
Dual dispersive extraction combined with electrothermal vaporization inductively coupled plasma mass spectrometry for determination of trace REEs in water and sediment samples
Abstract
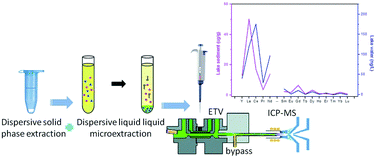
A simple and efficient two-step method based on dispersive solid phase extraction (D-SPE) and dispersive liquid–liquid microextraction (DLLME) has been developed for the separation and preconcentration of 15 rare earth elements (REEs) from environmental water and sediment samples, followed by electrothermal vaporization-inductively coupled plasma mass spectrometry (ETV-ICP-MS) detection. With Chelex 100 as the adsorbent of D-SPE, target REEs were firstly extracted and the retained REEs were then desorbed by 0.1 mol L−1 HNO3. After 125 mmol L−1 Tris and 40 mmol L−1 1-phenyl-3-methyl-4-benzoylpyrazolone (PMBP) were added into the above elution solution, target REEs were further preconcentrated into CCl4 by DLLME. The developed dual extraction technique exhibited high enrichment factors (234 to 566-fold) and good anti-interference ability. Various parameters affecting the extraction of target REEs by D-SPE and DLLME were investigated in detail. Under the optimal conditions, the limits of detection (LODs, 3σ) for target REEs were in the range of 0.003–0.073 ng L−1 with the relative standard deviations (CY,La,Ce,Pr,Nd,Gd,Dy = 1.0 ng L−1, CSm,Eu,Tb,Ho,Er,Tm,Yb,Lu = 0.2 ng L−1, n = 7) ranging from 6.7 to 11.5%. The proposed method of D-SPE-DLLME-ETV-ICP-MS was successfully applied to the determination of 15 REEs in water and sediment samples with the recoveries of 78–115% and 75–117% for the spiked water and sediment samples, respectively. To validate the accuracy of the method, a Certified Reference Material of GBW07301a stream sediment was analyzed and the determined values were in good agreement with the certified values.
 Please wait while we load your content...
Please wait while we load your content...

