Reduction intermediates of graphene oxide for low temperature reduction electrode material†
Sung Il Ahn*a,
Kukjoo Kimb,
Jun Young Kima,
Eun Se Kima,
Ji Ye Hana,
Ji Won Eoma,
Kyeong-Keun Choic,
Jung-Chul Parka,
Sung-Hoon Kima,
Euh Duck Jeongd and
Kyung Cheol Choib
aDepartment of Engineering in Energy and Applied Chemistry, Silla University, Busan 617-736, Republic of Korea. E-mail: siahn@silla.ac.kr
bDepartment of Electrical Engineering, KAIST, Daejeon 305-701, Republic of Korea
cNational Center for Nanomaterials Technology (NCNT), Pohang 790-784, Republic of Korea
dHigh-Technology Components & Materials Research Center & Busan Center, Korea Basic Science Institute (KBSI), Busan 609-735, Republic of Korea
First published on 12th May 2014
Abstract
Graphene oxide (GO) with an abnormal structure was isolated in an alcoholic solvent. An abnormally low C–O/C–C ratio and large red-shift of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration indicated that the GO was a reduction intermediate of graphene oxide, which could be reduced more easily under mild conditions, even without reducing chemicals.
O vibration indicated that the GO was a reduction intermediate of graphene oxide, which could be reduced more easily under mild conditions, even without reducing chemicals.
Considering the superior properties of graphene (i.e., giant carrier mobility,1,2 as well as outstanding mechanical,3 thermal,4 chemical,5 and optical properties6) discovered since it was first isolated from graphite using a simple taping method,7 the enormous current interest in it is only natural. Graphene oxide (GO), which is the highly oxygenated graphene of a single layer of graphite, has also been researched extensively due to the convenience of using it to make composites with polymers,8,9 thin films with a large area,10 paper-like materials,11,12 and the convenience of mass production at low cost. Although reports on GO related materials and their applications have reached tens of thousands during the last decade, many unknowns concerning structure and properties of GO are waiting to be resolved. There are two well-known preparation methods of GO: the Staudenmaier method based on KClO3 and HNO3 as oxidants, originated by Brodie who discovered the ability to oxidize graphite.13,14 The second method is that of Hummers and Offeman; using KMnO4 & H2SO4 to oxidize graphite.15 Most of the research on GO has been conducted by modifications of these methods. The overall process for the preparation of GO, whether modified from these methods or not, seems so simple that one can expect GO with reasonable uniformity. However, taking internal factors into account, for example the distribution of oxygen functional groups on GO, suggests that exfoliated GO cannot be a single compound, but instead a mixture of various sheets of GO having various oxygen groups and different carbons where those oxygen groups are placed. Consequently, we can affirm that the GO reported in tens of thousands of research papers are not exactly the same materials, but are statistically similar materials. How similar they are, is questionable, and is now believed to have made for confusion in the interpretation of structure and properties of GO. One of the experiments on GO by our research group was to investigate periodically exfoliated GO from the same graphite oxide to obtain purified GO. Based on reports revealing that GO consists of oxygenated and non-oxygenated regions of sp2-carbon-clusters (SPCCs),16,17 we considered that the chemical structure of GO varied according to the size of SPCCs (see ESI, Fig. S1†). We thought that if a GO sheet had different kinds of SPCCs, the variants should have different dispersion stabilities in a solvent.
Based on this simple concept, we analyzed for periodically exfoliated GO from the same graphite oxide in an alcoholic solvent and found that the exfoliated GO had an abnormal structure suspected as an oxidation intermediate of graphene, and/or a reduction intermediate of GO. In this study, we characterized the structure of the abnormal GO exfoliated in the alcoholic solvent (e-GO). In addition, partially reduced GO (rGO) was introduced to compare a reduction intermediate of GO. We then showed that the intermediates were made of an easily reducible GO sheet at low temperature, which could be applied to the printed electronics as an electrode material. Except for the direct reduction of GO film with hydrogen gas,18 which requires a high reaction temperature, the reduction of GO using chemical methods has a critical problem with the agglomeration of reduced GO (RGO; completely reduced form of GO). We had difficulty obtaining a high quality thin film with the coagulated RGO. For this reason, gaseous hydrazine has been applied to the reduction of thin film GO.19 However, it is hard to obtain complete RGO film due to the diffusion limit of gaseous hydrazine over a short reaction time.20 We considered how to obtain stable RGO or how to control the oxygen content of RGO using hydrazine in a solution without coagulation of the RGO sheet. If such a process is possible, we will be able to modify the electrical property of RGO with reasonable coating property for further applications. For this purpose, in this study, we developed and characterized e-GO and rGO with high stability in water.
A considerable amount of e-GO (about 0.11 g) was obtained, while the amount of exfoliated GO for 30 min sonication in distilled water from the same graphite oxide after the exfoliation of e-GO (ew-GO) was about 0.25 g. A dispersion study on graphite oxide in various solutions indicated that GO was hardly dispersed in the absolute ethanol.21 However, we found that some of GO from graphite oxide can be well dispersed in an ethanol solution with a small amount of distilled water as shown in Fig. 1f. Far from our expectation, the e-GO had an abnormally low C–O/C–C ratio compared to about 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (including CO) of GOs reported in the literature as shown in Fig. 1b.22,23 The same result was obtained several times at different oxidation conditions (see ESI, Fig. S2†). Apparently, the heterogeneity of reaction mixture is the main reason for the formation of e-GO. In Fig. 1e, synthesized XPS spectra of C 1s from e-GO and ew-GO by a weight factor after normalization to C–C had a similar curve compared to the Ref. GO, which means that the e-GO and ew-GO are hardly affected by the exfoliation conditions. What is more, e-GO is the earliest portion detached from GO sheets of graphite oxide by sonication for 5 min. Imagine the frayed surface of graphite after oxidation. A common sense on GO exfoliation would be that highly oxygenated GO should be the first detached from graphite oxide. Interestingly, the peak position of the CO vibration of e-GO in Fig. 2b is red-shifted about 28 cm−1 (from 1725 cm−1 to 1697 cm−1) compared to ew-GO.
:2 (including CO) of GOs reported in the literature as shown in Fig. 1b.22,23 The same result was obtained several times at different oxidation conditions (see ESI, Fig. S2†). Apparently, the heterogeneity of reaction mixture is the main reason for the formation of e-GO. In Fig. 1e, synthesized XPS spectra of C 1s from e-GO and ew-GO by a weight factor after normalization to C–C had a similar curve compared to the Ref. GO, which means that the e-GO and ew-GO are hardly affected by the exfoliation conditions. What is more, e-GO is the earliest portion detached from GO sheets of graphite oxide by sonication for 5 min. Imagine the frayed surface of graphite after oxidation. A common sense on GO exfoliation would be that highly oxygenated GO should be the first detached from graphite oxide. Interestingly, the peak position of the CO vibration of e-GO in Fig. 2b is red-shifted about 28 cm−1 (from 1725 cm−1 to 1697 cm−1) compared to ew-GO.
 | ||
| Fig. 1 (a) An image of graphite oxide, XPS spectra of C 1s from: (b) exfoliated GO for 5 min sonication in the 98% ethanol solvent (e-GO), (c) exfoliated GO for 30 min sonication in distilled water from the same graphite oxide after the exfoliation of e-GO (ew-GO), (d) exfoliated GO for 30 min sonication in distilled water from a graphite oxide using the same method as for preparation of (b) (Ref. GO), (e) synthesized XPS spectra of C 1s from e-GO and ew-GO after normalization to C–C and then input weight factor, (f) photographic image of e-GO and ew-GO samples. All the XPS spectra were fitted after a Shirley background correction. | ||
 | ||
| Fig. 2 (a) Normalized FT-IR spectra to CO vibration of various GO samples, (b) selected range of (a) showing a red-shift of e-GO, (c) normalized FT-IR spectra to CO vibration of gradually reduced ew-GO samples from rGO1 to rGO3 showing a red-shift, (d) normalized FT-IR spectra to CO vibration of gradually reduced ew-GO samples of rGO4 and rGO5 showing a partial blue-shift compared to rGO3. | ||
Considering the vibration of CO by unsaturated carboxylic group placed below 1700 cm−1, this result is indicating that e-GO is less oxidized than normal GO.
We had obtained intermediates of RGO through a weak reduction of GO using hydrazine and compared the peak position of CO. In Fig. 2c, we can clearly see the shifting of the CO vibration as the degree of reduction is increased by an increase of the reducing agent (added 0.034 (rGO1), 0.064 (rGO2), 0.102 (rGO3), 0.136 (rGO4), 0.170 (rGO5), and 0.34 ml (rGO6) of 0.8 wt% hydrazine to 30 ml of 0.02 wt% Ref. GO). Based on the FT-IR results, we consider the unusual e-GO as a kind of oxidation intermediate of graphene. In the cases of rGO4 and rGO5, reduced by more hydrazine, they showed a slightly blue-shifted (about 8 cm−1) CO vibration compared to rGO3 (Fig. 2d), which will be explained in the next section.
An uncommon behavior was observed during our investigation on the rGO. Reduction of GO in an aqueous solution gave only aggregated RGO due to the van der Waals forces between sheets of RGO, which is the same as with graphite24 and low polarity of RGO in polar aqueous media. Thus, we may be confident in our prediction that a reaction mixture of GO is more unstable in aggregation as the reductant increases. Naturally, we will say that a higher content of reducing agent will also result in faster aggregation of rGO. However, as shown in Fig. 3a, the lower end in a certain range of reducing agent content produces faster aggregation of rGO (added 0.034 (rGO1′), 0.064 (rGO2′), 0.102 (rGO3′), 0.136 (rGO4′), and 0.170 (rGO5′) of 8 wt% hydrazine to 30 ml of 0.2 wt% Ref. GO). The same experiments were carried out several times and had the same coagulation tendency. The surface image of spin coated rGO' samples showed a similar tendency of coagulation as seen in Fig. 3b. This interesting phenomenon appears to be related to the structure of reduction intermediates of GO, which have various SPCCs, the size of which is dependent on the concentration of the reducing agent.
 | ||
| Fig. 3 (a) Photographic image of rGO' sample showing a degree of coagulation as the hydrazine content and reaction time increase, (b) surface microscope image of spin coated rGO' sample according to reaction time. | ||
As we mentioned, GO consists of non-oxygenated and oxygenated regions.16,17 When a small amount of reducing agent is reacted with GO, we can image that the hydrazine starts to seek oxygenated sites to reduce where they become a more stable intermediate of RGO. The moderately slow reaction rate of hydrazine in the reaction mixture apparently allows enough time to find those sites (see ESI, Fig. S3†). Consequently, the most likely sites on GO to be reduced are the sites near preexisting sp2 carbon, since the reduced sites are easily conjugated with them and thus stabilized thermodynamically. This gives a reduction intermediate with large SPCCs on GO, which leads to coagulation of rGO quickly. Increasing the hydrazine content seems to reduce most of the oxygenated sites of GO and to form new small SPCCs.16 It still has oxygenated regions surrounding the sp2 carbons, which obstructs and delays the coagulation between sheets of rGO in the solution. The blue-shifted CO vibration in Fig. 2d and the increasing D band of Raman spectra in Fig. 4a for rGO4 and rGO5 as the hydrazine content increases appears related to the reduction intermediate of GO with small SPCCs. On rGO6, further reduction using hydrazine creates giant SPCCs on GO by combining the small SPCCs and leads to faster coagulation than rGO5 due to an increase of the van der Waals force between rGO sheets in the solution.
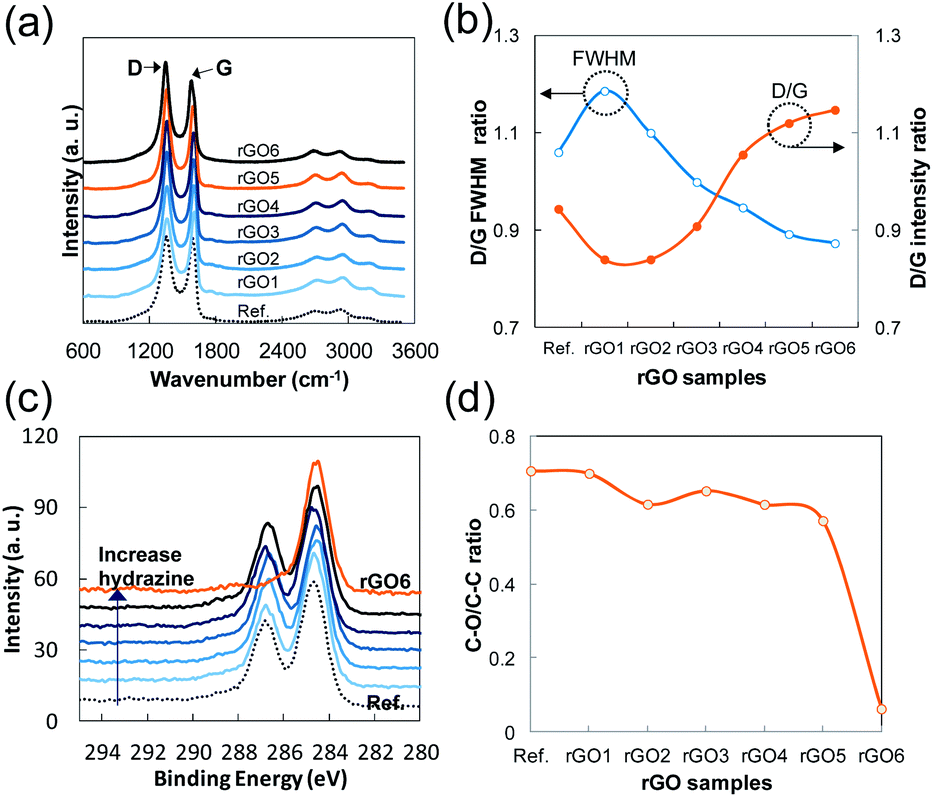 | ||
| Fig. 4 (a) Normalized Raman spectra of rGO to G band (dotted line indicates the initially exfoliated GO as a reference), (b) FWHM of D band of rGO sample compared to the reference GO and D/G band ratio of each rGO including the reference GO. (c) C 1s XPS spectra of rGO normalized to C–C bond energy, (d) C–O/C–C ratio of each rGO and the reference GO (note that minor hidden peaks such as C–N and CO are ignored). | ||
The broadness and intensity of the G and D bands in Raman spectra from GO or rGO is related to the oxide concentration, the structural defects, and/or the disordered carbon bonds as has been reported in many studies.25,26 Thus, the degree of oxidation in GO can be estimated by the D/G intensity ratio and the broadness of the bands. In the case of rGO, the intensity of the D band depends mostly on the size of SPCCs in a similar C–O/C–C ratio of rGO because a SPCC is surrounded by non-sp2 carbons including oxygen. In other words, the size of SPCCs is determined by the number of SPCC in a similar C–O/C–C ratio of rGO (Fig. 4d). Thus, the D band increases as the number of SPCC increases if the SPCC has Raman active six-folded symmetries of condensed benzene rings: breathing mode of A1g symmetry activated by disordering.25 In Fig. 4b, we observe a fluctuation in the D/G intensity ratio as the hydrazine content increases, while the full width at half maximum (FWHM) of the D band decreases greatly. The Raman spectra of rGO1 and rGO2 have slightly lower D/G ratios and broader D bands than those of the reference GO, which indicates that the size of preexisting SPCCs on rGO1 and rGO2 is increased by reduction rather than new SPCCs being created. In contrast, rGO4 and rGO5 have higher D/G ratios and sharper D bands than the rGO1, rGO2 or reference GO. Considering the increase of the D band and supposing the decrease of FWHM is directly related to the number of SPCCs with similar six-folded symmetries, this result confirms that a significant number of small sized SPCCs with a similar six-folded symmetries is created on rGO reacted with a high content of hydrazine in this study.
During the investigation on the reduction intermediates of GO, we also found an unusual result regarding the electrical conduction of rGO. When hydrazine is added to the GO dispersion, we would expect that the higher content of hydrazine would produce RGO film with higher electrical conduction in a certain range of hydrazine content (before adding an excess of hydrazine to the GO dispersion). As shown in Fig. 5a and b, however, all the rGO, regardless of hydrazine content, had similar conductivities when they were spin-coated immediately after the preparation of rGO' samples. This result suggests that a drying temperature of 120 °C can drive every hydrazine in the solution to react with oxygenated sites immediately and form a small non-oxygenated region on a rGO sheet, but in large numbers. If the oxygenated area is sufficiently close and/or small to conjugate with the non-oxygenated regions of SPCCs, they can easily be reduced by a drying temperature of 120 °C and stabilized as part of the honeycomb structure of graphene. The sudden drop of the C–O/C–C ratio of rGO6 in Fig. 4d is seemingly related to this interconnection between SPCCs. A similar phenomenon is observed in electrical conductance of e-GO film. Generally, GO has been known as a non-conductor due to fact that its oxygenated carbons are hardly reduced at temperatures below 200 °C in a few minute.
 | ||
| Fig. 5 (a) Sheet resistances of as-reacted rGO' films dependent on transmittance, (b) sheet resistances of rGO' films dependent on reaction time compared at around 72% transmittance of 550 nm (the insets are microscope images of the films), (c) sheet resistances of e-GO films according to heating temperature and time, (d) transmittance of e-GO films in (c). | ||
In the case of e-GO, it is easily reduced by a heat treatment below 200 °C for a few minutes as shown in Fig. 5c. In addition, the TEM image of e-GO in Fig. 6 also supports the hypothesis by showing small SPCCs placed irregularly on e-GO.
 | ||
| Fig. 6 TEM image of exfoliated GO in the alcoholic solvent for 5 min sonication (e-GO). Inset is enlarged photo indicating SPCCs (yellow circles) and highly oxidized areas (white circles). | ||
Conclusions
Until now, the C–O/C–C ratio of GO has been considered one of the most important factors that control properties of GO. We would like to suggest that the size of SPCCs and their distribution on GO are more important factors at a similar C–O/C–C ratio. Based on the controlled reduction of GO, the size of SPCCs was determined by a certain ratio between the hydrazine and oxygen contents on GO. At a low ratio, owing to the thermodynamic effect of conjugation with the preexisting SPCCs, the reduction of GO resulted in an enlargement of preexisting SPCCs on GO rather than the creation of new small SPCCs; this resulted in the coagulation of rGO. At a high ratio, all the oxygen group on GO are reacted to create new small SPCCs on the GO. This rGO has a lower attraction force between sheets and still has many non-sp2 bonds at the boundaries of the small SPCCs, which prohibit coagulation; this process seems to be a major reason for the high stability of rGO in the solution. Further reduction creates giant SPCCs on GO by combining the small SPCCs, leading to coagulation. Another discovery is that the proportion of oxidation intermediate of graphene can be estimated by the position of the CO vibration when we separate the intermediate using the method in this work. Based on the results, we should consider that the presence of intermediates in the GO mixture has affected somewhat the interpretation of GO in the substantial literature on this topic. In addition, we were able to figure out which structure of GO should be reduced more easily under mild conditions, even without reducing chemicals, and resulted in RGO with a similar or higher conductivity than normal RGO, which is highly desired property as an electrode material in the field of printed electronics.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (no. R11-2007-045-02001-0 and 2012R1A1A2006729).Notes and references
- S. V. Morozov, K. S. Novoselov, M. I. Katsnelson, F. Schedin, D. C. Elias, J. A. Jaszczak and A. K. Geim, Phys. Rev. Lett., 2008, 100, 016602 CrossRef CAS.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491 CrossRef CAS PubMed.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef CAS PubMed.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- M. Yoonessi, Y. Shi, D. A. Scheiman, M. Lebron-Colon, D. M. Tigelaar, R. A. Weiss and M. A. Meador, ACS Nano, 2012, 6, 7644 CrossRef CAS PubMed.
- X. Huang, X. Y. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- X. Cao, D. Qi, S. Yin, J. Bu, F. Li, C. F. Goh, S. Zhang and X. Chen, Adv. Mater., 2013, 25, 2957 CrossRef CAS PubMed.
- Z. Yan, J. Lin, Z. Peng, Z. Sun, Y. Zhu, L. Li, C. Xiang, E. L. Samuel, C. Kittrell and J. M. Tour, ACS Nano, 2012, 6, 9110 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457 CrossRef CAS PubMed.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1959, 149, 249 CrossRef.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481 CrossRef CAS.
- J. William, S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- C. T. Chien, S.-S. Li, W.-J. Lai, Y.-C. Yeh, H.-A. Chen, I.-S. Chen, L.-C. Chen, K.-H. Chen, T. Nemoto, S. Isoda, M. Chen, T. Fujita, G. Eda, H. Yamaguchi, M. Chhowalla and C.-W. Chen, Angew. Chem., Int. Ed., 2012, 51, 6662 CrossRef CAS PubMed.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467 CrossRef CAS PubMed.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538 CrossRef CAS PubMed.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463 CrossRef CAS PubMed.
- A. Mathkar, D. Tozier, P. Cox, P. J. Ong, C. Galande, K. Balakrishnan, A. L. M. Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577 CrossRef CAS.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- C. Mapelli, C. Castiglioni, G. Zerbi and K. Mullen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 12710 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supporting spectroscopic data. See DOI: 10.1039/c4ra01472c |
| This journal is © The Royal Society of Chemistry 2014 |