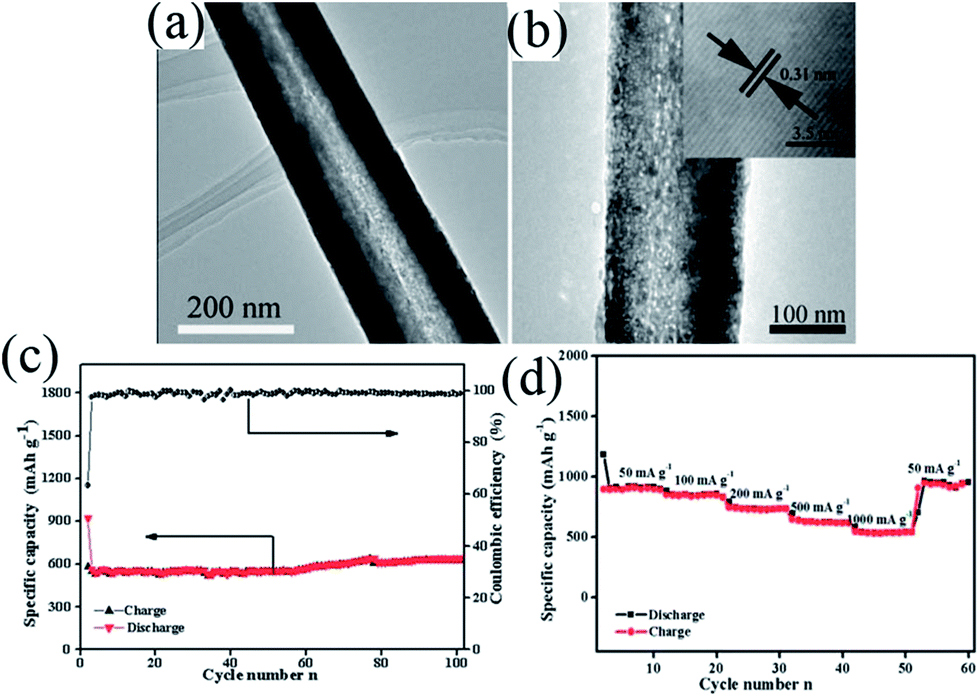 | ||
| Fig. 10 (a) TEM and (b) HRTEM images of mesoporous Mn3O4 nanotubes; (c) cycle performance and coulombic efficiency versus cycle number of Mn3O4 nanotubes at a current density of 500 mA g−1 and (d) rate capabilities with increasing current density. Reproduced with permission.84 Copyright 2013, RSC. | ||
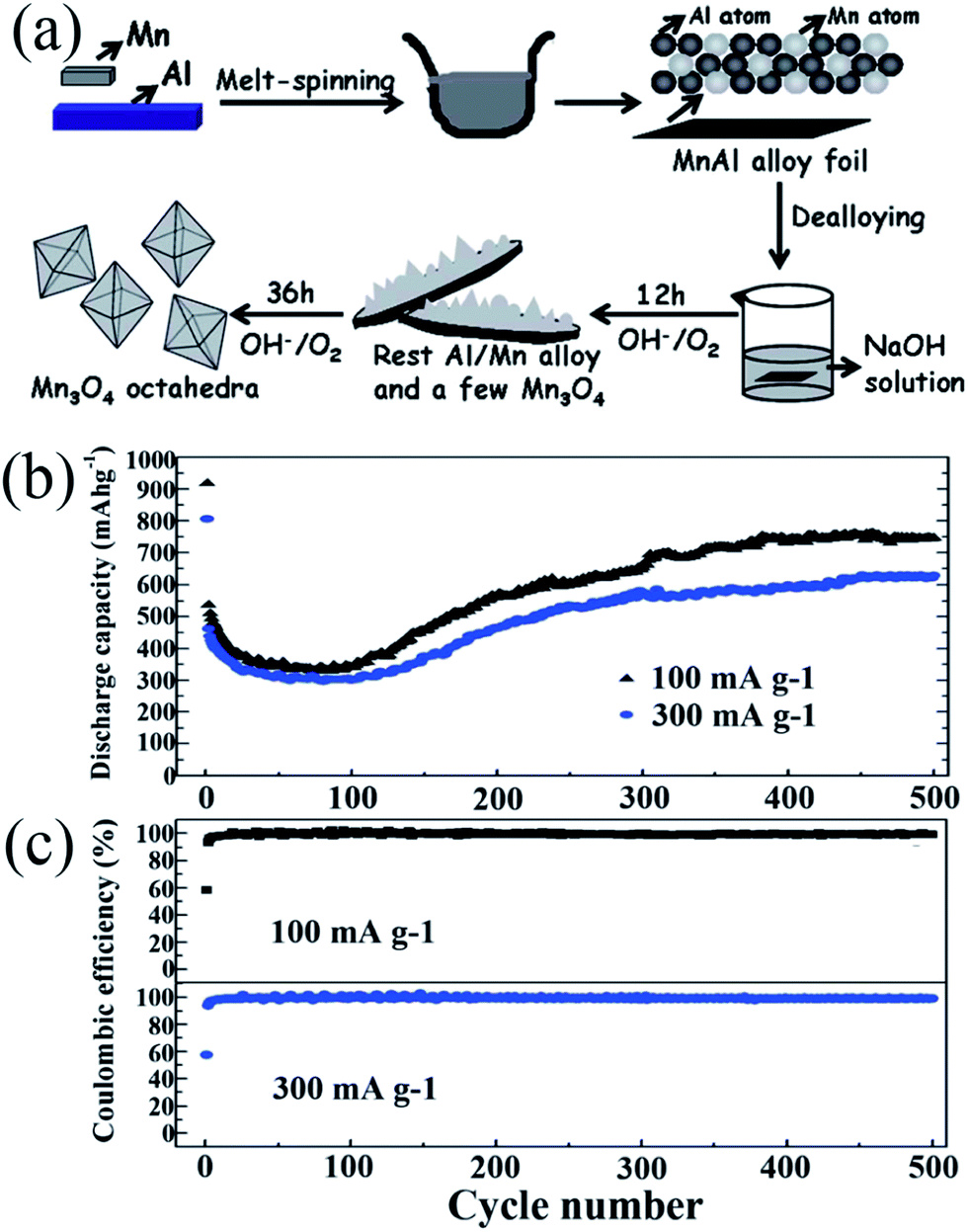 | ||
| Fig. 11 (a) Schematic for the fabrication of Mn3O4 octahedra; (b) cycling behaviour; and (c) coulombic efficiency of the Mn3O4 anode at different current densities. Reproduced with permission.86 Copyright 2014, RSC. | ||
In order to tackle the problem of the low capacity retention capability of pure Mn3O4 materials, researchers have designed M-doped Mn3O4 (M = Co, Fe and Cu) samples.76,87–89 In 2005, Pasero et al.76 synthesized a Co-doped Mn3O4 sample to improve the cycling performance of the pure Mn3O4 phase. The Co-doped Mn3O4 sample delivers a specific capacity of 400 mA h g−1, which is much higher than the specific capacity of pure Mn3O4 (∼200 mA h g−1). More recently, Ma et al.87 prepared Fe-doped MnxOy with connected macro- and mesopores to provide hierarchical porosity by a facile and scalable nanocasting technique followed by controlled calcination using a novel macroporous amine-functionalized bromomethylated poly(2,6-dimethyl-1,4-phenylene oxide) (BPPO) membrane as the sacrificial template (Fig. 12). The macropores in the final product were mainly formed by the reversible replication of the membrane porous structure and interconnected branches in the pore system. The macropores cushioned the volume change during the conversion reaction of the lithium during cycling, and also acted as reservoirs and thoroughfares to improve Li+ transport in the electrolyte phase. The mesopores between primary nanocrystalline particles were formed upon heat treatment and provided a large interfacial contact between electrolyte and electrode active material to support a high Li+ flux across the solid/liquid interface. The selection of iron as a dopant was intentional: the redox potential of iron (1.0 V for discharge/1.7 V for charge) is higher than that of manganese (0.5 V for discharge/1.2 V for charge).90 Therefore, the evenly-distributed iron remains in the zero-valent state during a large part of the MnxOy conversion reaction, conceptually serving the dual function of (1) electrical wiring of the electro-active material to lower the resistance for electron transport, and (2) promoting ionic conduction by creating narrowly-spaced interfaces at the oxide-grain boundary. As anticipated, the Fe-doped MnxOy composites show good capacity retention and high rate capabilities, delivering a capacity of ∼400 mA h g−1 at 1500 mA g−1 after 900 cycles (Fig. 13). The authors also synthesized manganese oxides doped with different amounts of iron and investigated the effect of Fe content on phase structures and electrochemical performance of the as-synthesized samples. These results indicate the importance of a moderate level of iron doping to balance its effects on interfacial charge transfer and the structural integrity of the mesopores. Further studies are required to explore the effect of metal dopants on the rate and long-term cycling performance as well as the columbic efficiency of Mn3O4 composites.
 | ||
| Fig. 12 (a) Schematic illustration of the preparation of hierarchical macro/mesoporous metal oxide structures; (b) FESEM images at low and high (inset) magnifications of Fe-doped MnxOy-2; (c) TEM images at low and high (inset) magnifications of Fe-doped MnxOy-2; (d) SEM image of Fe-doped MnxOy-2 and EDX element maps of Mn, Fe, and O; and (e) nitrogen adsorption–desorption isotherms of Fe-doped MnxOy-2 and the corresponding pore size distribution (inset). Reproduced with permission.87 Copyright 2013, Wiley. | ||
 | ||
| Fig. 13 (a) The 1st and 2nd discharge–charge curves of the Fe-doped MnxOy-2 electrode; (b) cycle stability of the Fe-doped MnxOy-0, Fe-doped MnxOy-1, Fe-doped MnxOy-2, Fe-doped MnxOy-3 and Fe-doped MnxOy-4 electrodes measured at 200 mA g−1; (c) cycling stability of the Fe-doped MnxOy-2 electrode at different current densities; and (d) cycling performance of the Fe-doped MnxOy-2 electrode at 1500 mA g−1. Reproduced with permission.87 Copyright 2013, Wiley. | ||
Mn3O4–graphene hybrids as high capacity anode materials for LIBs were first investigated by Cui's group.91 They developed a two-step solution-phase synthesis route to form hybrid materials of Mn3O4 nanoparticles with diameters around 10–20 nm on 10 wt% reduced graphene oxide (RGO) sheets (Fig. 14). A stable capacity of ∼900 mA h g−1 (∼810 mA h g−1 based on the total mass of the composite) was obtained for the first five cycles when cycled at a current of 40 mA g−1 in the voltage range of 0.1–3.0 V vs. Li/Li+. Very good rate capability and cycling stability were observed; for example, a capacity of ∼720 mA h g−1 at 400 mA g−1 was retained after 40 cycles of charge–discharge at various C-rates. However, the bare Mn3O4 nanoparticles exhibit inferior electrochemical performance, with an initial reversible specific capacity of 300 mA h g−1 at 40 mA g−1. The high capacity, good rate capability and cycling stability of this Mn3O4/RGO hybrid material are attributed to the precisely-synthesized nanocomposites of Mn3O4 (20 nm in diameter) wrapped in graphene sheets (GSs) via a facile, effective, energy-saving, and scalable microwave hydrothermal technique. The composite shows a high specific capacity of more than 900 mA h g−1 at 40 mA g−1, and an excellent cycling stability with nearly no capacity decay up to 50 cycles. They first give evidence that there is charge transfer between the graphene and the Mn3O4 nanoparticles by theoretical calculations. Li et al.95 developed a facile one-step route to generate interactions between the graphene substrates and the Mn3O4 nanoparticles grown directly on them, which makes Mn3O4 electrochemically active since charge carriers can be effectively and rapidly conducted back and forth from the Mn3O4 nanoparticles to the current collector through the highly conductive 3D graphene network. In addition, the graphene–nanoparticle interactions afford good dispersion of Mn3O4 nanoparticles on the RGO sheets, which could avoid Mn3O4 nanoparticle aggregation to result in better cycle stability.
 | ||
| Fig. 14 (a) Schematic of the two-step synthesis of Mn3O4/RGO; (b) SEM image of a Mn3O4/RGO hybrid; (c) XRD spectrum of a packed thick film of Mn3O4/RGO; (d) TEM image of Mn3O4/RGO; inset shows the electron diffraction pattern of the Mn3O4 nanoparticles on RGO; (e) high-resolution TEM image of an individual Mn3O4 nanoparticle on RGO; (f) charge (red) and discharge (blue) curves of Mn3O4/RGO for the first cycle at a current density of 40 mA g−1; (g) representative charge (red) and discharge (blue) curves of Mn3O4/RGO at various current densities; (h) capacity retention of Mn3O4/RGO at various current densities and (i) capacity retention of free Mn3O4 nanoparticles without graphene at a current density of 40 mA g−1. Reproduced with permission.91 Copyright 2010, ACS. | ||
This growth-on-graphene approach offers a new technique for the design and synthesis of high performance anode materials based on highly-insulating Mn3O4 materials. After this work, Mn3O4–carbon-based hybrid materials have been intensively studied due to their potential for improving their cycle stability and rate performance as LIB anode materials. The specific capacity values and cycling performances are listed in Table 3.92–101 For example, Li et al.93 successfully synthesized 3D hybrids with Mn3O4 nanoparticles homogeneously embedded in ordered mesoporous carbon (OMC). The Mn3O4/OMC hybrids display a high specific capacity up to 802 mA h g−1 and a high coulombic efficiency of up to 99.2% after 50 cycles at a high current density of 100 mA g−1. This capacity is 1.6 times higher than the discharge capacity for pure ordered OMC materials (512 mA h g−1), and more than 5.4 times higher than that of pure Mn3O4 nanoparticles (148 mA h g−1). The authors suggested that the enhanced capacity and cycling performance of the Mn3O4/OMC hybrids can be attributed to their unique, robust 3D composite structures and the synergistic effects between the Mn3O4 nanoparticles and OMC. The ordered mesostructured channels of Mn3O4/OMC hybrids are expected to effectively buffer against the local volume change during Li uptake/removal reactions, thus enhancing structural stability. The OMC matrix wall with a thickness of <10 nm greatly reduces the solid-state transport length for Li diffusion. Moreover, the hierarchically-ordered mesoporosity facilitates liquid electrolyte diffusion into the bulk of the electrode material, providing fast conductive ion transport channels for the conductive Li+ ions. The improved cycling performance can also be attributed to good electrical contact between Mn3O4 and OMC in the 3D nanocomposites during the phase transformation of Mn3O4 upon lithiation/delithiation that usually leads to capacity fading. Wang et al.97 developed a facile one-step solvothermal reaction route for the large-scale synthesis of carbon-homogeneously-wrapped manganese oxide (Mn3O4@C) nanocomposites for anode materials using manganese acetate monohydrate and polyvinyl pyrrolidone as precursors and reactants. The synthesized Mn3O4 with tetragonal structures (space group I41/amd) display nanorod-like morphology with widths of 200–300 nm and thicknesses of 15–20 nm. The Mn3O4@C nanocomposites display enhanced capacity retention on charge–discharge cycling, with a stable specific capacity of 473 mA h g−1 after 50 cycles (as much 3.05 times as that of pure Mn3O4 sample). Wang et al.99 fabricated colloidal Mn3O4 nanocrystals supported by graphene oxide (GO) and reduced graphene oxide (RGO) (Mn3O4–GO and Mn3O4/RGO nanocomposites) through a facile ultrasonic-assisted synthetic route in an ethanol amine (ETA)–water system (Fig. 15a). The integration of GO with abundant O-containing groups and colloidal Mn3O4 crystals endows this composite with excellent electrochemical performances including high reversible capacity, good cycle stability, and high rate performance, making it suitable for use as an electrode material in electrochemical capacitors (ECs). Compared with Mn3O4–GO nanocomposites, Mn3O4/RGO exhibits excellent electrochemical performance in rechargeable LIBs due to the high electrical conductivity of RGOs and the structure of Mn3O4 nanocrystals wrapped in RGOs (Fig. 15b). In addition, the authors explained how the synergetic structural composition of Mn3O4–GO and Mn3O4/RGO nanocomposites plays an important role in their properties for ECs or LIBs (Fig. 15c). Luo et al.100 recently reported the Li cycling behaviour of a Mn3O4–carbon nanotube hybrid material, which is a composite of Mn3O4 nanoparticles anchored on continuous super-aligned carbon nanotube (SACNT) films. They reported that the electrochemical performance of the Mn3O4/SACNT composite electrode is significantly affected by the Mn3O4 content and particle size. Mn3O4/SACNT composites with Mn3O4 sizes under 10 nm demonstrated higher capacity, lower polarization extent, and better cycle stability than those with Mn3O4 sizes of 165 nm. The optimized Mn3O4/SACNT composite displays a capacity of more than 700 mA h g−1 at 0.1 C (based on the total mass of the electrode). A large capacity of 342 mA h g−1 can still be obtained at a high rate of 10 C, and a capacity retention of 95% was achieved for 100 cycles at 1 C. Despite improvements in the anodic properties of Mn3O4 by conductive coating/composites, further studies on the effects of conductive coatings on cycling stability and rate performance are still needed.
 | ||
| Fig. 15 (a) Preparation routes for Mn3O4 nanoparticles, Mn3O4–GO and Mn3O4/RGO nanocomposites; (b) cycling performance of these samples at 40 mA g−1; (c) the charge mechanism of Mn3O4–GO; and (d) Mn3O4/RGO for ECs and LIBs. Reproduced with permission.99 Copyright 2013, RSC. | ||
2.3 Mn2O3 and its nanocomposites
There were only a few reports on Mn2O3 nanomaterial-based anodes for LIBs before 2010.102,103 Liu et al.102 reported preliminary studies of Li cycling behaviours on Mn2O3; they found a first discharge and charge capacity of 1080 and 480 mA h g−1, respectively, in the voltage range of 0.2–3.0 V vs. Li/Li+ at 0.25 mA cm−2. Lavela et al.103 prepared Mn2O3 by a sol–gel method followed by heat treatment at different temperatures. They reported that Mn2O3 showed a discharge potential (1st cycle) of ∼0.25 V vs. Li/Li+. The Mn2O3 delivered reversible capacities of 500–700 mA h g−1 depending on synthesis temperature when cycled in the voltage range of 0.005–3.0 V Li/Li+. However, capacity fading was noted in all cases with capacities ranging from 250 to 400 mA h g−1 after 20 cycles. Porous 1D Mn2O3 nanostructures containing nanofibers, nanowires and nanorods were successfully fabricated via a facile hydrothermal treatment and sequential thermal decomposition without any template or surfactant.104 These as-prepared porous Mn2O3 nanomaterials manifested high initial capacities; however, capacity fading was also observed in all cases with capacities of ∼250 mA h g−1 after 30 cycles. A similar phenomenon was also found by Shen's group.105 By means of morphology-conserved transformation, Qiu et al.106 synthesized hierarchically-structured Mn2O3 nanomaterials with different morphologies (oval- and strawsheaf-shaped) and pore structures by a hydrothermal method followed by heating in air at 600 °C. Both of the as-prepared mesoporous Mn2O3 nanomaterials deliver high reversible capacities and excellent cycling stabilities compared with the commercial Mn2O3 nanoparticles. The authors observed a reversible capacity of 380 and 320 mA h g−1 at the end of the 150th cycle for straw-sheaf- and oval-shaped nanostructured Mn2O3, respectively, at a current of 200 mA g−1 when cycled in the voltage range of 0.01–2.5 V vs. Li/Li+. The good capacity retention capability of the straw-sheaf-shaped Mn2O3 was attributed to the relatively high surface area and peculiar nanostrip structure, which resulted in a reduced length for lithium ion diffusion. Recently, our group107 has synthesized porous Mn2O3 microspheres by the morphology-controlled decomposition of spherical MnCO3 precursors at 600 °C. The porous Mn2O3 microspheres show a specific capacity as high as 796 mA h g−1 at a current density of 100 mA g−1 after 50 cycles. A high rate performance (∼600 mA h g−1 at 1200 mA g−1) was also observed. Based on the CV curves, discharge–charge curves and ex situ XRD analysis, a novel lithium storage mechanism for the synthesized porous Mn2O3 material after the first discharge process was suggested as follows:| Mn + xLi2O ↔ 2xLi+ + MnOx + 2xe− (1.0 < x < 1.5) |
After our work, porous Mn2O3 hierarchical microspheres108,109 and Mn2O3 nanocones,110 polyhedrons,111 porous sheets,112 cubes and spindles113 have been synthesized and investigated as anode materials by other groups. The results of these investigations are listed in Table 4. Although some advances in Mn2O3 anode materials have been made via morphology optimization, further studies on the improvement of the cycling stability and rate performance are still required.
| Materials | Preparation method | 1st discharge–charge capacity (mA h g−1) | Capacity retention | Rate performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| Mn2O3 nanorods | Hydrothermal + annealing | 1509/689 at 100 mA g−1 | 374 (30 cycles) | 104 | |
| Mn2O3 nanofibers | 1694/795 at 100 mA g−1 | 404 (30 cycles) | |||
| Mn2O3 nanowires | 1611/707 at 100 mA g−1 | 384 (30 cycles) | |||
| Mn2O3 nanorods | Hydrothermal + annealing | 998/349 at 100 mA g−1 | ∼50 (100 cycles) | 105 | |
| Straw-sheaf-shaped Mn2O3 | Solvothermal + annealing | 1179/680 at 200 mA g−1 (0.01–2.5 V) | 400 (150 cycles) at 400 mA g−1 | 270 at 800 mA g−1 | 106 |
| Oval-shaped Mn2O3 | 1159/∼650 at 200 mA g−1 (0.01–2.5 V) | 320 (150 cycles) at 400 mA g−1 | |||
| Porous Mn2O3 microsphere | Solvothermal + annealing | 1310/882 at 100 mA g−1 | 796 (50 cycles) at 100 mA g−1 | 470 at 2400 mA g−1 | 107 |
| Porous Mn2O3 microspheres | Solvothermal + annealing | 2134/1400 at 50 mA g−1 | 748 (45 cycles) | 423 at 1600 mA g−1 | 108 |
| Mesoporous Mn2O3 microspheres | Hydrothermal + annealing | 668/∼630 at 200 mA g−1 | 524 (200 cycles) | ∼150 at 50 mA g−1 | 109 |
| Hollow Mn2O3 nanocones | Hydrothermal + annealing | 1875/909 at 50 mA g−1 | 280 (200 cycles) at 200 mA g−1 | 380 at 400 mA g−1 | 110 |
| Porors Mn2O3 nanosheets | Solvothermal + annealing | 1518/987 at 300 mA g−1 | 521 (100 cycles) | 500 at 500 mA g−1 | 111 |
| Cubic Mn2O3 | Hydrothermal + annealing | 1680/780 at 100 mA g−1 | ∼300 (55 cycles) | 240 at 600 mA g−1 | 113 |
| Spindle Mn2O3 | 1530/620 at 400 mA g−1 | ∼200 (50 cycles) | |||
| Fusiform Mn2O3 | 1402/400 at 100 mA g−1 | 155 (40 cycles) | |||
| Cu-doped hollow Mn2O3 spheres | Hydrothermal + annealing | 1998/1190 at 100 mA g−1 | 642 (100 cycles) | 308 at 815 mA g−1 | 89 |
Recently, a good report on Cu-doped hollow spherical Mn2O3 samples by Li et al.113 indicated a higher specific capacity of 642 mA h g−1 at a current density of 100 mA g−1 after 100 cycles. They suggested that the significant enhancement of the electrochemical lithium storage performance can be attributed to improvements in the electronic conductivity and lithium diffusivity of electrodes.
Xia's group114 prepared Mn2O3 nanowires by thermally annealing MnOOH nanowire precursors, which were transformed from hydrothermally-grown MnO2 nanoflakes and directly attached on Ti foils via reaction with poly(vinyl pyrrolidone) (Fig. 16a). The Mn2O3 nanowires (Fig. 16b) show an initial discharge capacity of 815.9 mA h g−1 at 100 mA g−1 and maintains a capacity of 502.3 mA h g−1 after 100 cycles (Fig. 16c), and a good rate performance of 220 mA h g−1 at 1000 mA g−1 (Fig. 16d). Furthermore, the authors first fabricated a flexible Mn2O3//LiMn2O4 lithium ion full cell using the Mn2O3 nanowires as the anode and LiMn2O4 nanowires as the cathode (Fig. 17). The full cell has an output voltage of >3 V, a low thickness of 0.3 mm, high flexibility, and a specific capacity of 99 mA h g−1 based on the total weight of the cathode material. It also exhibits good cycling stability with a capacity of ∼80 mA h g−1 after 40 charge–discharge cycles. This successful example of the Mn2O3//LiMn2O4 full cell and further optimization and development of the nanowire design and fabrication will lead to new opportunities in high power-density energy storage devices.
 | ||
| Fig. 16 (a) Schematic of the synthesis of Mn2O3 nanowires (NMs) and fabrication the Mn2O3//LiMn2O4 LIB full cell; (b) SEM images of Mn2O3 NWs grown on Ti foils; (c) cycling performance of Mn2O3 NWs (red curve) for the first 100 cycles; and (d) rate performance of Mn2O3 NWs (1 C = 1000 mA g−1). Reproduced with permission.114 Copyright 2013, ACS. | ||
 | ||
| Fig. 17 Flexible Mn2O3//LiMn2O4 full cells. (a) 3D structure scheme and (b) optical image of a flexible Mn2O3//LiMn2O4 full cell; (c) optical images showing a light emitting diode (3 V, 10 mW) powered by a flexible Mn2O3//LiMn2O4 full cell; (d) optical image of the voltage output of a three-cell battery in tandem; (e) charge–discharge curves of a flexible Mn2O3//LiMn2O4 full cell at the first cycle and (f) cycling performance (red curve) and coulombic efficiency (black curve) of a representative flexible Mn2O3//LiMn2O4 full cell. Reproduced with permission.114 Copyright 2013, ACS. | ||
2.4 MnO2 and its nanocomposites
Manganese dioxide (MnO2) possesses a high theoretical capacity of 1233 mA h g−1 based on the conversion mechanism.44,115 Moreover, it is environmentally-benign and naturally abundant, making it a promising material for application in LIBs. However, due to the low conductivity of MnO2 and large volume expansion during battery operation, rather low coulombic efficiencies in the first cycle and rapid capacity decays with cycling were observed.116–119 For example, Li et al.117 synthesized novel α-MnO2 hollow urchins on a large scale by a facile and efficient low-temperature (60 °C) mild reduction route. The α-MnO2 hollow urchins show a high capacity of 746.0 mA h g−1 in the first discharge process, but the capacity of the 40th cycle is only 481 mA h g−1, about 64.4% of the initial capacity. As reported by Reddy et al.,119 a coaxial MnO2/CNT array material can give an initial capacity of approximately 2000 mA h g−1 at a rate of 50 mA g−1; however, the capacity sharply decays to 500 mA h g−1 after only 15 cycles. The authors suggested the initial high capacity and enhanced lithium storage capacity can be attributed to the in situ deposition of MnO2 on CNTs, while the fast capacity decay might result from a structural deterioration of a thick MnO2 layer with a compact structure deposited on the outside surface of the CNTs. One of the best results, which reported a capacity of 600 mA h g−1 after 100 cycles for a γ-MnO2 film deposited on nickel metal, was reported before 2010.118 Subsequently, systematic efforts have been dedicated to optimize MnO2 electrodes by combining materials synthesis, processing and engineering techniques to improve their electrochemical performance.Recently, attempts have also been made to fabricate MnO2 composites with unique nanostructures,120–122 MnO2-based nanocomposites consisting of MnO2 nanostructures and electrically conductive carbon such as carbon nanotubes123–125 amorphous carbon126 or graphene,127–131 its nanocomposites including MnO2 and other transition metal oxides,132–134 and MnO2@polymers.135 The results of these studies are listed in Table 5. Kundu et al.122 reported the direct growth of mesoporous MnO2 nanosheet (NS) arrays on a nickel (Ni) foam current collector by electro-deposition followed by low temperature thermal annealing (170 °C) in a nitrogen atmosphere. When using the Ni foam-supported mesoporous MnO2 NS arrays as an anode in LIBs, the electrode can deliver a reversible capacity as high as 1690 mA h g−1 even after 100 cycles at a charge–discharge current density of 100 mA g−1. Two anodic peaks located at 1.3 and 2.4 V vs. Li/Li+ are observed, and the re-oxidation of manganese was suggested to take place in two steps. It is also interesting to note that the capacity gradually increases with increasing cycle number, which is the opposite trend observed in previous studies.116–120,135 The increase in capacity might originate from the increased number of vacancies and grain boundaries, where Li ions could be stored, due to the pulverization of the MnO2 NSs upon cycling. This example suggests that the electrochemical performance of MnO2 may be optimized by nanostructural tuning. Guo et al.127 reported a novel approach to fabricate a hierarchically-nanostructured composite comprised of 1D rod-like layered birnessite-type manganese oxides (LMOs), 2D graphene nanosheets, and a 3D porous structure (LMO/PEDOT/graphene). As shown in Fig. 18a, the 3D graphene material is used as a supporting matrix on which in situ polymerized poly(3,4-ethylenedioxythiophene) (PEDOT) is assembled for the direct growth of uniformly-distributed 1D rod-like LMOs. The composites exhibit a large first discharge capacity of 1835 mA h g−1 and reversible discharge capacities of 1105 mA h g−1 and 948 mA h g−1 after 1 and 15 cycles, respectively, at a rate of 50 mA g−1 (Fig. 18b and c). The authors suggested that the enhanced performance of LMO/PEDOT/graphene in LIBs can be explained as follows: (1) the LMO in LMO/PEDOT/graphene has characteristics of an open structure with an interlayer space of approximately 7.2 Å, which promotes efficient lithium intercalation/deintercalation to achieve high specific capacity; (2) the use of highly conductive 3D graphene is favourable in electron transport, resulting in a fast charge–discharge rate and (3) the PEDOT coating directs the growth of relatively-ordered, textured LMO; it also prevents LMO aggregation to produce a uniform distribution, which can accommodate large volume expansion during battery operation.
| Materials | Preparation method | 1st discharge–charge capacity (mA h g−1) | Capacity retention | Rate performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| Phase-pure β-MnO2 nanorods | Hydrothermal + annealing | 1728/488 at 0.1 mA cm−2 | 43 | ||
| Interconnected MnO2 nanowires | Hydrothermal + annealing | 1650/800 at 85 mA g−1 | ∼580 (100 cycles) | 116 | |
| Hollowα-MnO2 urchin | Low-temperature (60 °C) mild reduction route | 746/650 at 270 mA g−1 | 481 (40 cycles) | 117 | |
| Nanoporous γ-MnO2 hollow microspheres | Hydrothermal + annealing | 1289/1071 at 100 mA g−1 | 657 (20 cycles) | 118 | |
| Nanoporous γ-MnO2 nanocubes | 1992/1042 at 100 mA g−1 | 602 (20 cycles) | |||
| Coaxial MnO2/carbon nanotube | Templates + infiltration + annealing | 2500/880 at 50 mA g−1 | 500 (16 cycles) | 119 | |
| α-MnO2 nanotubes | Exfoliation and scrolling approach | ∼1200/1000 at 200 mA g−1 | 512 (300 cycles) at 800 mA g−1 | 120 | |
| Hierarchical hollow microspheres constructed with MnO2 nanotubes | A Bubble template-based self-scrolling method | 983/830 at 300 mA g−1 | 700 (30 cycles) | 121 | |
| Mesoporous MnO2 nanosheet | Electrodeposition + annealing | 1643/1225 at 50 mA g−1 | 1690 (100 cycles) at 100 mA g−1 | 900 at 1000 mA g−1 | 122 |
| Nanoflaky MnO2/carbon nanotube | Hydrothermal method | 1402/816 at 200 mA g−1 | 620 (50 cycles) | 320 at 4000 mA g−1 | 123 |
| Mesoporous γ-MnO2 particles/carbon nanotube | Ultrasound irradiation | 1278/741 at 50 mA g−1 | 934 (150 cycles) | ∼380 at 1000 mA g−1 | 124 |
| Coaxial MWNTs@MnO2@PPy | Immersing + in situ polymerization | 1250/730 at 100 mA g−1 | 820 (120 cycles) | 530 at 1000 mA g−1 | 125 |
| MnO2 nanosheets/core-leaf onion-like carbon | Hydrothermal method | 1278/854 at 50 mA g−1 | 541 (100 cycles) | 102 at 2000 mA g−1 | 126 |
| MnO2/conjugated polymer/graphene | In situ polymerization + Immersing | 1835/1050 at 50 mA g−1 | 948 (15 cycles) | 698 at 400 mA g−1 | 127 |
| MnO2 nanotube/graphene | Ultra-filtration technique | 1250/686 at 100 mA g−1 | 495 (40 cycles) | 208 at 1600 mA g−1 | 128 |
| Graphene-wrapped MnO2–graphene nanoribbons | Hydrothermal + electrostatic interaction | 880/600 at 100 mA g−1 | 612 (250 cycles) at 400 mA g−1 | 580 at 1000 mA g−1 | 129 |
| α-MnO2–graphene 3D network | Hydrothermal approach | 1875/1150 at 60 mA g−1 | 998 (30 cycles) | 590 at 12 A g−1 | 130 |
| Porous MnO2–3D graphene nanocomposites | A redox process + annealing | 1554/917 at 100 mA g−1 | 836 (200 cycles) | 358 at 1600 mA g−1 | 131 |
| Porous α-Fe2O3 branches on β-MnO2 nanorods | Hydrothermal + annealing | 1480/1147 at 100 mA g−1 | 1028 (200 cycles) at 1000 mA g−1 | 881 at 4000 mA g−1 | 132 |
| TiO2–C/MnO2 core−double-shell nanowire | In situ chemical redox | 865/500 at 33.5 mA g−1 | 218 (150 cycles) at 3350 mA g−1 | 230 at 3350 mA g−1 | 133 |
| ZnO/MnO2 sea urchin-like sleeve array | Electro-deposition | 2023/878 at 200 mA g−1 | 1259 (100 cycles) | 150 at 5000 mA g−1 | 134 |
| Polythiophene (PTh)-coated ultrathin MnO2 nanosheets | A Redox process + in situ polymerization | 700 at 500 mA g−1 | 500 (100 cycles) | 135 |
 | ||
| Fig. 18 (a) Schematic representation of the hierarchically-nanostructured composite fabrication; (b) cycling variation in charge–discharge capacity vs. cycle number for different LIBs; and (c) capacity of LMO/PEDOT/graphene batteries under various current densities. Reproduced with permission.127 Copyright 2011, Wiley. | ||
Li et al.129 designed a unique, hierarchical, sandwiched-structured graphene–MnO2–GNRs (GMG) composite in which the graphene wraps the porous MnO2 that is grown directly from graphene nanoribbons (GNRs). Fig. 19a depicts the synthetic procedures for the GMG composites. Graphene and GNRs are in good contact with MnO2, improving the electrical conductivity of the GMG composites. More importantly, graphene and GNRs can buffer the volume changes and prevent the loss of MnO2 during the Li ion conversion reaction with Li, improving the composite's electrochemical stability. As an anode material, the GMG composites demonstrate a reversible specific discharge capacity of 890 mA h g−1 at 0.1 A g−1 after 180 cycles with various current rates ranging from 0.1 to 1.0 A g−1; the anode shows about 24% improvement compared to its initial capacity after 245 cycles at a current density of 0.4 A g−1 (Fig. 19b).
 | ||
| Fig. 19 (a) Schematic illustration of the synthesis of the GMG composite; (b) rate performance of GMG at various current rates from 0.1 to 1.0 A g−1; and (c) cycling performance of MnO2, MG, and GMG at 0.1 A g−1 for the first five cycles and at 0.4 A g−1 for the following cycles. Reproduced with permission.129 Copyright 2013, Wiley. | ||
In a recent paper, Li et al.131 prepared MnO2/3D porous graphene-like (PG) (3D PG–xMn) composites via a simple and cost-effective redox process. Typically, the self-controlled reaction between 3D PG and KMnO4 under neutral pH conditions led to the homogeneous deposition of MnO2 on the 3D PG networks. They suggested that the porosities of the 3D PG–xMn composites can be adjusted by modulating the initial concentration of KMnO4. The surface areas and total pore volumes of 3D PG–xMn composites are gradually decreased with the increase of the concentration of KMnO4. In addition, after the deposition of MnO2 on 3D PG, there was no obvious decrease in powder conductivity of the 3D PG–xMn (x = 0.5 and 1.0) composites, while the 3D PG–1.5Mn composite shows an obvious decrease. Among the three 3D PG–xMn composites, the 3D PG–1Mn composite displayed the best electrochemical performance. It delivered a discharge capacity of 988 mA h g−1 in the second cycle, and the discharge capacity remained as high as 836 mA h g−1 after the 200th cycle (∼84.6% capacity retention). The excellent electrochemical performances of the as-prepared 3D PG–1Mn composites can be attributed its reasonable MnO2 content (62.7 wt% MnO2) and high conductivity. Branched nanocomposites with β-MnO2 nanorods as the backbone and porous α-Fe2O3 nanorods as the branches were synthesized by Gu et al.132 using a high-temperature annealing process to achieve epitaxial growth of FeOOH on the β-MnO2 nanorods. The branched nanorods of β-MnO2/α-Fe2O3 exhibit reversible specific capacities of 1028 mA h g−1 and 881 mA h g−1 at current densities of 1000 mA g−1 and 4000 mA g−1, respectively, up to 200 cycles. The presence of α-Fe2O3 in the branched nanorods greatly promotes the charge transfer at the electrode/electrolyte interface, which is beneficial to electrochemical performance. These results indicate the importance of the selection of proper chemical components and the control on their construction. The combination of advantages in structure and components would effectively improve the performance of electrode materials in LIBs.
3. Ternary Mn-based oxides as anode materials
Ternary Mn-based oxides containing AMn2O4 (A = Co and Zn) with a spinel or spinel-like crystal structure have been investigated as anode materials for LIBs. The Li cycling mechanism involves the “alloying and conversion reaction” for ZnMn2O4 and the “conversion reaction” for CoMn2O4.8 Depending on the structure, morphology, particle size and composition, large and stable reversible capacities have been reported.3.1 ZnMn2O4-based anodes
ZnMn2O4 has been proposed as a practical alternative anode material for LIBs because of its high theoretical capacity of 1008 mA h g−1 (the Zn metal can form an alloy with Li and participate in the conversion reaction along with manganese) and the low oxidation potentials (i.e., delithiation potential) of zinc and manganese (1.2 and 1.5 V vs. Li/Li+, respectively). These low oxidation potentials could eventually increase the battery output voltage compared to ZnFe2O4 and ZnCo2O4, allowing higher energy density delivery. Furthermore, zinc and manganese are abundant, environmentally-friendly, and relatively inexpensive compared to cobalt.45,136Yang et al.45 synthesized ZnMn2O4 nanocrystals by a polymer pyrolysis method and first used the obtained nanocrystals as lithium storage anode materials. The nanocrystalline ZnMn2O4 electrode delivers an initial reversible capacity of 766 mA h g−1 when cycled at 100 mA g−1 in the voltage range of 0.01–3.0 V vs. Li/Li+. Cycling studies up to 50 cycles show almost stable cycling performance between 10 and 50 cycles with an average capacity fading of only 0.20% per cycle. The electrode still maintains a capacity of 569 mA h g−1 after 50 cycles. Subsequently, ZnMn2O4 with different particle sizes and morphologies including flower-like ZnMn2O4 superstructures,136 ZnMn2O4 nanoparticles,44,137,138 ZnMn2O4 nanowires,139 ZnMn2O4 nanoplates,140 ZnMn2O4 hollow microspheres,141–143 ball-in-ball ZnMn2O4 hollow microspheres,144 loaf-like ZnMn2O4 nanorods,145 ZnMn2O4 nanofibers146 and ZnMn2O4 mesoscale tubular arrays147 have been studied and applied as anode materials for LIBs. The corresponding results are listed in Table 6. Specifically, the ball-in-ball ZnMn2O4 hollow microspheres143 were prepared by a novel template-free method (Fig. 20a–d). The hollow microspheres typically consist of small nanoparticles with an average size of 30 nm, and exhibit mesoporous features. When used as an anode material for LIBs, the ZnMn2O4 hollow microspheres show a high specific capacity of 750 mA h g−1 after 120 cycles at 400 mA g−1 (Fig. 20e and f). Despite the improved cyclic and rate performance of ZnMn2O4 nanomaterials obtained by different strategies, further studies are still required to achieve long cycling stability and the high rate performance for these materials.
| Materials | Preparation method | 1st discharge–charge capacity (mA h g−1) | Capacity retention | Rate performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| Nanocrystalline ZnMn2O4 | Polymer pyrolysis route | 1302/766 at 100 mA g−1 | 569 (50 cycles) | 45 | |
| Flower-like ZnMn2O4 | Solvothermal method | 1350/763 at 100 mA g−1 | 626 (50 cycles) | 136 | |
| Nanocrystalline ZnMn2O4 | Coprecipitation method | 1145/800 at 100 mA g−1 | 690 (70 cycles) | 365 at 1000 mA g−1 | 44 |
| Nanocrystalline ZnMn2O4 | A single-source precursor route | 1088/680 at 100 mA g−1 | 650 (200 cycles) | 405 at 600 mA g−1 | 137 |
| Nanocrystalline ZnMn2O4 | Hydrothermal method | 1200/680 at 100 mA g−1 | 430 (50 cycles) | 75 at 2000 mA g−1 | 138 |
| ZnMn2O4 nanowires | Solid-state reaction | 1400/891 at 60 mA g−1 | 650 (40 cycles) | 350 at 1000 mA g−1 | 139 |
| ZnMn2O4 nanoplates | An “escape-by-crafty-scheme” method | 1277/730 at 100 mA g−1 | 502 (30 cycles) | 324 at 1800 mA g−1 | 140 |
| ZnMn2O4 hollow microspheres | Coprecipitation + annealing method | 1325/772 at 400 mA g−1 | 607 (100 cycles) | 361 at 1600 mA g−1 | 141 |
| ZnMn2O4 hollow microspheres | Solvothermal method | 1335/750 at 100 mA g−1 | 433 (50 cycles) | 245 at 400 mA g−1 | 142 |
| ZnMn2O4 hollow microspheres | Coprecipitation + annealing method | 1260/830 at 100 mA g−1 | 599 (50 cycles) | 450 at 1000 mA g−1 | 143 |
| Ball-in-ball ZnMn2O4 hollow microspheres | Solvothermal + annealing method | 945/6622 at 400 mA g−1 | 790 (120 cycles) | 396 at 1200 mA g−1 | 144 |
| Loaf-like ZnMn2O4 | Hydrothermal + annealing | 1357/839 at 100 mA g−1 | 517 (100 cycles) at 500 mA g−1 | 457 at 1000 mA g−1 | 145 |
| ZnMn2O4 nanorods | Electrospinning | 1257/680 at 60 mA g−1 | 318 (50 cycles) | 146 | |
| ZnMn2O4 nanofibers | 1469/716 at 60 mA g−1 | 705 (50 cycles) | |||
| ZnMn2O4 nanowires | 1526/891 at 60 mA g−1 | 530 (50 cycles) | |||
| ZnMn2O4 mesoscale tubular arrays | A Reactive template route + annealing | 1198/784 at 100 mA g−1 | 784 (100 cycles) | 364 at 1600 mA g−1 | 147 |
| rGO–ZnMn2O4 composite | A bottom-up wrapping route | 1412/960 at 100 mA g−1 | 707 (100 cycles) | 450 at 2000 mA g−1 | 148 |
 | ||
| Fig. 20 Typical (a), (b) FESEM and (c), (d) TEM images of ZnMn2O4 ball-in-ball hollow microspheres; (e) cycling performance of the ZnMn2O4 hollow microcubes in the voltage range of 0.01–3.0 V vs. Li/Li+ at current densities of 400 mA g−1 and (f) rate performance at different current densities for the same cell after 120 cycles. Reproduced with permission.143 Copyright 2012, Wiley. | ||
Recently, in an attempt to improve the electrochemical properties of ZnMn2O4, Zheng et al.148 prepared thermally-reduced graphene oxide (rGO)-wrapped ZnMn2O4 nanorods via a facile bottom-up approach. They suggested that this unique architecture can provide 1D interconnected ZnMn2O4 nanoparticle networks to facilitate rapid diffusion of lithium ions within the electrode material. Meanwhile, the rGO sheets could act as a conductive layer for electron transfer and an efficient buffer layer to enable the structural stabilization of ZnMn2O4 crystals. The results indicate a high and stable reversible capacity (707 mA h g−1 at 100 mA g−1 over 50 cycles) and an excellent rate capability (440 mA h g−1 at 2000 mA g−1) for this composite.
As mentioned in the introduction, the reasonable selections of binders and electrolytes are necessary to improve the rate and cyclic performance of metal oxides-based electrodes. Courtel et al.44 have studied the effects of binder and electrolyte on the ZnMn2O4 cycling performance in a recently-published paper. The investigation of the electrochemical performance of ZnMn2O4 electrodes using five binders (PVDF dissolved in NMP and four other water-soluble binders: NaCMC, LiCMC, XG and Baytron; CMC = carboxymethylcellulose) show that LiCMC and NaCMC are the best binders (Fig. 21). Specifically, the electrode fabricated by ZnMn2O4 nanoparticles (<150 nm) sintered at 800 °C with the LiCMC binder exhibits a capacity of 690 mA h g−1 after 70 cycles at C/10 and a good rate capability (450 mA h g−1 for 0.5 C, 340 mA h g−1 for 1 C, 230 mA h g−1 for 2 C, and 150 mA h g−1 for 3 C). In addition, studies on the effect of the electrolyte on ZnMn2O4 electrode cycling performance showed that a carbonate (EC)–dimethyl carbonate (DEC) mixture (3:7 by vol) is the best electrolyte, while PC (propylene carbonate) is the worst (Fig. 22). These results could be explained by the fact that PC does not form a polymeric organic layer, or that the layer formed with PC is inactive for lithium storage. Furthermore, the authors assembled a full cell for the first time using ZnMn2O4 as the anode material. LiMn1.5Ni0.5O4 was the cathode material, and 1 M LiPF6 in EC–DEC (3:7 by vol) was the electrolyte. The full cell was cycled between 1.5 V and 4.7 V at 0.1 C, and the capacity was reported versus the limiting electrode (cathode). The initial capacity at a cycling rate of 0.1 C is 108 mA h g−1, and there is a plateau at 4.1 V during the first charge. However, in the subsequent discharge and charge cycles, the charge vs. voltage curve exhibits a sloping voltage between 3.5 V and 3 V and 3.5 and 4.1 V, respectively. Unfortunately, after 20 cycles, the battery exhibits a capacity of only 75 mA h g−1.
 | ||
| Fig. 21 Discharge capacities of ZnMn2O4 electrodes made using five different binders: PVDF, NaCMC, LiCMC, XG, and Baytron. Reproduced with permission.44 Copyright 2011, RSC. | ||
 | ||
| Fig. 22 Discharge capacities of ZnMn2O4 electrodes. The cells were cycled at room temperature using the same lithium salt, 1 M LiPF6, in three different electrolytes: EC–DMC (1:1 by vol), EC–DEC (3:7 by vol), and PC. Reproduced with permission.21 Copyright 2011, RSC. | ||
3.2 CoMn2O4 anodes
CoMn2O4 has an incomplete normal-type spinel with a tetragonal structure (c/a = 1.14) because of the Jahn–Teller effect of Mn3+ (3d4). Co2+ mainly occupies the A sites, while Mn3+ mainly occupies the B sites. It can store Li+ through the oxidation of metallic Co and Mn to CoO and MnO, respectively, leading to a high initial discharge capacity of 921 mA h g−1 and a reversible capacity of 691 mA h g−1.44,149 Courtel et al.44 first reported sub-micrometer-sized CoMn2O4 particles with a reversible capacity of about 515 mA h g−1 at a current density of 69 mA g−1. However, a capacity of only 330 mA h g−1 after 50 cycles was obtained for this material.Zhou et al.150 prepared double-shelled CoMn2O4 hollow microcubes via a facile co-precipitation and annealing method (600 °C with a ramping rate of 2 °C min−1) using Co0.33Mn0.67CO3 as the precursor. These materials had initial discharge and charge capacities of 1282 and 806 mA h g−1, respectively, and a reversible capacity of 624 mA h g−1 after 50 cycles at 200 mA g−1 (Fig. 23). It is interesting that the initial charge capacity is much higher than the theoretical value (691 mA h g−1) based on the oxidation reactions of metallic Co and Mn nanoparticles to CoO and MnO: 3Li2O + Co + 2Mn ↔ 2MnO + CoO + 6Li+ + 6e−. They suggested that the extra reversible capacity in their studies is attributed to the reversible formation/dissolution of polymeric gel-like films. Single-crystalline CoMn2O4 nano/submicrorods with approximately 100 nm diameters and lengths up to tens of micrometers were assembled151 using hydrothermally-synthesized β-MnO2 nanorods as templates and cobalt hydroxide as the cobalt source; the samples were subsequently annealed at 650 °C in air for 10 h with a heating rate of 5 °C min−1. Reversible capacities of 512 and 400 mA h g−1 at the current densities of 200 and 1000 mA g−1, respectively, were obtained after 100 cycles. Recently, Kim et al.152 synthesized yolk–shell and hollow CoMn2O4 powders by spray pyrolysis from aqueous spray solutions of the cobalt and manganese components with different concentrations of sucrose. The single and double-shelled yolk–shell powders prepared from spray solutions with 0.4 and 0.7 M sucrose have high discharge capacities of 519 and 573 mA h g−1, respectively, in the voltage range of 0.01–3 V vs. Li/Li+ after 40 cycles at a high discharge rate of 800 mA g−1. The group of Lou153 obtained hierarchical CoMn2O4 array micro-/nanostructures with tunable morphologies on conductive stainless steel using a facile solvothermal route with subsequent annealing. The CoMn2O4 nanowires show reversible capacities of 530–215 mA h g−1 when cycled at 1–10 C (1 C = 700 mA g−1). Additional recent studies on CoMn2O4 as anode materials include investigations of multiporous CoMn2O4 spinel quasi-hollow spheres reported by Li et al.154 and hierarchical nanosheets reported by Hu et al.155
 | ||
| Fig. 23 (a) Illustration of the fabrication process for double-shell CoMn2O4 hollow microcubes; typical (b), (c) FESEM and (d), (e) TEM images; (f) discharge–charge profiles; and (g) cycling performance of the double-shelled CoMn2O4 hollow microcubes in the voltage range of 0.01–3.0 V vs. Li/Li+ at current densities of 200 and 800 mA g−1. Reproduced with permission.150 Copyright 2013, Wiley. | ||
4. Conclusion and outlook
Recent progress in the design, synthesis and electrochemical evaluation of Li storage and cycling properties of binary, ternary, and complex Mn-based metal oxides have been reviewed. The motivations of the reviewed studies came from the eager desire to replace the graphite anode to increase energy density, decrease cost, and improve operational safety of the existing LIBs.Two mechanisms were reviewed: the “conversion reaction” mechanism for binary Mn-based oxides (MnO, Mn3O4, Mn2O3 and MnO2) and some ternary oxides (CoMn2O4), as well as the “conversion reaction involving alloying” mechanism for ZnMn2O4. As discussed previously, many strategies including morphology control of the micro-/nanostructures, carbon-coating and composition optimization of the Mn-based oxides have been extensively developed with the aims of improving rate and cycling performance and advancing the practical application of these oxides in LIBs. Despite some important advances in cycling and rate performance of Mn-based oxides as anode materials, the majority of the promising reported results relating to the applications of these oxides in LIBs are experimental results. Furthermore, different experimental results have been obtained even when the same synthetic methods were used to prepare the Mn-based anode materials with the same morphologies. A comprehensive and in-depth understanding of the relationship between the composition, micro/nano-structures and electrochemical Li storage performance of these oxides is therefore still demanded. Achieving this understanding requires in situ characterizations including in situ X-ray diffraction (XRD), in situ X-ray absorption spectroscopy (XAS) and in situ high-resolution transmission electron microscopy (HTEM) to verify the fine structure and reaction pathways of anode materials, and also to assign electrochemical features within specific reactions.
Because of the large electrode polarization and solid electrolyte interphase (SEI) film formation, Mn-based oxides suffer from significant irreversible capacity loss (ICL) during the first cycle. Moreover, the mechanical and chemical properties of SEI films have significant impacts on columbic efficiency, cycling performance and the operational safety of LIBs. Efforts should be made to modify the mechanical properties of SEI films by adding electrolyte additives and/or modifying electrode surfaces with the goal of forming flexible SEI films which can accommodate large volume variation during charging–discharging cycles to achieve improved cycling performance and columbic efficiencies.
The fabrication of full cells using Mn-based oxides as anode materials has not been thoroughly investigated, although several examples have been reported in recent years. Mn-based oxides are suitable to assemble full cells by combining with 4 V and 5 V cathodes. To obtain safe full cells with high cycling and rate performance, significant developments need to be made to optimize anode preparation parameters, cathodes and electrolytes along with their interfaces. Such progress will boost the development of full cells based on Mn-based oxides as anode materials in the coming years.
In spite of the aforementioned challenges, the research area of Mn-based oxide anodes, especially MnO, Mn3O4 and ZnMn2O4, for LIBs has a very bright future due to the extensive growth and application of nanotechnology and in situ characterization techniques.
Acknowledgements
This work was supported by the special financial grant from the China postdoctoral science foundation (2013T60795), the Guangzhou Scientific and Technological Planning Project (2013J4100112), the Guangdong Province Science & Technology Bureau (Industry-Education-Research Project, grant no. 2011B050300008, 2012B050300004), and the Fok Ying Tung Foundation (NRC07/08.EG01).Notes and references
- http://www.isuppli.com/semiconductor-value-chain/pages/strong-growth-to-drive-lithium-ion-battery-market-to-61-billion-by-2020.aspx, accessed on February 29, 2012.
- M. G. Kim and J. Cho, Adv. Funct. Mater., 2009, 19, 1497 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- J. M. Tarascon, N. Recham, M. Armand, J. N. Chotard, P. Barpanda, W. Walker and L. Dupont, Chem. Mater., 2010, 22, 724 CrossRef CAS.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638 CAS.
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565 RSC.
- L. W. Ji, Z. Lin, M. Alcoutlabi and X. W. Zhang, Energy Environ. Sci., 2011, 4, 2682 CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364 CrossRef CAS PubMed.
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc and C. Wang, ACS Nano, 2013, 7, 760 CrossRef CAS PubMed.
- J. Zheng, J. Xiao, X. Yu, L. Kovarik, M. Gu, F. Omenya and J. G. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 13515 RSC.
- M. Gu, I. Belharouak, A. Genc, Z. Wang, D. Wang, K. Amine and C. Wang, Nano Lett., 2012, 12, 5186 CrossRef CAS PubMed.
- A. Manthiram, K. Chemelewski and E. S. Lee, Energy Environ. Sci., 2014, 7, 1399 Search PubMed.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Adv. Mater., 1998, 10, 725 CrossRef CAS.
- Z. G. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. H. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588 CrossRef CAS PubMed.
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353 CrossRef CAS PubMed.
- M. Gu, Y. Li, X. Li, S. Hu, X. Wang, W. Hu and C. Wang, ACS Nano, 2012, 6(9), 8439 CrossRef CAS PubMed.
- M. Gu, Z. Wang, J. G. Connell, D. E. Peaea, L. J. Lauhon, F. Gao and C. Wang, ACS Nano, 2013, 7(7), 6303 CrossRef CAS PubMed.
- R. A. Huggins, J. Power Sources, 1999, 81, 13 CrossRef.
- X. Wen Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536 CrossRef.
- J. Y. Huang, L. Zhong, C. M. Wang, J. P. Sullivan, W. Xu, L. Q. Zhang and J. Li, Science, 2010, 330, 1515 CrossRef CAS PubMed.
- J. S. Chen and X. W. Lou, Small, 2013, 9, 1877 CrossRef CAS PubMed.
- M. Gu, A. Kushima, Y. Shao, J. G. Zhang, J. Liu, N. D. Browning and C. Wang, Nano Lett., 2013, 13, 5203 CrossRef CAS PubMed.
- R. Wang, C. Xu, J. Sun, L. Gao and H. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3427 CAS.
- D. J. Xue, S. Xin, Y. Yan, K. C. Jiang, Y. X. Yin, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 2512 CrossRef CAS PubMed.
- L. F. Cui, R. Ruffo, C. K. Chan, H. L. Peng and Y. Cui, Nano Lett., 2009, 9, 491 CrossRef CAS PubMed.
- M. N. Obrovac, L. Christensen, D. B. Le and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A849 CrossRef CAS PubMed.
- C. M. Park, J. H. Kim, H. Kim and H. J. Sohn, Chem. Soc. Rev., 2010, 39, 3115 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170 CrossRef CAS PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526 RSC.
- H. Wang and H. Dai, Chem. Soc. Rev., 2013, 42, 3088 RSC.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 1388 CAS.
- W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L. J. Wan, Adv. Funct. Mater., 2008, 18, 3941 CrossRef CAS.
- R. Wang, C. Xu, M. Du, J. Sun, L. Gao, P. Zhang and C. Lin, Small, 2014 DOI:10.1002/smll.201303371.
- L. Zhang, H. B. Wu and X. W. Lou, Adv. Energy Mater., 2014, 4, 1300958 Search PubMed.
- Y. Yu, C. H. Chen, J. L. Shui and S. Xie, Angew. Chem., Int. Ed., 2005, 44, 7085 CrossRef CAS PubMed.
- R. Wang, C. Xu, J. Sun, Y. Liu, L. Gao and C. Lin, Nanoscale, 2013, 5, 6960–6967 RSC.
- G. Y. Huang, S. M. Xu, J. L. Wang, L. Y. Li and X. J. Wang, Acta Chim. Sin., 2013, 71, 1589 CAS.
- H. Liu, G. X. Wang, J. Liu, S. Z. Qiao and H. J. Ahn, J. Mater. Chem., 2011, 21, 3046 RSC.
- B. Wang, J. L. Cheng, Y. P. Wu, D. Wang and D. N. He, Electrochem. Commun., 2012, 23, 5 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A1212 CrossRef CAS PubMed.
- J. Gao, M. A. Lowe and H. D. Abruńa, Chem. Mater., 2011, 23, 3223 CrossRef CAS.
- X. P. Fang, X. Lu, X. W. Guo, Y. Mao, Y. S. Hu, J. Z. Wang, Z. X. Wang, F. Wu, H. K. Liu and L. Q. Chen, Electrochem. Commun., 2010, 12, 1520 CrossRef CAS PubMed.
- F. M. Courtel, H. Duncan, Y. Abu-Lebdeh and I. J. Davidson, J. Mater. Chem., 2011, 21, 10206 RSC.
- Y. Y. Yang, Y. J. Zhao, L. F. Xiao and L. Z. Zhang, Electrochem. Commun., 2008, 10, 1117 CrossRef CAS PubMed.
- K. F. Zhong, X. Xia, B. Zhang, H. Li, Z. X. Wang and L. Q. Chen, J. Power Sources, 2010, 195, 3300 CrossRef CAS PubMed.
- X. D. Liu, C. Z. Chen, Y. Y. Zhao and B. Jia, J. Nanomater., 2013, 736375 Search PubMed.
- G. L. Xu, Y. F. Xu, H. Sun, F. Fu, X. M. Zheng, L. Huang, J. T. Li, S. H. Yang and S. G. Sun, Chem. Commun., 2012, 48, 8502 RSC.
- X. W. Li, D. Li, L. Qiao, X. H. Wang, X. L. Sun, P. Wang and D. Y. He, J. Mater. Chem., 2012, 22, 9189 RSC.
- T. Kokubu, Y. Oaki, E. Hosono, H. S. Zhou and H. Imai, Adv. Funct. Mater., 2011, 21, 3673 CrossRef CAS.
- D. Larcher, S. Beattie, M. Morcrette, K. Edstroem, J. C. Jumas and J. M. Tarascon, J. Mater. Chem., 2007, 17, 3759 RSC.
- Y. F. Deng, Q. M Zhang, Z. C. Shi, L. J. Han, F. Peng and G. H. Chen, Electrochim. Acta, 2012, 76, 495 CrossRef CAS PubMed.
- K. F. Zhong, B. Zhang, S. H. Luo, W. Wen, H. Li and X. J. Huang, J. Power Sources, 2011, 196, 6802 CrossRef CAS PubMed.
- Z. H. Cui, X. X. Guo and H. Li, J. Power Sources, 2013, 244, 731 CrossRef CAS PubMed.
- J. C. Guo, Q. Liu, C. S. Wang and M. R. Zachariah, Adv. Funct. Mater., 2012, 22, 803 CrossRef CAS.
- C. J. Chae, J. H. Kim, J. M. Kim, Y. K. Sun and J. K. Lee, J. Mater. Chem., 2012, 22, 17870 RSC.
- W. Luo, X. L. Hu, Y. M. Sun and Y. H. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 1997 CAS.
- Y. Xia, Z. Xiao, X. Dou, H. Huang, X. H. Lu, R. J. Yan, Y. P. Gan, W. J. Zhu, J. P. Tu, W. K. Zhang and X. Y. Tao, ACS Nano, 2013, 7, 7083 CrossRef CAS PubMed.
- X. F. Sun, Y. L. Xu, P. Ding, M. R. Jia and G. Ceder, J. Power Sources, 2013, 224, 690 CrossRef PubMed.
- C. T. Hsieh, C. Y. Lin and J. Y. Lin, Electrochim. Acta, 2011, 56, 8861 CrossRef CAS PubMed.
- D. F. Qiu, L. Y. Ma, M. B. Zheng, Z. X. Lin, B. Zhao, Z. Wen, Z. B. Hu, L. Pu and Y. Shi, Mater. Lett., 2012, 84, 9 CrossRef CAS PubMed.
- K. J. Zhang, P. X. Han, L. Gu, L. X. Zhang, Z. H. Liu, Q. S. Kong, C. J. Zhang, S. M. Dong, Z. Y. Zhang, J. H. Yao, H. X. Xu, G. L. Cui and L. Q. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 658 CAS.
- Y. J. Mai, D. Zhang, Y. Q. Qiao, C. D. Gu, X. L. Wang and J. P. Tu, J. Power Sources, 2012, 216, 201 CrossRef CAS PubMed.
- Y. M. Sun, X. L. Hu, W. Luo, F. F. Xia and Y. H. Huang, Adv. Funct. Mater., 2013, 23, 2436 CrossRef CAS.
- Y. L. Ding, C. Y. Wu, H. M. Yu, J. Xie, G. S. Cao, T. J. Zhu, X. B. Zhao and Y. W. Zeng, Electrochim. Acta, 2011, 56, 5844 CrossRef CAS PubMed.
- B. Sun, Z. X. Chen, H. S. Kim, H. Ahn and G. X. Wang, J. Power Sources, 2011, 196, 3346 CrossRef CAS PubMed.
- X. W. Li, S. L. Xiong, J. F. Li, X. Liang, J. Z. Wang, J. Bai and Y. T. Qian, Chem.–Eur. J., 2013, 19, 11310 CrossRef CAS PubMed.
- X. N. Li, Y. C. Zhu, X. Zhang, J. W. Liang and Y. T. Qian, RSC Adv., 2013, 3, 10001 RSC.
- L. W. Su, Y. R. Zhong, J. P. Wei and Z. Zhou, RSC Adv., 2013, 3, 9035 RSC.
- Y. M. Sun, X. L. Hu, W. Luo and Y. H. Huang, J. Mater. Chem., 2012, 22, 19190 RSC.
- S. R. Li, Y. Sun, S. Y. Ge, Y. Qiao, Y. M. Chen, I. Lieberwirth, Y. Yu and C. H. Chen, Chem. Eng. J., 2012, 192, 226 CrossRef CAS PubMed.
- X. Zhang, Z. Xing, L. Wang, Y. Zhu, Q. Li, J. Liang, Y. Yu, T. Huang, K. Tang, Y. Qian and X. Shen, J. Mater. Chem., 2012, 22, 17864 RSC.
- C. J. Chae, H. Y. Park, D. W. Kim, J. S. Kim, E. S. Oh and J. K. Lee, J. Power Sources, 2013, 244, 214 CrossRef CAS PubMed.
- G. L. Xu, Y. F. Xu, J. C. Fang, F. Fu, H. Sun, L. Huang, S. H. Yang and S. G. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6316 CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 46 CrossRef.
- D. Pasero, N. Reeves and A. R. West, J. Power Sources, 2005, 141, 156 CrossRef CAS PubMed.
- Q. Fan and M. S. Whittingham, Mater. Res. Soc. Symp. Proc., 2007, 972 DOI:0972-AA07-03-BB08-03.
- Q. Fan and M. S. Whittingham, Electrochem. Solid-State Lett., 2007, 10, A48 CrossRef CAS PubMed.
- P. Li, C. Y. Nan, Z. Wei, J. Lu, Q. Peng and Y. D. Li, Chem. Mater., 2010, 22, 4232 CrossRef CAS.
- X. P. Shen, Z. Y. Ji, H. J. Miao, J. Yang and K. M. Chen, J. Alloys Compd., 2011, 509, 5672 CrossRef CAS PubMed.
- W. Xiao, J. S. Chen and X. W. Lou, CrystEngComm, 2011, 13, 5685 RSC.
- D. P. Dubal and R. Holze, RSC Adv., 2012, 2, 12096 RSC.
- Z. C. Bai, B. Sun, N. Fan, Z. C. Ju, M. H. Li, L. Q. Xu and Y. T. Qian, Chem.–Eur. J., 2012, 18, 15049 CrossRef PubMed.
- Z. C. Bai, N. Fan, Z. C. Ju, C. L. Guo, Y. T. Qian, B. Tang and S. L. Xiong, J. Mater. Chem. A, 2013, 1, 10985 CAS.
- J. Z. Wang, N. Du, H. Wu, H. Zhang, J. X. Yu and D. R. Yang, J. Power Sources, 2013, 222, 32 CrossRef CAS PubMed.
- Q. Hao, J. P. Wang and C. X. Xu, J. Mater. Chem. A, 2014, 2, 87 CAS.
- Y. Ma, C. L. Fang, B. Ding, G. Ji and J. Y. Lee, Adv. Mater., 2013, 25, 4646 CrossRef CAS PubMed.
- S. M. Oh, I. Y. Kim, S. J. Kim, W. Jung and S. J. Hwang, Mater. Lett., 2013, 107, 221 CrossRef CAS PubMed.
- T. Li, Y. Y. Wang, R. Tang, Y. X. Qi, N. Lun, Y. J. Bai and R. H. Fan, ACS Appl. Mater. Interfaces, 2013, 5, 10975 Search PubMed.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682 CAS.
- H. L. Wang, L. F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Y. Liang, Y. Cui and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS PubMed.
- N. Lavoie, P. R. L. Malenfant, F. M. Courtel, Y. Abu-Lebdeh and I. J. Davidson, J. Power Sources, 2012, 213, 249 CrossRef CAS PubMed.
- L. Li, Z. P. Guo, A. J. Du and H. K. Liu, J. Mater. Chem., 2012, 22, 3600 RSC.
- S. Y. Liu, J. Xie, Y. X. Zheng, G. S. Cao, T. J. Zhu and X. B. Zhao, Electrochim. Acta, 2012, 66, 271 CrossRef CAS PubMed.
- Z. Q. Li, N. N. Liu, X. K. Wang, C. B. Wang, Y. X. Qi and L. W. Yin, J. Mater. Chem., 2012, 22, 16640 RSC.
- Z. H. Wang, L. X. Yuan, Q. G. Shao, F. Huang and Y. H. Huang, Mater. Lett., 2012, 80, 110 CrossRef CAS PubMed.
- C. B. Wang, L. W. Yin, D. Xiang and Y. X. Qi, ACS Appl. Mater. Interfaces, 2012, 4, 1636 CAS.
- I. Nam, N. D. Kim, G. P. Kim, J. Park and J. Yi, J. Power Sources, 2013, 244, 56 CrossRef CAS PubMed.
- L. Wang, Y. H. Li, Z. D. Han, L. Chen, B. Qian, X. F. Jiang, J. Pintoc and G. Yang, J. Mater. Chem. A, 2013, 1, 8385 CAS.
- S. Luo, H. C. Wu, Y. Wu, K. L. Jiang, J. P. Wang and S. S. Fan, J. Power Sources, 2014, 249, 463 CrossRef CAS PubMed.
- C. Chen, H. Jian, X. X. Fu, Z. M. Ren, M. Yan, G. D. Qian and Z. Y. Wang, RSC Adv., 2014, 4, 5367 RSC.
- X. J. Liu, H. Yasuda and M. Yamachi, J. Power Sources, 2005, 146, 510 CrossRef CAS PubMed.
- P. Lavela, J. L. Tirado and C. Vidal-Abarca, Electrochim. Acta, 2007, 52, 7986 CrossRef CAS PubMed.
- Y. Cai, S. Liu, X. M. Yin, Q. Y. Hao, M. Zhang and T. H. Wang, Phys. E, 2010, 43, 70 CrossRef CAS PubMed.
- X. P. Shen, Z. Y. Ji, H. J. Miao, J. Yang and K. M. Chen, J. Alloys Compd., 2011, 509, 5672 CrossRef CAS PubMed.
- Y. C. Qiu, G. L. Xu, K. Y. Yan, H. Sun, J. W. Xiao, S. H. Yang, S. G. Sun, L. M. Jin and H. Deng, J. Mater. Chem., 2011, 21, 6346 RSC.
- Y. F. Deng, Z. E. Li, Z. C. Shi, H. Xu, F. Peng and G. H. Chen, RSC Adv., 2012, 2, 4645 RSC.
- L. Hu, Y. K. Sun, F. P. Zhang and Q. W. Chen, J. Alloys Compd., 2013, 576, 86 CrossRef CAS PubMed.
- L. Chang, L. Q. Mai, X. Xu, Q. Y. An, Y. L. Zhao, D. D. Wang and X. Feng, RSC Adv., 2013, 3, 1947 RSC.
- Y. H. Dai, H. Jiang, Y. J. Hu and C. Z. Li, RSC Adv., 2013, 3, 19778 RSC.
- S. L. Chen, F. Liu, Q. J. Xiang, X. H. Feng and G. H. Qiu, Electrochim. Acta, 2013, 106, 360 CrossRef CAS PubMed.
- X. Zhang, Y. T. Qian and Y. C. Zhu, Nanoscale, 2014, 6, 1725 RSC.
- M. W. Xu, Y. B. Niu, S. J. Bao and C. M. Li, J. Mater. Chem. A, 2014, 2, 3749 CAS.
- Y. H. Wang, Y. H. Wang, D. S. Jia, Z. Peng, Y. Y. Xia and G. F. Zheng, Nano Lett., 2014, 14, 1080 CrossRef CAS PubMed.
- J. X. Dai, S. F. Y. Li, K. S. Siow and Z. Q. Gao, Electrochim. Acta, 2000, 45, 2211 CrossRef CAS.
- M. S. Wu, P. C. J. Chiang, J. T. Lee and J. C. Lin, J. Phys. Chem. B, 2005, 109, 23279 CrossRef CAS PubMed.
- B. Li, G. Rong, Y. Xie, L. Huang and C. Feng, Inorg. Chem., 2006, 45, 6404 CrossRef CAS PubMed.
- J. Zhao, Z. Tao, J. Liang and J. Chen, Cryst. Growth Des., 2008, 8, 2799 CAS.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9, 1002 CrossRef CAS PubMed.
- L. H. Li, C. Y. Nan, J. Lu, Q. Peng and Y. D. Li, Chem. Commun., 2012, 48, 6945 RSC.
- J. Wang, J. Liu, Y. C. Zhou, P. Hodgson and Y. C. Li, RSC Adv., 2013, 3, 25937 RSC.
- M. Kundu, C. C. A. Ng, D. Y. Petrovykh and L. F. Liu, Chem. Commun., 2013, 49, 8459 RSC.
- H. Xia, M. Lai and L. Lu, J. Mater. Chem., 2010, 20, 6896 RSC.
- Y. S. Yun, J. M. Kim, H. H. Park, J. Lee, Y. S. Huh and H. J. Jin, J. Power Sources, 2013, 244, 747 CrossRef CAS PubMed.
- J. X. Lia, M. Z. Zou, Y. Zhao, Y. B. Lin, H. Lai, L. H. Guan and Z. G. Huang, Electrochim. Acta, 2013, 111, 165 CrossRef PubMed.
- Y. Wang, Z. J. Han, S. F. Yu, R. R. Song, H. H. Song, K. Ostrikov and H. Y. Yang, Carbon, 2013, 64, 230 CrossRef CAS PubMed.
- C. X. Guo, M. Wang, T. Chen, X. W. Lou and C. M. Li, Adv. Energy Mater., 2011, 1, 736 CrossRef CAS.
- A. P. Yu, H. W. Park, A. Davies, D. C. Higgins, Z. W. Chen and X. C. Xiao, J. Phys. Chem. Lett., 2011, 2, 1855 CrossRef CAS.
- L. Li, A. R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298 CrossRef CAS PubMed.
- Y. Zhang, H. Liu, Z. H. Zhu, K. W. Wong, R. Mi, J. Mei and W. M. Lau, Electrochim. Acta, 2013, 108, 465 CrossRef CAS PubMed.
- Y. Y. Li, Q. W. Zhang, J. L. Zhu, X. L. Wei and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3163–3168 CAS.
- X. Gu, L. Chen, Z. C. Ju, H. Y. Xu, J. Yang and Y. T. Qian, Adv. Funct. Mater., 2013, 23, 4049 CrossRef CAS.
- J. Y. Liao, D. Higgins, G. Lui, V. Chabot, X. C. Xiao and Z. W. Chen, Nano Lett., 2013, 13, 5467 CrossRef CAS PubMed.
- J. Fang, Y. F. Yuan, L. K. Wang, H. L. Ni, H. L. Zhu, J. L. Yang, J. S. Gui, Y. B. Chen and S. Y. Guo, Electrochim. Acta, 2013, 112, 364 CrossRef CAS PubMed.
- M. S. Wu, P. C. J. Chiang, J. T. W. Xiao, J. S. Chen, Q. Lu and X. W. Lou, J. Phys. Chem. C, 2010, 114, 12048 Search PubMed.
- L. F. Xiao, Y. Y. Yang, J. Yin, Q. Li and L. Z. Zhang, J. Power Sources, 2009, 194, 1089 CrossRef CAS PubMed.
- Y. F. Deng, S. D. Tang, Q. M. Zhang, Z. C. Shi, L. T. Zhang, S. Z. Zhan and G. H. Chen, J. Mater. Chem., 2011, 21, 11987 RSC.
- F. M. Courtel, Y. Abu-Lebdeh and I. J. Davidson, Electrochim. Acta, 2012, 71, 123 CrossRef CAS PubMed.
- S. W. Kim, H. W. Lee, P. Muralidharan, D. H. Seo, W. S. Yoon, D. K. Kim and K. Kang, Nano Res., 2011, 4, 505 CrossRef CAS PubMed.
- P. F. Teh, Y. Sharma, S. S. Pramana and M. Srinivasan, J. Mater. Chem., 2011, 21, 14999 RSC.
- L. Zhou, H. B. Wu, T. Zhu and X. W. Lou, J. Mater. Chem., 2012, 22, 827 RSC.
- H. B. Wang, F. Y. Cheng, Z. L. Tao, J. Liang and J. Chen, Chin. J. Inorg. Chem., 2011, 27, 816 CAS.
- X. F. Chen, L. Qie, L. L. Zhang, W. X. Zhang and Y. H. Huang, J. Alloys Compd., 2013, 559, 5 CrossRef CAS PubMed.
- G. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609 CrossRef CAS PubMed.
- Z. C. Bai, N. Fan, C. H. Sun, Z. C. Ju, C. L. Guo, J. Yang and Y. T. Qian, Nanoscale, 2013, 5, 2442 RSC.
- P. F. Teh, Y. Sharma, Y. W. Ko, S. S. Pramana and M. Srinivasan, RSC Adv., 2013, 3, 2812 RSC.
- J. G. Kim, S. H. Lee, Y. M. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 11321 CAS.
- Z. M. Zheng, Y. L. Cheng, X. B. Yan, R. T. Wang and P. Zhang, J. Mater. Chem. A, 2014, 2, 149 CAS.
- L. J. Wang, B. Liu, S. H. Ran, L. M. Wang, L. N. Gao, F. Y. Qu, D. Chen and G. Z. Shen, J. Mater. Chem. A, 2013, 1, 2139 CAS.
- L. Zhou, D. Y. Zhao and X. W. Lou, Adv. Mater., 2012, 24, 745 CrossRef CAS PubMed.
- L. J. Wang, B. Liu, S. H. Ran, L. M. Wang, L. N. Gao, F. Y. Qu, D. Chen and G. Z. Shen, J. Mater. Chem. A, 2013, 1, 2139 CAS.
- M. H. Kim, Y. J. Hong and Y. C. Kang, RSC Adv., 2013, 3, 13110 RSC.
- L. Yu, L. Zhang, H. B. Wu, G. Q. Zhang and X. W. Lou, Energy Environ. Sci., 2013, 6, 2664 CAS.
- J. F. Li, S. L. Xiong, X. W. Li and Y. T. Qian, Nanoscale, 2013, 5, 2045 RSC.
- L. Hu, H. Zhong, X. R. Zheng, Y. M. Huang, P. Zhang and Q. W. Chen, Sci. Rep., 2012, 2, 986 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |