Fabrication, modification, and biomedical applications of anodized TiO2 nanotube arrays
Kaifu Huo
*ab,
Biao Gao
c,
Jijiang Fuc,
Lingzhou Zhaod and
Paul K. Chu
*a
aDepartment of Physics and Materials Science, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China. E-mail: paul.chu@cityu.edu.hk
bWuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: kfhuo@hust.edu.cn
cSchool of materials and metallurgy, Wuhan University of Science and Technology, Wuhan 430081, China
dState Key Laboratory of Military Stomatology, Department of Periodontology, School of Stomatology, The Fourth Military Medical University, No. 145 West Changle Road, Xi'an 710032, China
First published on 31st March 2014
Abstract
Titanium dioxide (TiO2) nanotubes have attracted increasing attention due to their outstanding properties and potential applications in photocatalysis, dye-sensitized solar cells, and biomedical devices. In this paper, recent research progress on TiO2 nanotube arrays (NTAs) produced by anodic oxidation of Ti in fluoride-containing electrolytes is reviewed with emphasis on the modification methods and biomedical applications. The fabrication protocol and growth mechanism are first discussed and common modification methods used to improve the optical, electronic, and biomedical properties of TiO2 NTAs are reviewed. Photo/electro-chemical biosensors based on TiO2 NTAs dedicated to the detection of glucose, hydrogen peroxide, and other biomolecules are described and recent examples of using TiO2 NTAs to improve the cellular response in vitro and accelerate osseointegration in vivo are provided. The incorporation and delivery of inorganic bioactive agents such as Ag, Sr, and Zn to achieve antibacterial and/or osteogenesis inducing ability are described and finally, the outlook and future development of TiO2 nanotubes pertaining to biomedical devices are briefly discussed.
 Kaifu Huo | Kaifu Huo received his B.S. in Applied Chemistry from China University of Petroleum in 1997 and a Ph.D. in Physical Chemistry from Nanjing University (China) in 2004. He is currently a Professor in the National Laboratory for Optoelectronics at Huazhong University of Science and Technology. He is an associate editor of Nanoscience and Nanotechnology Letters (NNL). He has authored/co-authored more than 100 papers in international refereed journals. His main research activities encompass bioactive nanomaterials and nanostructured electrode materials for electrochemical biosensors and energy storage devices. |
 Biao Gao | Biao Gao received his B.S. and M.S. in Material Science from Wuhan University of Science and Technology in 2009 and 2012. He is currently a doctoral student in the School of Materials and Metallurgy at Wuhan University of Science and Technology. His main research activities include bioactive nanomaterials, electrochemical sensors and electrochemical energy storage devices. |
 Paul K. Chu | Paul K. Chu received his Ph.D. in chemistry from Cornell University and is presently Chair Professor of Materials Engineering in City University of Hong Kong. His research activities encompass plasma surface engineering and materials science. He is chairman of the Plasma-Based Ion Implantation (PBII&D) International Committee, a member of the Ion Implantation Technology (IIT) International Committee and IEEE Nuclear and Plasma Science Society Fellow Evaluation Committee, senior editor of IEEE Transactions on Plasma Science, and associate editor of Materials Science & Engineering Reports. He is a Fellow of the APS, AVS, IEEE, MRS, and HKIE. |
1. Introduction
Titanium dioxide (TiO2) has long been used as a white pigment in paints and polymers. Following the discovery of photocatalytic water splitting on a TiO2 electrode under ultraviolet (UV) light in 1972 by Fujishima and Honda,1 enormous efforts have been devoted to the research of TiO2 materials as well as application to a myriad of areas including photovoltaics, photocatalysis, photo-/electrochromics, energy storage, and sensors.2–7 The discovery of carbon nanotubes (CNTs) by Iijima in 1991 (ref. 8) illustrates the importance and structural elegance of tubular nanomaterials and has spurred immense research activity leading to the discovery of many other types of materials having a nanotubular morphology.9–15 TiO2 nanotubes (NTs), one of the most extensively studied nanostructured oxides, possess remarkable physical and chemical properties and diverse applications that include, but not limited to, photocatalysis, solar cells, photoelectrochemical water splitting, supercapacitors, sensors, drug delivery, and biological coatings.15–29In contrast to CNTs, TiO2 NTs are readily synthesized by simple solution-based techniques such as template-assisted processes,30,31 sol–gel method,32,33 hydro/solvothermal means,34–36 and electrochemical anodization.15–22 TiO2 NTs were first prepared by Hoyer30 by the template-assisted method. In 1998, Kasuga and colleagues illustrated the hydrothermal alkaline synthesis of TiO2 NTs (ref. 34) and in 1999, Zwilling et al. reported the formation of nanoporous TiO2 by simple electrochemical anodization on Ti in a hydrofluoric acid solution.37 In 2001, Gong and co-workers fabricated self-organized TiO2 nanotube arrays (NTAs) by anodizing Ti in a diluted hydrogen fluoride (HF) electrolyte.38 However, only NTs with a length of 500 nm were obtained and it was difficult to produce self-supporting TiO2 NTs. In subsequent experiments, highly ordered TiO2 TNAs with a length of up to approximately 1000 μm (ref. 39) as well as free-standing TNAs films40 were produced. In contrast to the sol–gel32,33 and hydro/solvothermal techniques,34–36 electrochemical anodization is an easy and simply method to produce ordered TiO2 NTAs. In addition, the diameter, wall thickness, length, and even density of the NTs can be precisely controlled by tailoring the anodization parameters such as the electrolyte composition, applied voltage, electrolyte pH, temperature, and anodizing time.15–20,41,42 The TiO2 NTAs are vertically aligned to the Ti surface having a well-defined opening top and large internal surface area without the concomitant decrease in geometry and structural order. They thus constitute a good platform for drug delivery and size-dependent cell interactions in biomedical applications.19–22,28,29 Furthermore, TiO2 NTAs offer the unique combination of wide band gap semiconducting properties, large aspect ratio, large surface area, and vertical alignment, rendering them promising candidates in many applications such as photocatalysis, photovoltaics, sensing, and energy storage.15–20,23–25 Moreover, anodization enables the growth self-ordered NT layer on a Ti surface with virtually any shape and the technique can be readily modified and extended to other metals such as V,43 Zr,44,45 Hf,46 Nb,47,48 Ta,49–51 W,52–54 Fe (ref. 55 and 56) and binary TiAl,57 TiZr,58,59 TiNb,60,61 TiTa,62 TiW,63 ternary Ti–Zr–Nb,64 and Ti–6Al–4V (ref. 65) to produce highly aligned oxide NTAs or nanoporous structures with improved properties to cater to individual applications.
Since the first report on the fabrication of highly ordered TiO2 NTAs by anodic oxidation of Ti, the field has expanded tremendously. The number of scientific and technical publications on this topic has been going up exponentially and more than 1800 papers were published in the last decade as shown in Fig. 1. Many excellent reviews15–22,66 covering the preparation and applications of anodized TiO2 NTAs are available. However, the field has grown so rapidly that it is difficult to summarize all the different topics in one paper. In particular, Ti and its alloys are widely used in biomedical implants due to the good mechanical properties, excellent corrosion resistance, and biocompatibility.67,68 It has been shown that the TiO2 NTA coatings prepared on Ti or Ti alloys by anodization enhances the cell response and osseointegration significantly by accelerating apatite formation and improving osteoblast adhesion, proliferation, and differentiation.69–73 Moreover, TiO2 NTAs produced on Ti provide the multi-dimensional space for enzymes or drugs to be loaded and immobilized and form a good platform for biomedical diagnosis and drug delivery.19,28,29,66,74 In this paper, recent research activities on the surface modification and biomedical applications of TiO2 NTAs are reviewed. This paper is composed into three sections. The electrochemical anodization process to form TiO2 NTAs and corresponding growth mechanism are first described. The common modification methods applicable to as-anodized TiO2 TNAs are reviewed and finally, recent examples of using TiO2 NTAs in biomedical applications, for instance, cellular response in vitro, osseointegration in vivo, drug loading and delivery, as well as photo-/electrochemical biosensing are discussed.
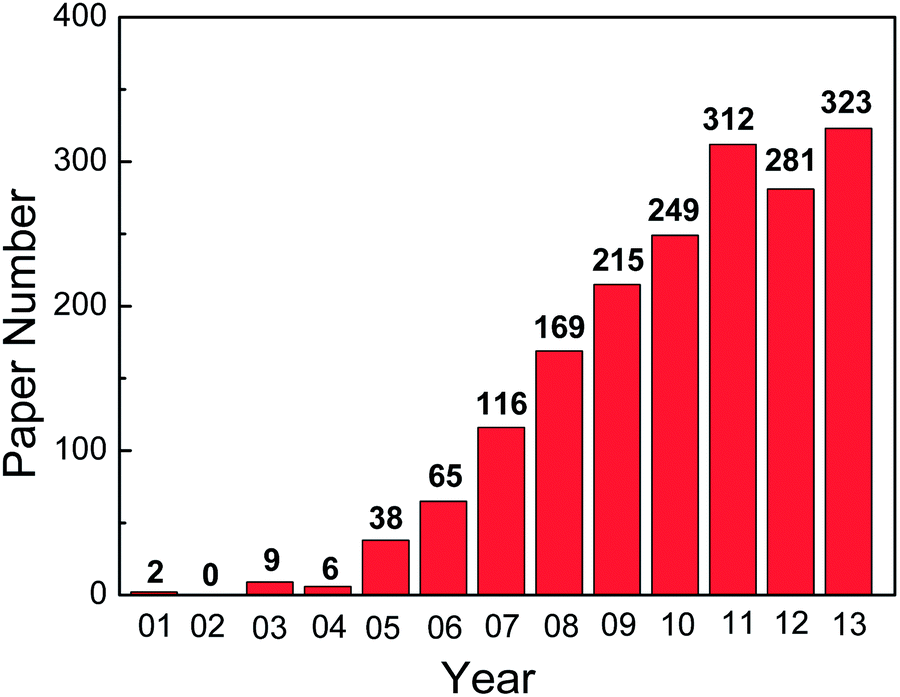 | ||
| Fig. 1 The number of papers related to anodized TiO2 nanotubes as a function of the year of publication from 2001 to 2013. (Data were collected from the Science Citation Index Expanded (SCI-EXPANDED) database using nanotub* and anodi* and “Ti or Titanium” as keywords). | ||
2. Fabrication and growth mechanism of anodized TiO2 NTAs
Electrochemical anodization of the suitable metals to create protective or decorative oxide layers on metal surfaces have been conducted for almost a century. However, only recently have self-organized oxide nanotube layers or ordered nanopore assembly been fabricated. Anodization is generally carried out on a two-electrode configuration (Fig. 2a). The metal foil/sheet undergoing anodizing is connected to the positive terminal of a DC power supply as the anode and placed in the electrolyte. The counter electrode (cathode) is normally a plate or rod of platinum, although materials such as carbon are also used. The electrolyte composition primarily determines if the oxide film is porous or compact.18,19 When the anodic oxide is insoluble in the electrolyte, a compact oxide film forms but if the anodic oxide is moderately soluble in the electrolyte and suitable anodization conditions are used, nanotubular/nanoporous oxide is formed (Fig. 2b). In general, the morphology and structure of the porous layers are affected by the electrochemical conditions, especially the anodizing voltage and solution parameters including the electrolyte composition, pH, and water content.42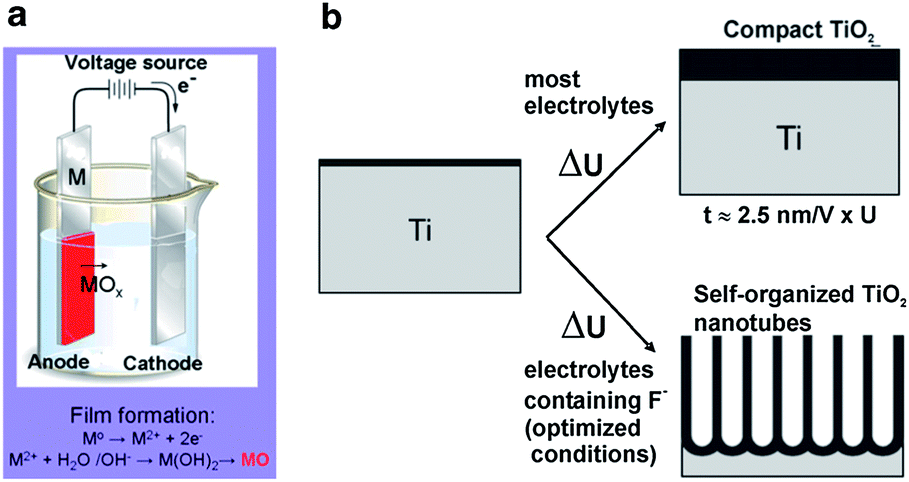 | ||
| Fig. 2 Schematic set-up for anodization experiments (a). Depending on the anodization conditions, the anodized oxide layer can be either compact, or nanotubular (nanoporous) oxide layers (b). Reproduced from ref. 18, Copyright (2007), with permission from Elsevier. | ||
Anodization of self-organized hexagonal porous aluminum oxide was demonstrated in an acidic solution by Masuda and Fukuda in 1995.75 The nanoporous anodized alumina arrays have since been widely used as a template to synthesize orderly metal, polymer or nonmetal NTs or nanorods.31,76–78 The first report on anodized TiO2 dates back to 1984 when Assefpour-Dezfuly and colleagues produced porous TiO2 by performing etching in alkaline peroxide followed by anodization in chromic acid.79 Electrochemical anodization of Ti is usually conducted at a constant voltage between 1 and 30 V in an aqueous electrolyte or 5–150 V in a non-aqueous electrolyte containing 0.05–0.5 M (0.1–1.0 wt%) fluoride ions to produce TiO2 NTAs. In 2001, Gong et al. reported the preparation of self-organized TiO2 NTAs by anodization of a Ti foil in H2O–HF at room temperature.38 However, the length of the TiO2 NTAs is limited to a few hundred nanometers.38 Second generation TiO2 NTAs with a length of several micros were produced in neutral electrolytes containing fluoride ions (F−) such as Na2SO4/NaF or (NH4)2SO4/NH4F (ref. 80–82) by taking advantage of limited dissolution of the mouth of the NTs. Usually, the NTs produced in an aqueous electrolyte have a rough external surface and rings (ripples) on the walls because of current oscillation during anodization.83 In later work, third generation TiO2 NTAs with lengths of up to approximately 1000 μm were produced in a non-aqueous, polar organic electrolyte such as formamide, dimethylsulfoxide (DMSO), ethylene glycol (EG), or diethylene glycol containing fluoride ions.39,84,85 The TiO2 NTAs produced in the viscous organic electrolytes are smooth and have larger aspect ratios without ripples or rings.19 Some attempts have been made to produce TiO2 NTAs in a fluorine-free electrolyte such as chlorine-containing electrolyte and they are commonly considered as the fourth generation materials.66,86–90 Fig. 3 displays the typical field-emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM) images of a representative TiO2 NTAs prepared in an HF electrolyte or ethylene glycol/fluoride.
 | ||
| Fig. 3 SEM and TEM images showing TiO2 NTAs grown by different anodization processes of Ti. (a and b) Typical morphology produced in acidic fluoride or HF electrolytes, (c and d) ethylene glycol/fluoride electrolytes. (e) A typical TEM images of anatase TiO2 NT. The inset in (d) is the bottom-view of TiO2 NTAs after peeling off the Ti substrate. The inset in (e) is the high resolution-TEM image of the nanotube wall. | ||
The growth mechanism of aligned TiO2 NTAs under anodic conditions involves the competition and equilibrium between field-assisted electrochemical oxidation and dissolution of the TiO2 layer.18,91 As shown in Fig. 4a, the initial oxide layer forms on the Ti surface as anodization commences via the interaction between Ti and O2− (from deprotonation of H2O) or OH− ions according to reactions ((1)–(3))18,19
| Ti → Ti4+ + 4e− | (1) |
| Ti + 2H2O → TiO2 + 4H+ + 4e− | (2) |
| Ti4+ + 2H2O → TiO2 + 4H+ | (3) |
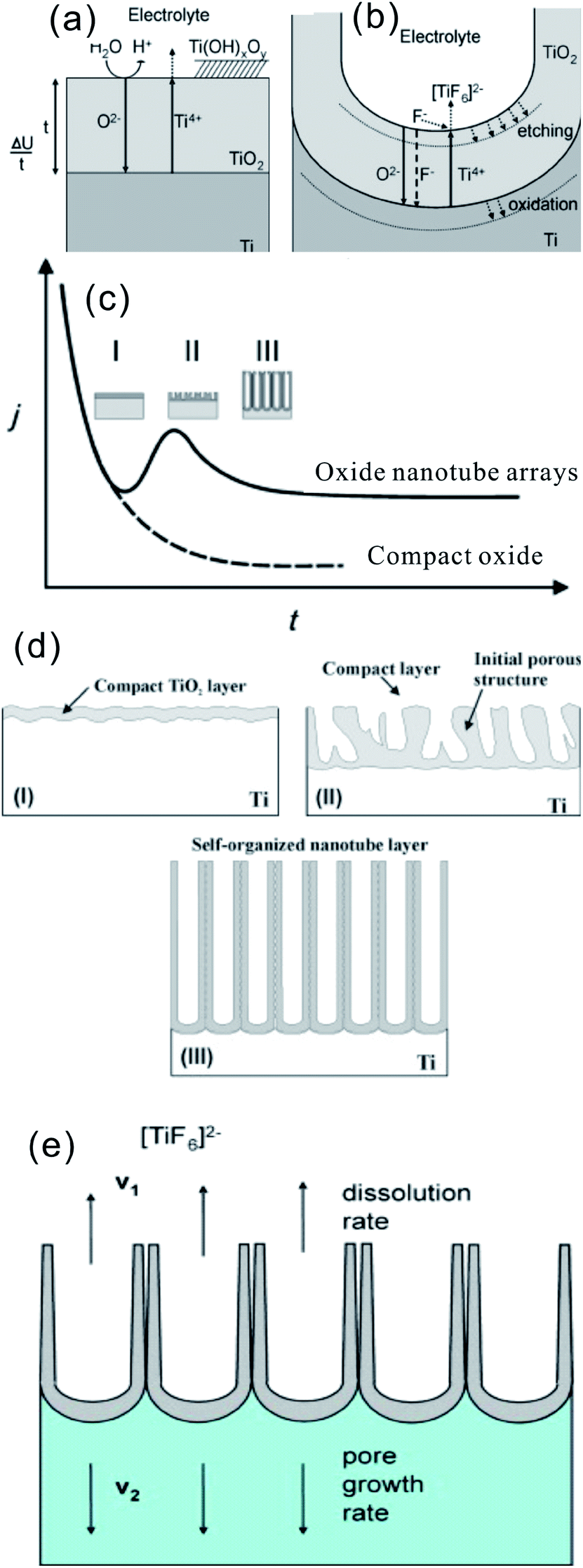 | ||
| Fig. 4 (a and b) schematic representation of the Ti anodization characteristics of the Ti anodization. (c) Typical current–time (j–t) with and without fluorides in the electrolyte, reproduced with permission from ref. 19. Copyright (2012) Wiley-VCH. (d) Corresponding evolution of the TiO2 morphology by different morphological stages; (e) steady state growth situation characterized by equal rates of TiO2 dissolution (v1) and formation (v2). (a, b, d and e) Reproduced from ref. 18, Copyright (2007), with permission from Elsevier. | ||
Simultaneously, hydrogen (H2) evolution occurs on the cathode according to reaction (4)
| 4H2O + 4e− → 2H2 + 4OH− | (4) |
After the formation of an initial oxide layer on the Ti anode, O2− migrates through the oxide layer reaching the metal/oxide interface where it further reacts with Ti and Ti4+ forming a TiO2 layer under an electric field. At the same time, Ti4+ is released from the metal at the metal/oxide interface and moves towards the oxide/electrolyte interface (Fig. 4a). In the absence of fluoride ions, the current–time curves usually show an exponential decay (Fig. 4c) as a result of the formation of a thick oxide barrier layer with low conductivity. However, the presence of HF or another source of fluoride ions strongly affects the anodization process of Ti because F− can etch/attack the oxide layer to form water soluble [TiF6]2− species [reaction (5)], as schematically shown in Fig. 4b. On the other hand, Ti4+ being mobile in the anodic layer reacts with fluoride ions to form soluble [TiF6]2− [reaction (6)].
| TiO2 + 6F− + 4H+ → [TiF6]2− + 2H2O | (5) |
| Ti4+ + 6F− → [TiF6]2− | (6) |
Thus, the current–time curves of Ti anodization in the presence of fluoride ions (Fig. 4c) usually show an initial exponential decrease in current (stage I) followed by an increase (stage II), and then to the quasi steady-state level (stage III). The behavior is associated with different stages in the NT/nanopore formation process during anodization. In the initial stage of anodization (stage I), the curve is essentially similar to that of the fluoride-free case and a compact oxide layer is formed, leading to a decay in the current because of the reduced electrical conductivity of the oxide layer. In stage II, the TiO2 layer is locally etched by reaction (5) and small pits acting as pore forming centers are formed randomly due to the difference in the roughness of the as-formed TiO2 layer or defects stemming from impurities, dislocation, and grain boundaries. As a consequence, irregular nanoscale pores are initially formed on the oxide layer surface at the oxide/electrolyte interface, as schematically shown in Fig. 4d. This process is usually accompanied by a rise in the current due to the decreased resistance of the anodic oxide film as more pathways are available for the ions. As the pores grow deeper, the electric field density across the remaining barrier layer increases and the individual pores start interfering with each other and competing for the available current. This leads to a situation in which the pores equally share the available current and self-ordering under steady state conditions is established (III). Consequently, uniform self-assembled nanopores are formed. At the base of the pore/tubes, the electric field is stronger resulting in enhanced field-assisted oxide growth and oxide dissolution. When the pore growth rate of oxide formation at the metal/oxide interface is identical to the rate of oxide dissolution at the pore-mouth/electrolyte interface (Fig. 4e), the current passing through the electrode attains a constant value and the NT layer is continuously etched from the Ti substrate but the oxide layer does not get thicker. The overall rate in the steady-state phase is limited by the transport (diffusion) of F− inside the channel from the solution to the growing TiO2 cap and the transport of [TiF6]2− in the opposite direction (Fig. 4b and e). Both effects can limit the total current. The pore initiation phase and evolution processes have been observed and confirmed by SEM.92,93 The as-anodized TiO2 NTAs are effectively immobilized on the Ti surface with an open top and a bottom closed by a barrier layer of Ti oxide. The walls of the NTs typically consist of amorphous TiO2 or Ti suboxide (TiOx) and are generally thinner at the mouth facing the electrolyte and thicker at the end contacting the Ti substrate. It does shows a V-shape morphology due to slow chemical dissolution of NT walls during anodization.19 The reason for the formation of TiO2 nanotubes as opposed to a nanoporous structure may be ascribed to the accumulation of fluoride species at the tube bottom thus establishing an anion containing weaker (and more soluble) TiO2 structure between neighbors.18,91
During anodization, the concentration of fluoride ions, electrolyte pH, water content, electrolyte viscosity, anodization voltage and duration, as well as temperature have important influence on the geometry and structure of the NTAs such as the NT diameter, length, and wall thickness. As a result of continuous breakthroughs associated with anodized TiO2 NTAs, precise control of the geometric parameters including the diameter, wall thickness, length, and even density of TiO2 NTs can be accomplished by tailoring the electrochemical anodization parameters such as the electrolyte composition, applied voltage, electrolyte pH value, temperature and time. Several review papers addressing the factors that affect the geometry, morphology, and structures of as-anodized TiO2 NTAs are available15–19,42,94 and so this topic will be not further elaborated in this review.
3. Modification of as-anodized TiO2 NTAs
The application of TiO2 NTAs is closely related to their electrical, chemical and optical properties. The as-anodized TiO2 NTAs are generally amorphous and the conductivity of native TiO2 is very low thus hampering many applications pertaining to biosensors and photoelectrochemical cells. On the other hand, the band gap of TiO2 is in the UV regime (3.0 eV for the rutile phase and 3.2 eV for the anatase phase)4 and corresponds to only a small fraction of the sun's energy (<5%). To achieve efficient visible light photocatalytic processes, the optical activity in the visible light region must be increased. In addition, biomolecule or drug loading that offers both biocompatibility as well as drug eluting can increase the functionality of TiO2 NTAs in biomedical applications. Therefore, in recent years, many studies have focused on the modification of TiO2 NTAs to improve the electrical, chemical, and optical properties.15–19 The common modification methods include (1) thermal treatment, (2) doping, (3) decoration or filling of TiO2 NTs with other species, and (4) conversion of TiO2 NTAs into nanoporous MTiO3 (M = Ba, Sr, Ca, Pb, Zn, etc.) or perovskite NTAs.3.1 Thermal treatment of anodized TiO2 NTAs
TiO2 crystallizes into three different polymorphs: anatase, rutile, and brookite.2–4 Rutile is generally considered to be the thermodynamically most stable bulk phase. The as-anodized TiO2 NTs are generally amorphous in nature, although some reports indicate the presence of anatase nanocrystallites in the tube wall if anodization is conducted at a high voltage.95 The amorphous TiO2 NTAs can be converted into anatase or rutile by a thermal treatment in air or O2.19,96,97 Post-growth annealing and the resulting crystal phases have a significant impact on the mechanical, electronic, optical, biomedical and chemical properties of the NTAs.15–24,66,96–98 For example, anatase TiO2 NTAs usually possess better properties as photovoltaic devices and photocatalysts than amorphous TiO2 NTAs.16,96 Glancing angle X-ray diffraction (GAXRD) patterns acquired from anodized TiO2 NTAs annealed at different temperature in dry oxygen are displayed in Fig. 5. Conversion of the amorphous TiO2 NTAs into anatase begins at around 280 °C.97,99 The relative intensity of the anatase (A) peaks increases with annealing temperature from 280 to 430 °C. The rutile (R) crystals appear at 430 °C, and a mixture of A and R crystals is detected between 480 and 620 °C. Complete transformation to rutile occurs at 680 °C and the anatase (A) peaks could not be observed. At higher temperature, the tubular structure collapses leaving dense rutile crystallites.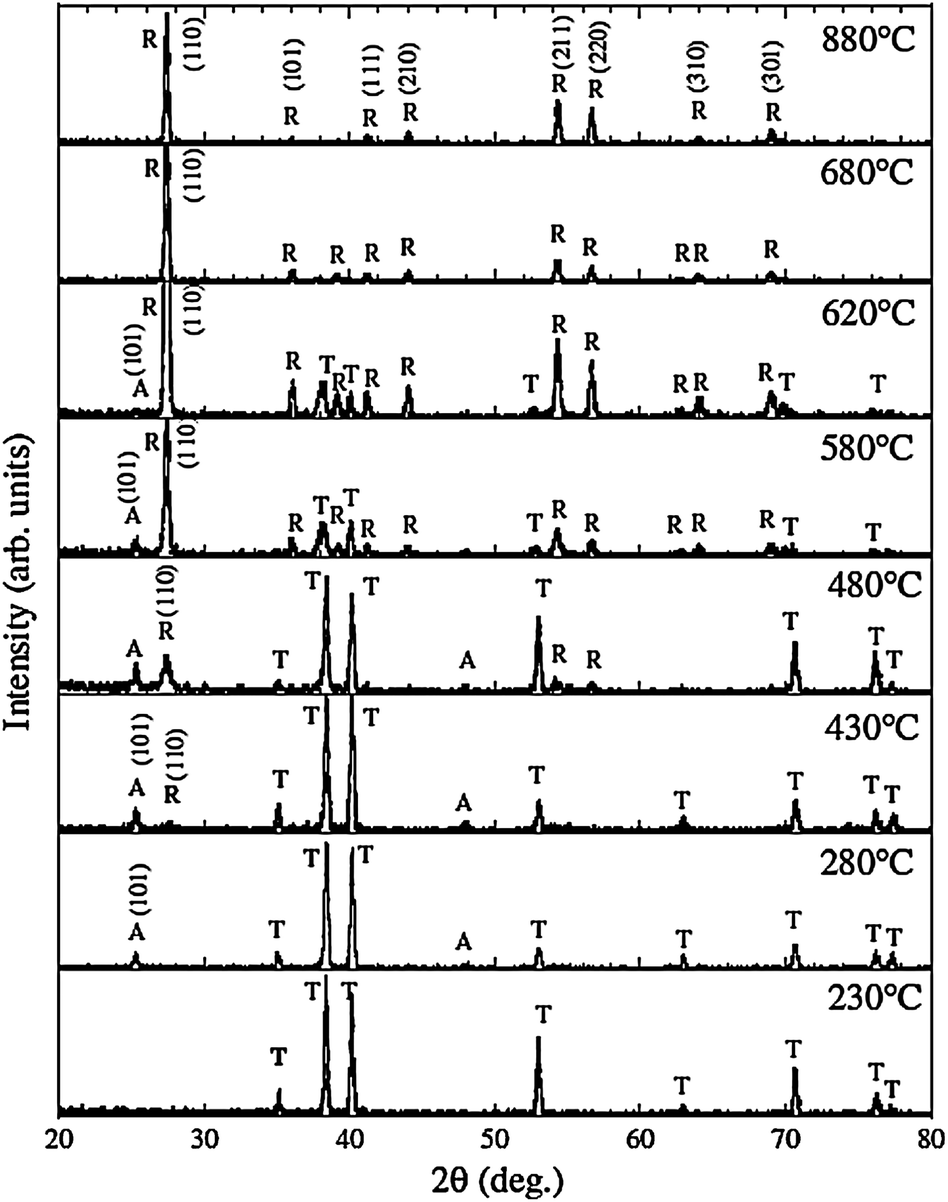 | ||
| Fig. 5 X-Ray diffraction (GAXRD) spectra of TiO2 NTs annealed at temperatures ranging from 230 to 880 °C in dry oxygen. A, R and T indicates anatase, rutile TiO2 and Ti (substrate), respectively. Reproduced from ref. 97, Copyright (2003), with permission from Materials Research Society. | ||
It is generally accepted that amorphous TiO2 NTAs can be fully converted into anatase TiO2 NTAs at 450 °C by a thermal treatment in air. When the annealing temperature is higher than 500 °C, anatase and rutile TiO2 coexist. However, according to the study by Yang et al.,100 the amorphous-to-anatase phase transition occurs initially at 300 °C and the NTs retain the anatase phase during the calcination process until being merged by the rutile layer grown from the NT–Ti interface.
Thermal treatment affects the length and morphology of TiO2 NTAs. We investigated the temperature-dependent length and morphology evolution of the TiO2 NTs during calcination.96 As shown in Fig. 6a, the as-anodized TiO2 NTs have a length of 13.6 μm and the length of the NTAs calcined between 300 and 450 °C shows no obvious change. However, at 500 °C, the length of the NTs decreases slightly to 12.6 μm and a thin rutile layer is formed beneath the NTA. At higher temperature of 550 and 600 °C, the length of the NTAs further decreases to 10.1 and 6.6 μm, respectively. When the annealing temperature is raised to 750 °C, the NTAs collapse and nanotubular structure vanishes. According to the Raman scattering spectra, the calcined NTAs show the vibration modes of anatase but there are no rutile signals. Transmission electron microscopy (TEM) and selected-area electron diffraction patterns disclose that the specimen is still anatase even after calcination at 650 °C and the result is in agreement Yang's study.100 At 300 °C, crystallization of anatase from amorphous TiO2 NTs occurs and crystallization of anatase takes place in both the nanotubes and oxide barrier layer. However, the anatase-to-rutile phase transition occurs preferentially at the interface between the NTs and Ti substrate during annealing in an oxygen-containing atmosphere at high temperature (>500 °C). The rutile crystallites formed on the bottom of the TiO2 NTs can grow along the anatase NTs and merge/consume the anatase NTs gradually forming a thick rutile film if the annealing temperature is further increased. This process results in a dramatic decrease in the NT length as shown in Fig. 6a and is schematically illustrated in Fig. 6b. However, the TiO2 NTs retain the anatase phase during calcination until fully merged by rutile. This is also supported by the fact that the diameter and wall thickness of NT have no obviously change during calcination and the photocatalytic activity of the TiO2 NTAs increases from 250 to 500 °C and then decreases at higher calcination temperature. The trend in the photocatalytic activity is attributed to the phase transformation as well as length associated with the surface area of the NTs.96
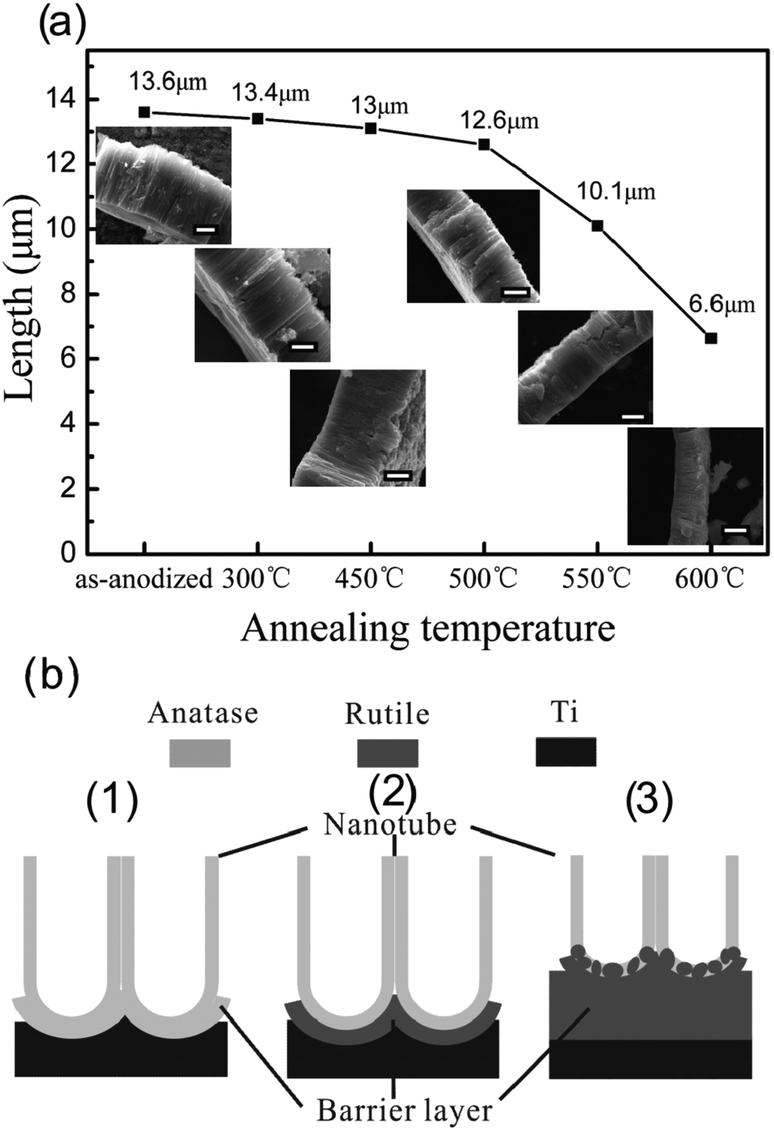 | ||
| Fig. 6 (a) Evolution of length of NTAs at different calcination temperature from 300 to 600 °C for 13.6 μm long TiO2 NTAs. The insets are corresponding profile images and the scale bar is 5 μm. (b) Schematic of the TiO2-NTAs annealed at (1) 450 °C, (2) 500 °C and (3) 600 °C in air. Reprinted with permission from ref. 96. Copyright @ American Scientific Publisher. | ||
The conductivity along the NTs is also affected to a large extent by the thermal treatment.101 As shown in Fig. 7, the resistivity increases with temperature after a low temperature (<200 °C) treatment because of evaporation of water from the surface. At about 300 °C, higher conductivity is observed due to conversion of amorphous NT into anatase. The resistivity is the smallest when amorphous NTs are totally converted into anatase TiO2 NTs at 350–450 °C. At a higher temperature (>500 °C), the resistivity of the NTAs increases again due to the formation of the rutile layer underneath the NTs. The presence of the rutile layer between the NT and Ti substrate negatively affects some applications of TiO2 NTAs on account of the inferior electronic properties of the rutile layers.
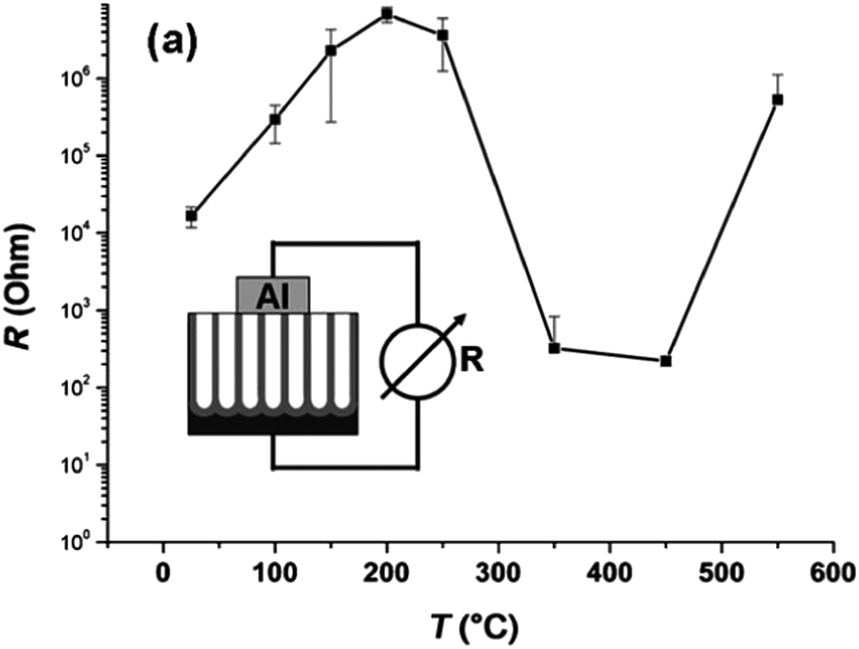 | ||
| Fig. 7 Two-point conductivity measurements for TiO2 NT layers annealed for 2.5 h at different temperatures in air. Reproduced from ref. 101, Copyright (2007), with permission from Elsevier. | ||
Thermal treatment of the as-anodized amorphous TiO2 NTs in vacuum or different gas can form unsaturated Ti cations such as Ti3+, Ti2+, and Ti+ or O2− vacancies in TiO2 NTAs. The Ti defect or oxygen defect result in different light absorption, conductivity photocatalytic activity, photoelectrochemical properties and biological functionality.102–109 For example, the as-anodized TiO2 annealed in nitrogen, argon or Ar/H2 will produce crystallined TiO2 NTAs with oxygen defects, when annealed in ammonia, N-doped TiO2 NTAs will be formed, which will be further discussed in Section 3.2. All in all, comprehensive consideration of the crystallization process of amorphous TiO2 NTAs is needed to optimize the performance and extend the application of TiO2 NTAs.
3.2 Doped-TiO2 NTAs
The electronic and chemical properties of materials are closely related to the chemical composition, atomic arrangement, and physical dimensions. Asahi et al. reported that N doped TiO2 exhibited improved photoelectrochemical reactivity under visible light illumination.110–112 Subsequently, other non-metals such as C, B, S, and P and metals such as V, Cr, and Fe have been incorporated into TiO2 by various means.113–119 These doped TiO2 NTAs usually exhibit enhanced visible light absorption and photoelectrochemical conversion efficiency or sensing performance.19,112,120,121 The typical methods to prepare doped TiO2 NTAs include (1) thermal treatment of TiO2 NTAs in a gas atmosphere containing the dopants, (2) plasma ion implantation or sputtering in an atmosphere with the doping species, and (3) Ti alloy anodization.Plasma ion implantation and sputtering conducted in an atmosphere containing the dopants are reliable methods to incorporate N or other species into the TiO2 lattice,122 but vacuum and special equipment are required and the implantation depth is usually limited to several hundred nanometers. C doping is typically achieved by thermal treatment in CO (ref. 108) or acetylene123 or flame annealing.113,124 The carbothermal reduction methods require a carbon source operated at a high temperature and it may cause the C concentration to diminish with depth in lieu of a uniform C distribution along the NT. A high temperature may also destroy the structure of the TiO2 NTAs. A simple and easy process to fabricate C-doped TiO2 NTAs has been developed125 by annealing the as-anodized TiO2 NTAs in Ar in the absence of foreign carbonaceous precursors at 400–500 °C (Fig. 8). The residual ethylene glycol (EG) absorbed on the NT wall during anodization serves as the carbon source and the NT provides the favorable nanoscale space to enhance the carbon doping reaction. The in situ C doping reaction is confirmed by thermal analysis (TG-DTA) combined with mass spectrometry (MS), as well as X-ray photoelectron spectroscopy (XPS). The morphology of the NTAs does not change significantly after annealing in air or Ar but the nanotube length is reduced by about 0.3 μm compared to the as-anodized one with a length of 5.4 μm due to partial sintering. This self-doping approach leads to C atoms being uniformly distributed along the entire NT (Fig. 8) thus forming C-doped TiO2 NTAs with high conductivity. The highly-ordered C-doped TiO2-NTAs have a large surface area, well-defined structure, high conductivity, and improved electron transfer (ET) efficiency and have immense potential in high-sensitivity and selectivity biosensor devices. Furthermore, the high photocatalytic activity of the C-doped TiO2 NTAs enables regeneration of the fouled surface simply by UV/visible light irradiation without damaging the microstructure. The surface can recover the high selectivity and sensitivity, thus proving a refreshable platform for high-sensitivity biosensor devices. This will be further discussed in Section 4.2. Alloy anodization is a widely used method for the fabrication of metal-doped TiO2 NTAs. Dopants such as N,126 W,63 Mo,127,128 Al,57 Ta,62 Zr (ref. 58) and Nb (ref. 60) are incorporated into the TiO2 NTAs by anodization of the corresponding Ti alloy substrate. Compared to pure TiO2 NTAs, the doped NT layers show faster photocatalytic kinetics and enhanced photocatalytic activity. Anodization of the Ti alloy in fluoride-containing electrolytes allows the fabrication of metal-doped TiO2 NTAs on an extremely wide range of alloys with enhanced properties and therefore has technological potential for biomedical applications.
 | ||
| Fig. 8 (a–c) FE-SEM images of the top-surface and cross-section (inset) morphology of as-anodized NTAs, TiO2-NTAs annealed in Air or Ar at 500 °C, (d) spatially resolved EDS elemental maps depicting the distribution of the constituting elements within the C-doped TiO2 NTAs (annealed in Ar). Reproduced from ref. 125. Copyright (2011) American Chemical Society. | ||
3.3 Decoration or filling of TiO2 NTs
Another suitable approach is decoration with noble metal nanoparticles such as, Ag, Au, Pt, Pd, or alloys,129–132 semiconductor nanoparticles such as CdS, CdSe, or PbS,133–135 or polymers.136,137 Noble metal decoration has been shown to be an effective way to restrain the recombination of photogenerated electron/hole pairs to produce photoelectrocatalytic activity enhancement. The Fermi levels of some noble metals such as Ag, Au, and Pt are lower than the conduction band of TiO2 and thus, the photogenerated electron produced by visible or UV illumination can be transferred from the conduction band to the metal nanoparticles deposited on the TiO2 surface. The recombination probability of electron and hole decreases dramatically resulting in improved photocatalytic activity in decomposing environmental pollutants and higher solar conversion efficiency in solar cells.15,138–140 Moreover, noble metal nanoparticles such as Ag, Au, and Pt, can improve the photoresponse of TiO2 in the visible region based on surface plasmon resonance (SPR).138,141–143 Metal nanoparticles decorated TiO2 have specific applications in energy or biomedical devices. For example, Ag has been reported to be a good antimicrobial species and Ag decorated TiO2 NTAs are antibacterial coatings in biomedicine.144 Pt, Ru, or Pd modified TiO2 NTAs are used as anode catalysts in direct methanol fuel cells (DMFC)129 and Pt or Au decorated TiO2 NTAs are excellent electrodes in electrochemical detection of glucose.145Many strategies have been developed to construct metal nanoparticles decorated TiO2 hybrid nanocomposites, typically by electrodeposition and photoreduction.130,146,147 Ag nanoparticles decorated TiO2 NTAs (NT–Ag) have been fabricated by soaking the anatase TiO2 NTAs in AgNO3 solutions and then photocatalytically reduced under UV illumination.144 The amount of Ag introduced to the NTs can be controlled by changing the processing parameters such as the AgNO3 concentration and immersion time. The NT–Ag can kill all the planktonic bacteria in the suspension during the first few days and the ability of the NT–Ag to prevent bacterial adhesion is maintained without obvious decline for 30 days. The materials are thus potential antibacterial coatings on biomedical implants. TiO2 NTAs can also be modified by covalent attachment of polymers, organic layers, or biomolecules. Some bio-molecules or linker molecules such as silane (APTES), Arg–Gly–Asp (RGD) peptide,148 Pluronic F127 (a common triblock polymer),149 and DNA150 have been incorporated into TiO2 NTs to adjust the hydrophilic property, promote cellular initial attachment and proliferation, and control the release kinetics of loaded drugs.149–151
3.4 Conversion of TiO2 NTAs into pervskite MTiO3/TiO2 heterostructured NTAs
TiO2 NTAs can be easily transformed into perovskite NTAs such as BaTiO3, SrTiO3, PbTiO3, and ZnTiO3 by hydrothermal treatment in the respective cation-containing solutions.152–157 The titanate-based perovskite materials with morphological regularity on a Ti substrate have many applications such as capacitors, actuators, electrochromics, gas-sensors, photocatalysts, and biological coatings.19,157,158 However, the perovskite materials reported so far have failed to maintain the high-quality morphology of the original anodic TiO2 NTAs.Recently, heterostructured anatase TiO2 NP/NTAs and nanorod arrays (NRAs)159 have been produced spontaneously from as-anodized amorphous TiO2 NTAs in water at a temperature as low as 90 °C by water-assisted dissolution and precipitation in the absence of an additive or Ti precursor (Fig. 9). The morphology and phase evolution from amorphous TiO2 NTAs to NPs/NTAs and finally NRAs under the hydrothermal conditions are related to water-induced dissolution and recrystallization.159,160 As schematically shown in Fig. 10, when the amorphous titania NTAs are hydrothermally treated in water, the amorphous and hydrothermally unstable TiO2 or TiOx (1 < x ≤ 2) species absorb water molecules to form soluble species of Ti(OH)62−, which can be further dehydrated and precipitated by bridging and sharing faces to form anatase TiO2. The overall reaction is described by eqn (7) and (8).
| TiOx + (1 − x/2)O2 + 4H2O → Ti(OH)62− + 2H+ (1 < x ≤ 2) | (7) |
| Ti(OH)62− + 2H+ → TiO2 + H2O | (8) |
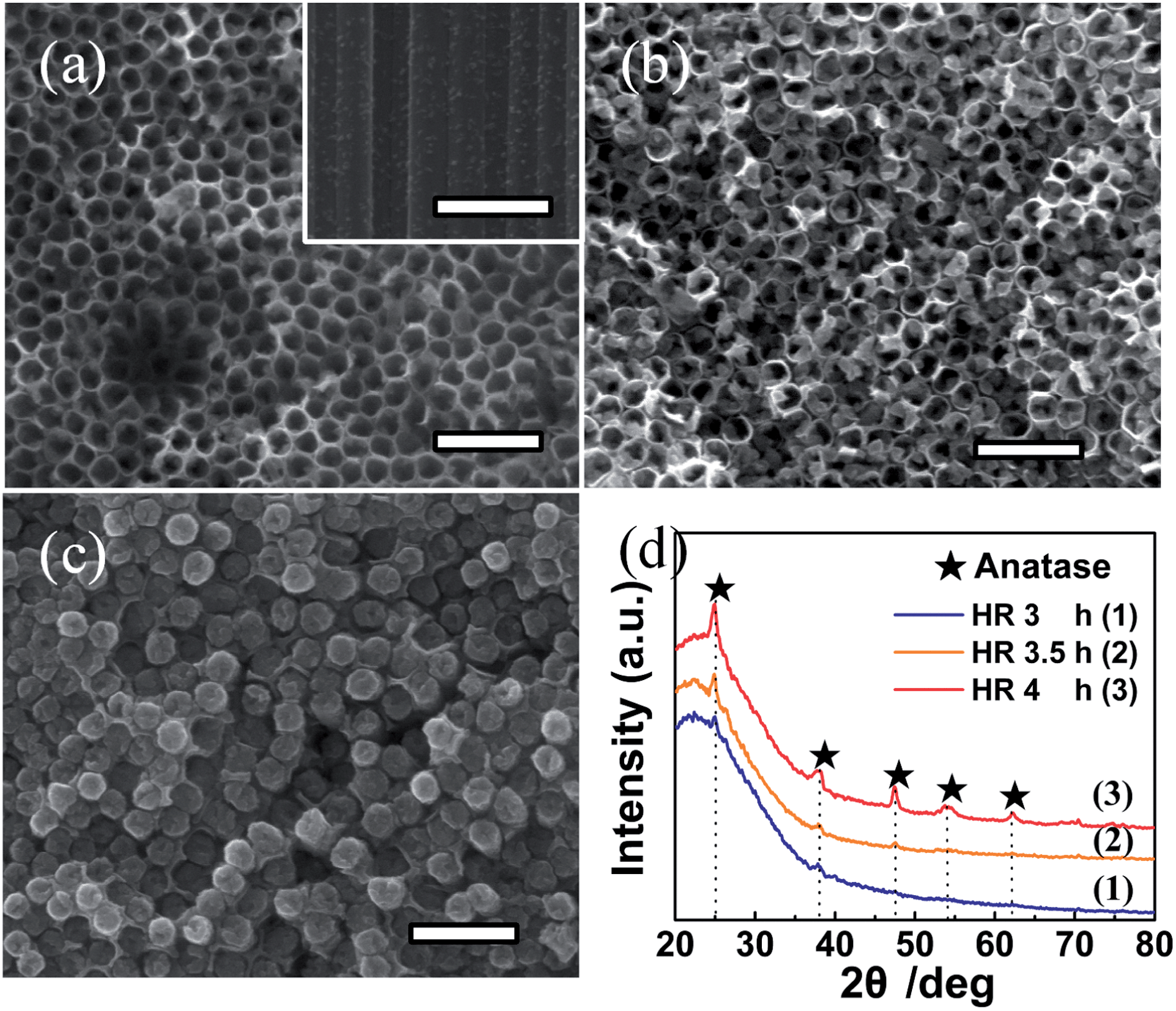 | ||
| Fig. 9 FE-SEM images of amorphous TiO2 NTAs formed by hydrothermal reaction (HR) in water at 90 °C for (a) 3 h, (b) 3.5 h, and (c) 4 h. (d) Corresponding XRD patterns of the three sample in (a)–(c). The scale bar is 500 nm. Reproduced with permission from ref. 159, Copyright (2012) Wiley-VCH. | ||
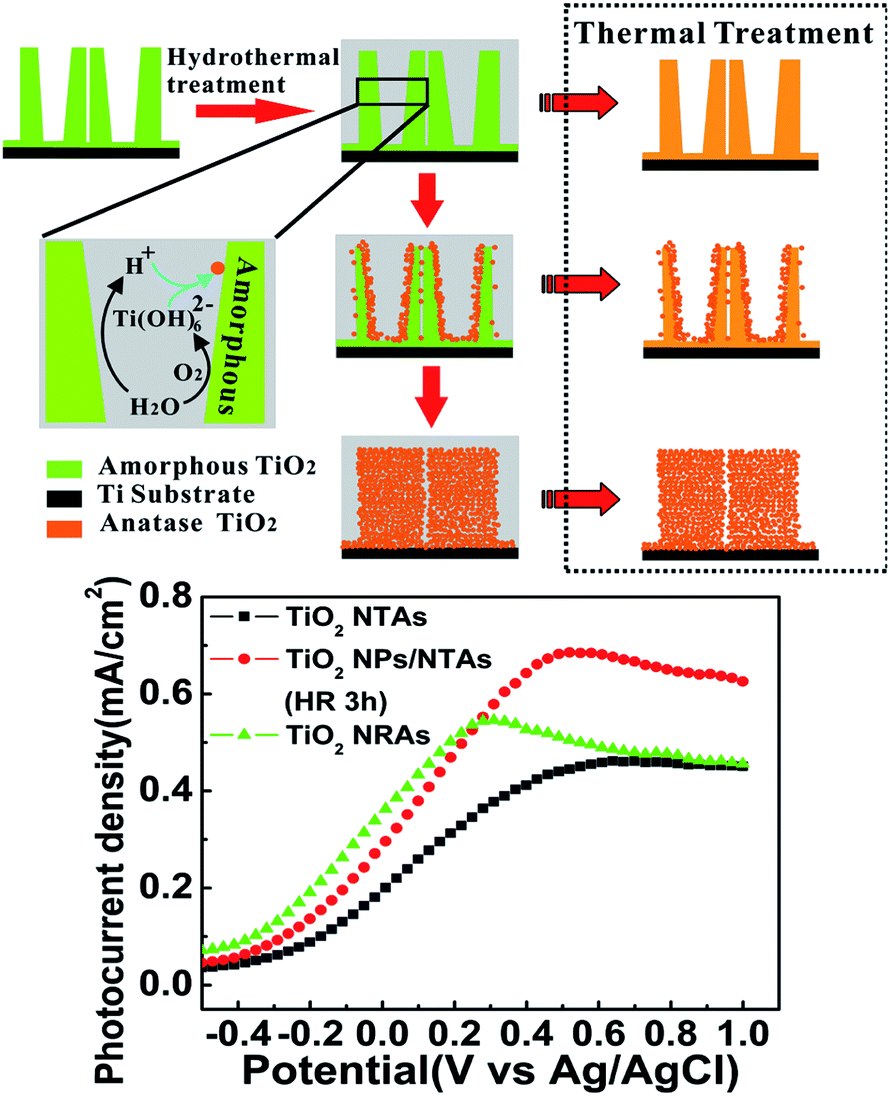 | ||
| Fig. 10 Schematic diagram of water molecule induced phase and morphological evolution of the amorphous NTs under hydrothermal conditions and the corresponding photocurrent density as a function of applied potential (vs. Ag/AgCl) in a 0.5 M Na2SO4 solution under UV irradiation for the samples of annealed TiO2 NTAs, TiO2 NPs/NTAs and TiO2 NRAs electrodes. Reproduced with permission from ref. 159, Copyright (2012) Wiley-VCH. | ||
The hydrothermal transformation from amorphous TiO2 NTAs to NPs/NTAs and finally NRAs does not require an additive or Ti precursor except water. Therefore, it is expected that amorphous TiO2 can be converted into perovskite oxide/TiO2 heterostructured NTAs and even perovskite NTAs if the hydrothermal reaction is carried out in metal cation-containing solutions according to reaction (9). This provides a simple method to fabricate heterostructured perovskite MTiO3/TiO2 NTAs (M = Ba, Sr, Ca, Zn, Co, etc.) and even MTiO3 NTAs on a large area while the nanotubular morphology of the initial anodic TiO2 can be preserved.
| x/2Ti(OH)62− + Mx+ → MTiO3 + H2O (M = Ba, Sr, Ca, Zn, Co, etc.) | (9) |
Heterostructured SrTiO3/TiO2 NTAs, ZnTiO3/TiO2 NTAs, and BaTiO3 NTAs, have been prepared by this simple hydrothermal technique.153–155,157 The SrTiO3/TiO2 NTAs and ZnTiO3/TiO2 NTAs NTA coatings exhibit enhanced osteogenic effects due to in situ delivery of trace elements (Zn and Sr). The materials have many applications to biomedical implants and will be discussed in Section 4.1.
4. Biomedical applications of TiO2 NTAs
Ti and Ti alloys are widely used in dental and bone implant materials due to their good mechanical properties, excellent corrosion resistance, and biocompatibility.68,69 The long-term normal functions of implants are related to early and rigid osseointegration. Although the native TiO2 layer provides some corrosion resistance and biocompatibility, it is not able to induce bone formation. If bonding between an Ti implant and bone cannot be formed initially, it is typically encapsulated by fibrous tissues instead of bonding directly to juxtaposed bones, leading to implant dislocation and premature loosening.161,162 Hence, considerable efforts focus on surface modification of Ti-based implants to improve osteointegration. Bone is a nanostructured composite matrix composed of non-stoichiometric inorganic calcium phosphate minerals and organic proteins and collagen.163 Formation of a nanostructured coating on a implant is a good strategy to achieve enhanced osseointegration from the viewpoint of bionics. A TiO2 NTAs coating produced on a Ti implant by anodization provides a well-ordered nanotubular topography that mimics the dimensions of collagen fibril in bones and has elasticity similar to that of bones to enhance a wide range of cellular responses.164,165Therefore, the cellular interaction on the NT coatings in a biologically relevant environment is of high significance. The large surface area of TiO2 NTs and ability to precisely tune the pore size and NT length render it a good platform to load or immobilize functional drugs, trace elements, or growth factors in the realization of in situ controlled delivery to the implant–tissue interfaces to accelerate tissue reconstruction and healing. In addition, TiO2 NTAs are electrically connected to the underlying Ti substrate and the structure is a favorable choice as an electrode in a chemical and biosensor for biomedical diagnosis and environmental analysis due to high surface area, favorable transport pathways for the analytes, and multi-dimensional space for proteins, enzymes, and DNA immobilization. Another significant advantage of TiO2 NTA or modified TiO2 NTAs in biosensors is the self-cleaning ability under light irradiation. The medical applications of TiO2 NTAs and biologically relevant responses are of high significance. Fig. 11 provide a highly vivid picture about recent activities of TiO2 NTAs in biomedical applications.
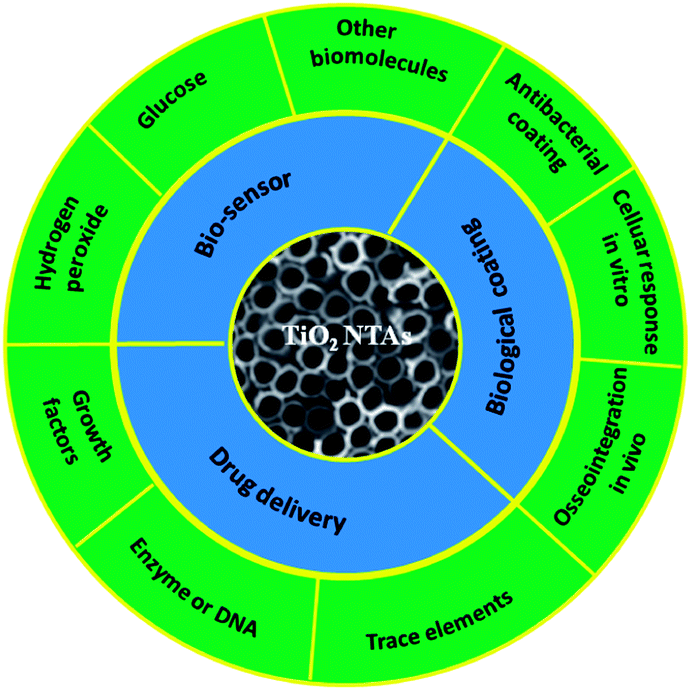 | ||
| Fig. 11 Schematical diagram of the recent activities of TiO2 NTAs in biomedical applications. | ||
4.1 Biological coatings of TiO2 NTAs on Ti or Ti alloys
 | ||
| Fig. 12 Patterned samples after immersion in 1.5× SBF. (a and b) FE-SEM micrographs showing selective apatite nucleation exclusively on nanotube regions. Reproduced from ref. 168, Copyright (2011), with permission from Elsevier. | ||
 | ||
| Fig. 13 SEM images of the as-formed and annealed nanotubes after soaking in SBF for 3 (left images) and 5 days (right images): (a) as-anodized; (b) 450 °C; (c) 600 °C. Adapted with permission from ref. 169, Copyright (2012) Wiley-VCH. | ||
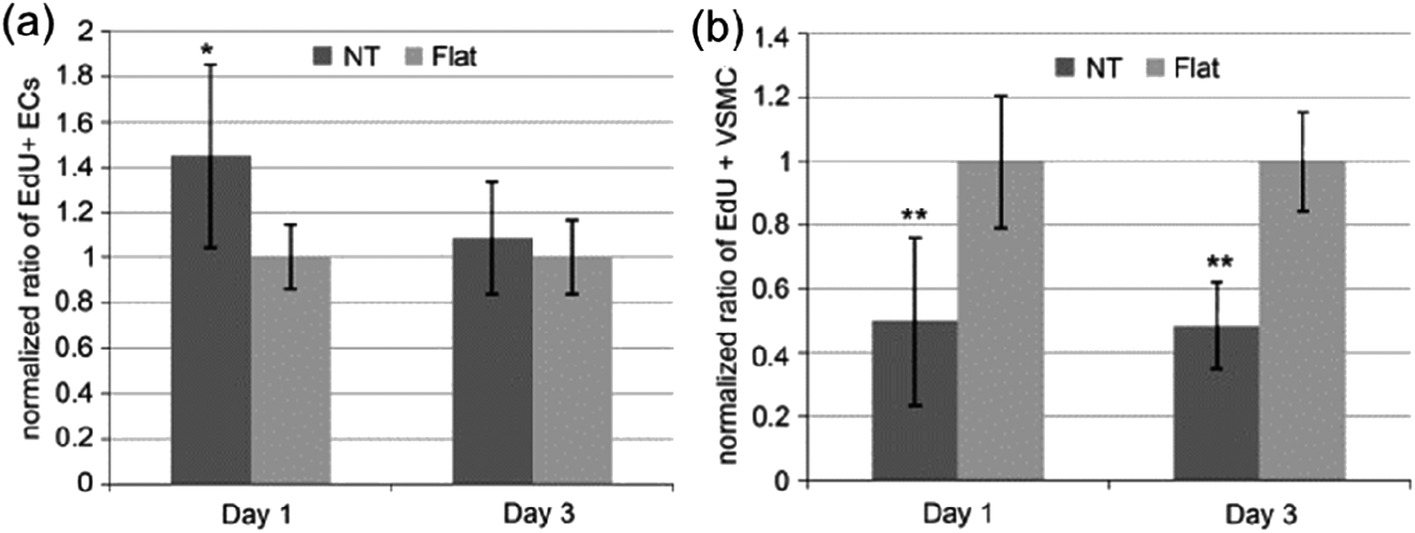 | ||
| Fig. 14 Ratio of EdU positive (a) ECs and (b) VSMCs on flat or NT substrate normalized by the average proportion of positive cells on flat surfaces on day 1 and 3. Data are presented as average ± standard deviation. *p < 0.05, **p < 0.01 versus same day flat control. Reproduced from ref. 176, Copyright (2009), with permission from Elsevier. | ||
Osteoblasts are responsible for bone formation and the effects of NTs on osteoblast functions have been observed using primary rat calvarial osteoblasts and MC3T3-E1 mouse osteoblasts.77,171,172,180,181 Recent reports indicate TiO2 NTA coatings on Ti significantly enhances extracellular matrix secretion, mineralization,167 adhesion of osteoblasts in vitro,174 bone growth in vivo,182 and bone bonding compared to conventional micro-roughened Ti produced by sand blasting.
Yu et al. evaluated the osteoblast behavior on anatase and mixed anatase–rutile NTAs produced by annealing as-anodized TiO2 nanotubes at 450 and 550 °C, respectively. Proliferation and mineralization of MC3T3-E1 preosteoblasts cultured on the anatase or anatase–rutile NT layers were significantly higher than those on the amorphous NT layers.183 Lee et al. examined the growth of MC3T3-E1 osteoblasts on polished Ti, TiO2 NTAs, and TiO2 NTAs embedded with gelatin stabilized gold nanoparticles (TN–AuNPs–gelatin surfaces). The adhesion and propagation of the osteoblast cells were improved significantly on TiO2 NTAs and the cells actually penetrate the nanotube pores producing an interlocked cell structure.184
The diameter of the NTs also impacts the cellular behavior although there is controversy on the effects of different NT size on some of the osteoblast functions due to the different experimental conditions. Brammer et al. prepared TiO2 NTs with different diameters (30–100 nm) on Ti by anodization and investigated the nanosize effect on osteoblast cell adhesion, morphology, and osteogenic functionality.185 The 30 nm TiO2 NTs exhibited the largest degree of cell adhesion and proliferation but the NTs with larger diameters (70–100 nm) showed a smaller cell population with an elongated cellular morphology and higher alkaline phosphatase (ALP) levels. The large NTs (100 nm) induced more osteoblast elongation and higher up-regulated level of ALP activity than smaller ones (30–70 nm) and possess greater bone-forming ability. Our recent study indicates that the hierarchical hybrid micro/nano-textured NTs produced by simple acid etching Ti followed by anodization mimics the hierarchical structure of bone tissues, thereby inducing more collagen secretion compared to the microrough or flat Ti.171
Mesenchymal stem cells (MSCs) play a crucial role in bone regeneration and bony fixation of implanted biomaterials.186,187 Most of the osteoblastic cells that colonize the implant surface to induce bone growth originate from MSCs and hence, in order to accomplish good osseointegration, it is critical to induce differentiation of MSCs preferentially towards osteogenitor cells and then into osteoblasts in lieu of other cell lineages. Oh et al. have observed that small NTs (30 nm) promote MSC adhesion without noticeable differentiation, whereas larger ones (70–100 nm) elicit selective MSC differentiation to osteoblasts.188 Moon et al. have recently assessed the effects of the NT size on the behavior and osteogenic functionality of human MSCs.189 After incubation for 2 weeks, expression of ALP, osteopontin, integrin-β, and protein kinase R-like ER kinase genes are significantly higher in cells cultured on the 70 nm NTs than that those cultured on the 30, 50 and 100 nm NTs as well as pure Ti. Park et al. have reported that 15 nm NTs provide substantially stronger stimulation of differentiation of mesenchymal cells to endothelial cells and smooth muscle cells than 70–100 nm NTs, while high rates of apoptosis are observed from 100 nm NTs. Endothelial cell adhesion, proliferation, and motility are several folds higher on the 15 nm NTs than 100 nm ones. These findings indicate that fine-tuning of the TiO2 surface on the nanoscale is crucial to the response of endothelial and smooth muscle cells on vascular implants.190 TiO2 NTs with a suitable tube size have positive cell interaction and osteogenesis inducing ability.
The osteogenesis inducing ability of the NTs arises from the modulating effects on the cell shape and focal adhesion. This leads to changes in the mechano-transduction including indirect ones, namely integrin dependent signal pathways, and direct ones involving gene expression originating from distortion of the cell nucleus by force transferred via the cytoskeleton. The shape of the MSCs on the Ti implants is related to the high cytoskeletal tension such as well-spread stem cells.191 Our recent study shows that the NTs significantly promote MSC attachment and spreading (Fig. 15), collagen secretion, ECM mineralization, as well as osteogenesis-related gene expression in the absence of extra osteogenic supplements (OS) and the osteogenesis inducing ability of the 80 nm NTs is higher than that of the 25 nm ones.170
 | ||
| Fig. 15 SEM pictures showing the morphology of MSCs after 2 days of culture on samples: (a) and (b) flat Ti control, (c) and (d) 25 nm NTs, (e) and (f) 80 nm NTs. Reproduced from ref. 170, Copyright (2012), with permission from Elsevier. | ||
The good bone favoring properties of NTs with the suitable size has been verified in vivo. Bjursten et al. investigated bone bonding between 80 nm NTs and grit blasted TiO2 in vivo.182 Four weeks after implantation into rabbit tibias, the NTs improved the bone bonding strength by as much as nine folds compared to the grit blasted TiO2. The histological analysis confirmed larger bone–implant contact areas, new bone formation, and higher calcium and phosphorus levels on the NTs. Wang, et al. investigated the effects of the NTs with different diameters of 30, 70, and 100 nm on the attachment mechanism to bone in vivo.192 Compared to the machined titanium implants, a significant increase in bone–implant contact (BIC) (Fig. 16) and gene expression levels was found from the bone attached to the implants with the NTs, especially the 70 nm ones. The result demonstrates the ability of TiO2 NTs to induce better osseointegration and NTs with a size of about 70 nm are the optimal ones from the perspective of osseointegration.
 | ||
| Fig. 16 Values of bone–implant contact (BIC) for all implant surfaces at 3, 5 and 8 weeks after implantation. Asterisk (*) shows a significant difference in comparison with machined implant (p < 0.05). Double asterisks (**) show a significant difference in comparison with all other groups in experiment (p < 0.05). Reproduced from ref. 192, Copyright (2011), with permission from Elsevier. | ||
Desai's group evaluated the suitability of the NTs as a potential drug loading and delivering platform.29,193 They used bovine serum albumin and lysozyme as model proteins to investigate the loading and release efficiencies from the TiO2 NTA. Various amounts of drugs were incorporated into the NTs and their release was adjusted by varying the NT length, diameter, and wall thickness.29 There are recent reports on the incorporation of growth factors or antibiotics into the NTs and resulting bioactivity and antibacterial ability. It is believed that NTs are suitable for loading and delivering targeted inorganic agents such as silver, strontium, and zinc. First of all, these inorganic species are smaller than growth factors and antibiotics and function at low effective doses. Long-lasting activity can thus be achieved by increasing the loaded amounts and controlling the release rate appropriately. Secondly, these agents are stable due to their inorganic nature, thereby facilitating the use of loading processes and loading methods that tend to be carried out under harsh conditions. Thirdly, the stable agents permit storage for a relatively long time after fabrication and it is an important point from the commercial point of view. Hence, Ag,144,194 Sr,154,155 and Zn (ref. 157) have been introduced to NTs to obtain long-term antibacterial ability and osteogenesis induction.
Implant-associated infection, an important issue impairing the normal function of implants, is usually difficult to treat and sometimes requires implant removal and repeated surgeries67. Infections associated with implants are characterized by bacterial colonization and biofilm formation on the implanted device as well as infection of adjacent tissues, and so a surface with long-term antibacterial ability is highly desirable in order to prevent implant-associated infection. Ag is one of the strong bactericides and has attracted attention because of benefits such as a broad antibacterial spectrum including antibiotic-resistant bacteria, non-cytotoxicity at suitable doses, satisfactory stability, and small possibility to develop resistant strains.
It has been reported that a certain range of Ag concentrations can kill bacteria without impairing mammalian cell functions and Ag-containing coatings that can resist biofilm formation without cytotoxicity are desirable for implants. Ag nanoparticles (NPs) have been incorporated into NTs (NT–Ag) on Ti implants to achieve this purpose.144 The SEM images (Fig. 17) show that NT–Ag retains the original nanotubular morphology after Ag loading. The amount of Ag introduced to the NTs can be controlled by changing processing parameters such as the AgNO3 concentration and immersion time. The NT–Ag can kill all the planktonic bacteria in the suspension during the first several days and the capability of the NT–Ag to prevent bacterial adhesion is maintained without obvious decline for 30 days which are normally long enough to prevent post-operation infection in the early and intermediate stages and perhaps even late infection around the implant. The ability of the NT–Ag to prevent viable bacteria colonization is assessed by fluorescence staining and the results are presented in Fig. 18. After 7 days of repeated bacteria invasion every 24 h, there are large amounts of viable bacteria on the flat Ti and smaller amounts on the TiO2-NTs. In comparison, the amounts of viable bacteria are obviously smaller on the NT–Ag samples. Although the NT–Ag structure shows some cytotoxicity, it can be reduced by properly controlling the Ag release rate. This NT–Ag structure with relatively long-term antibacterial ability has promising applications in bone implants after minimizing the cytotoxicity by properly controlling Ag release and can deliver both excellent bone bonding and long-lasting antibacterial ability.
 | ||
| Fig. 17 SEM images of the samples: (a) TiO2 NTs, (b–e) Ag incorporating NTs formed in AgNO3 solutions of different concentrations (0.5, 1, 1.5 and 2 M). The inset in (a) is the side-view SEM image revealing that the length of the nanotubes is about 7 μm. The red arrows in (b–e) indicate the Ag nanoparticles. (f) TEM image acquired from the Ag incorporating NTs formed in 1 M AgNO3 shows that the Ag nanoparticles attached to the inner wall of the NTs have a diameter of about 10–20 nm. Reproduced from ref. 144, Copyright (2011), with permission from Elsevier. | ||
 | ||
| Fig. 18 Representative images showing viability of the bacteria after 7 days of incubation on samples: (a) Ti, (b) NTs, (c–f) Ag incorporating NTs formed in AgNO3 solutions of different concentrations (0.5, 1, 1.5 and 2 M). The live bacteria appear green while dead ones are orange. Reproduced from ref. 144, Copyright (2011), with permission from Elsevier. | ||
Sr has aroused tremendous clinical interests especially after the development of the anti-osteoporosis drug strontium ranelate which exhibits pronounced effects to decrease the bone fracture risk in osteoporotic patients. Sr modulates bone turnover towards osteogenesis by enhancing osteoblast functions, inhibiting osteoclast functions, and increasing bone matrix mineralization. Sr has also been recently reported to direct the MSC commitment to the bone lineage via the Wnt/β-catenin and MAPK pathways as well as induction of cyclooxygenase (COX)-2 and prostaglandin E2 expressions while simultaneously repressing their commitment to other lineages such as adipocytes. Sr loaded NT coatings (NT–Sr) that combine the functions of nanotubular topography and Sr controlled and long-term release are expected to yield favorable osteogenic effects.
NT–Sr coatings have been prepared on Ti implants by transforming the TiO2 NTs into heterostructured SrTiO3/TiO2 NTs by simple hydrothermal treatment of anodized TiO2 NTAs in a 0.02 M Sr(OH)2 solution while the original nanotubular structure is retained,153–155 as schematically shown in Fig. 19. The NT samples produced by anodization at 10 and 40 V (NT10 and NT40) are subjected to hydrothermal treatment in 0.02 M Sr(OH)2 for 1 and 3 h to form different Sr-containing NT samples (denoted as NT10–Sr1, NT10–Sr3, NT40–Sr1, and NT40–Sr3). The Sr amounts can be adjusted by the tube length, diameter, and hydrothermal reaction time (Fig. 19). After the hydrothermal treatment, the nanotubular architecture is preserved but the tube diameter decreases with treatment time due to volume expansion in the transformation of TiO2 to SrTiO3. The XPS spectra acquired from the NT–Sr demonstrate that the Sr contents increase with the hydrothermal treatment time and tube length. Water contact angle measurements reveal that all the NT coatings are hydrophilic and Sr incorporation does not alter the water contact angles significantly.
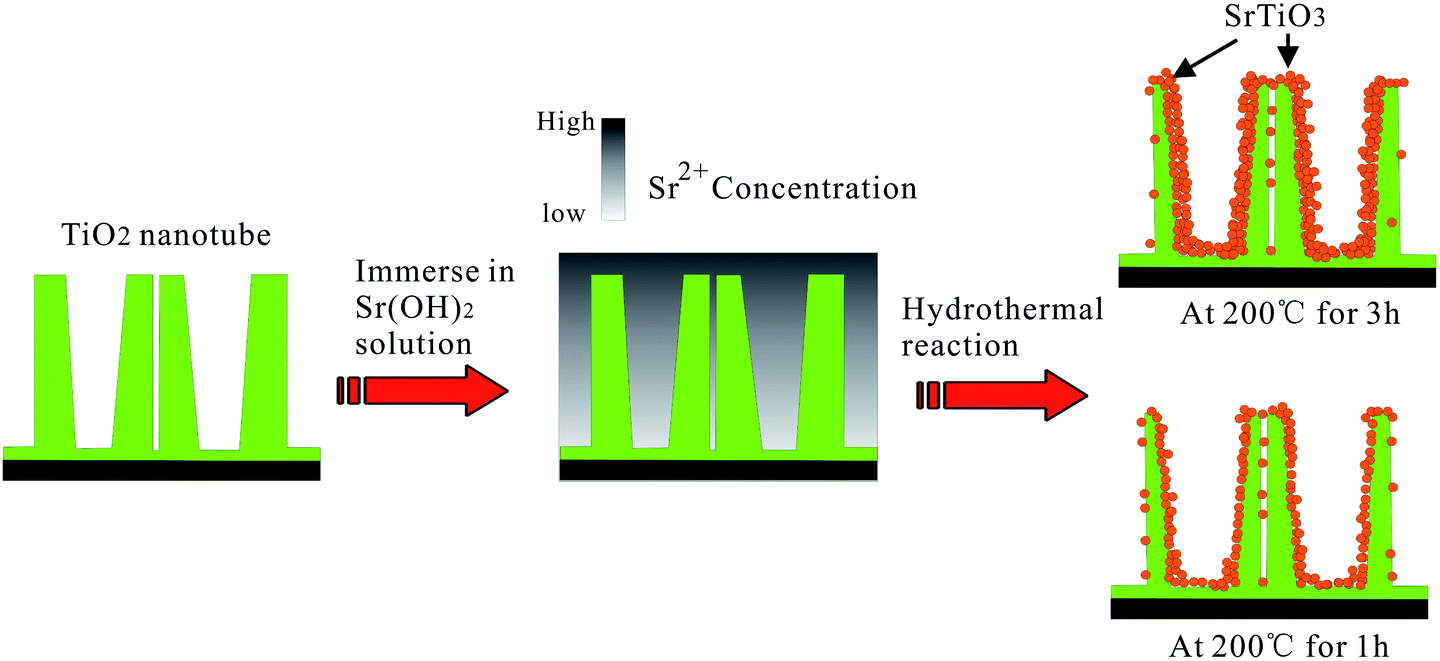 | ||
| Fig. 19 Schematic diagram illustrating the transformation of TiO2-NTAs into heterojunction SrTiO3/TiO2-NTAs by hydrothermal reaction in 0.02 M Sr(OH)2 solution. Adapted with permission from ref. 153, Copyright (2010) Wiley-VCH. | ||
The Sr release kinetics is assessed by immersing the NT–Sr in 5 mL of PBS for as long as 1 month and the data are depicted in Fig. 20A. Generally, NT–Sr shows an initial Sr release burst but after four days, the released Sr amounts are relatively constant and in fact exhibit a slight decline. The average Sr amounts released from NT10–Sr1, NT10–Sr3, NT40–Sr1, and NT40–Sr3 daily are 0.025, 0.039, 0.042, and 0.053 ppm, respectively, with the exception of the burst release at day 1. The total Sr contents leached from the four samples are 45.48, 54.95, 88.20, and 118.00 mg based on the 1 cm2 coatings, respectively (Fig. 20B). Theoretically, it is estimated that Sr may be delivered from the NT–Sr for a period longer than 1 year.
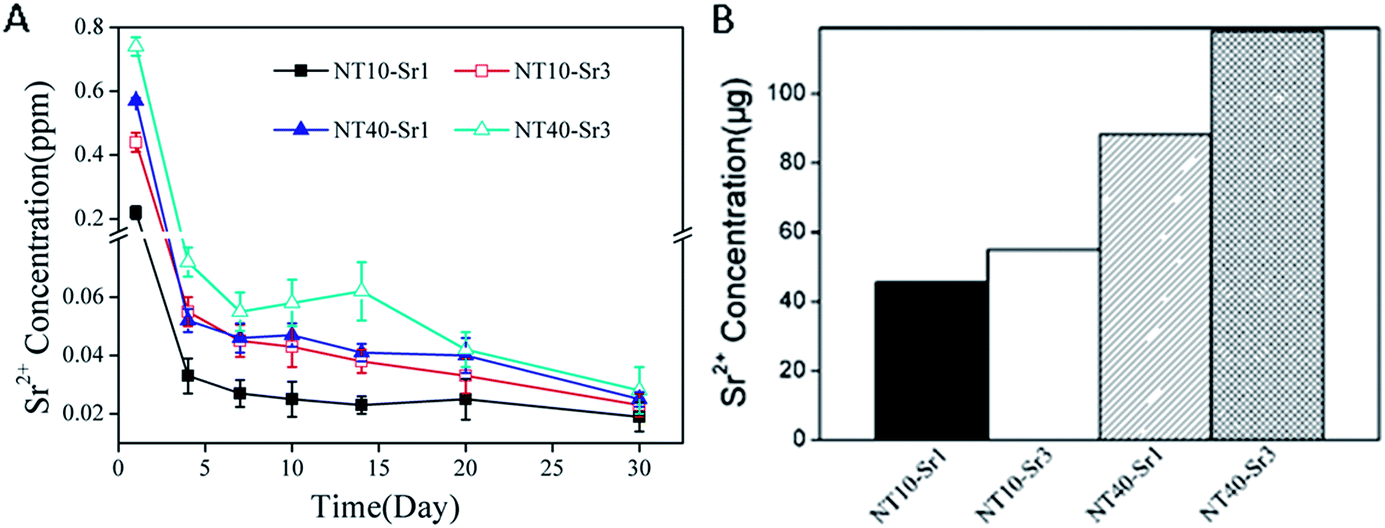 | ||
| Fig. 20 (A) Non-cumulative Sr release time profiles from NT–Sr into PBS and (B) total Sr content of NT–Sr. Reproduced from ref. 155, Copyright (2013), with permission from Elsevier. | ||
The activity of lactate dehydrogenase (LDH) in the culture media released by the cells when co-cultured with the samples is evaluated as an indication of cytotoxicity. All the NT and NT–Sr coatings exhibit no cytotoxicity compared to the Ti control. The LDH release from NT10–Sr is slightly smaller than that from the Ti control, indicating even better cytocompatibility on NT10–Sr. In vitro SBF experiments reveal that the NT–Sr arrays possess good biocompatibility and can induce precipitation of HAP from SBF.154 In vitro cell cultures indicate that the NT–Sr coating can dramatically improve MSC spreading as well as proliferation and enhance MSC differentiation towards osteogenitor cells and subsequently osteoblasts. The good cytocompatibility is verified by the good attachment, spreading, and proliferation of cells after incubation for 8 days as revealed by the FE-SEM (Fig. 21) and fluorescence images. The MSCs on the flat Ti spread poorly into a spindle shape indicative of differentiated quiescent cells. On the other hand, the MSCs on NT10 are more extended exhibiting a typical polygonal osteoblastic shape. Sr incorporation enhances proliferation of MSCs on the NT, especially NT10–Sr which promotes the spreading of the MSCs into an appreciably polygonal osteoblastic shape. Obvious improvement in cell spreading on NT–Sr over NT is attributed to Sr incorporation. Both the NT and NT–Sr samples promote osteogenesis to varying degrees as indicated by gene expression and among the various samples, NT10–Sr3 and NT40–Sr significantly up-regulate the expression of the osteogenesis related genes in the absence of an extra osteogenic agent. NT10 and NT10–Sr generate big nodular ALP products and induce extracellular matrix (ECM) mineralization, and the effects on NT10–Sr3 are most obvious due to the multiple scaled nanostructure and proper amount of incorporated Sr. As shown in Fig. 22, the surface architecture of NT–Sr combines the functions of nanoscaled topography and Sr release. It gives rise to excellent osteogenic properties and favorable osteogenic effects. The Ti implants with the NT–Sr coating can be fabricated easily and have good stability thereby facilitating large-scale industrial production, storage, transport, sterilization, and clinical use. The materials are attractive to bone implants, especially osteoporotic bone implants, in clinical applications.
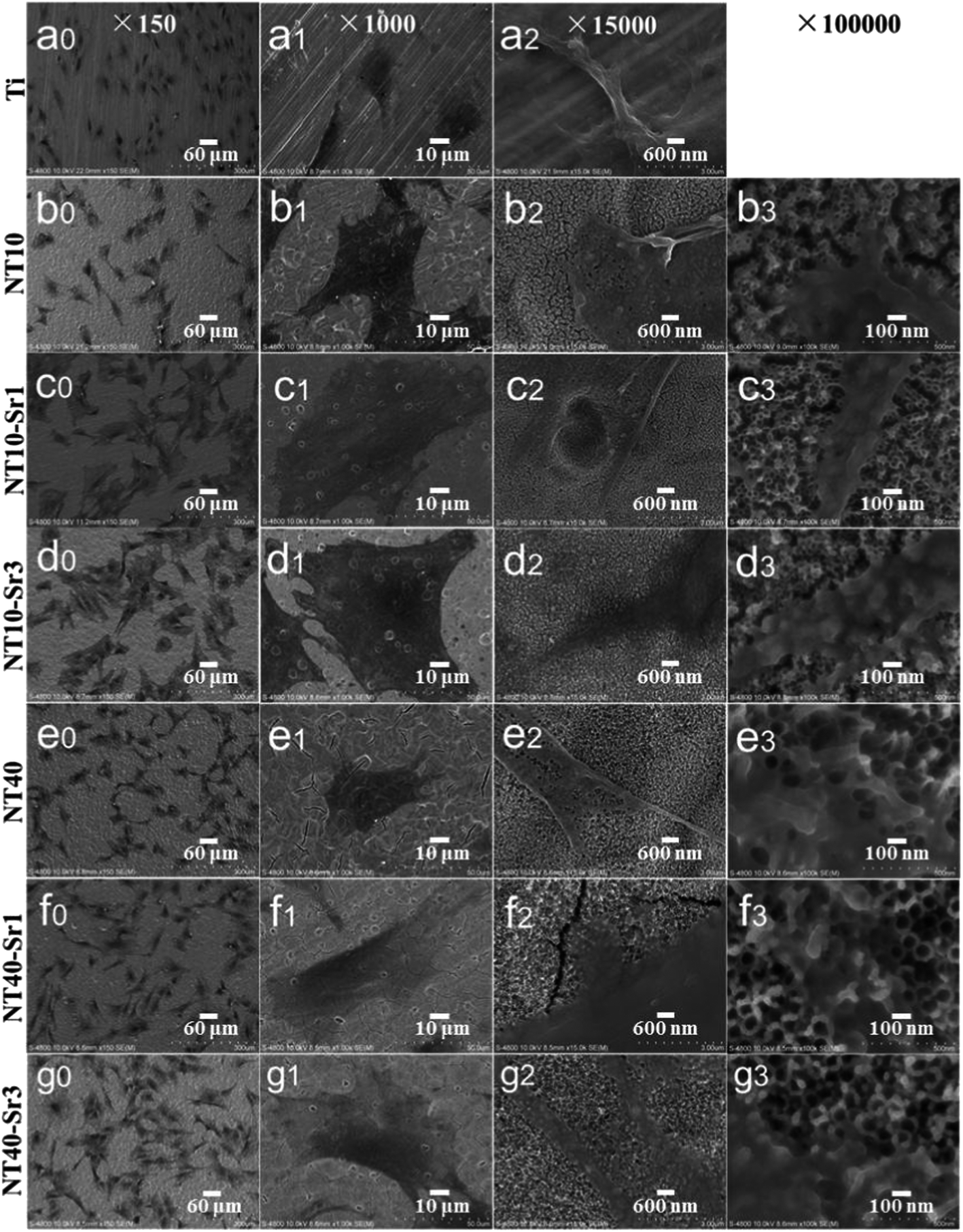 | ||
| Fig. 21 SEM views of MSCs after 2 days of culture on different samples: (a) Ti, (b) NT10, (c) NT10–Sr1, (d) NT10–Sr3, (e) NT-40, (f) NT40–Sr1 and (g) NT40–Sr3. Reproduced from ref. 155, Copyright (2013), with permission from Elsevier. | ||
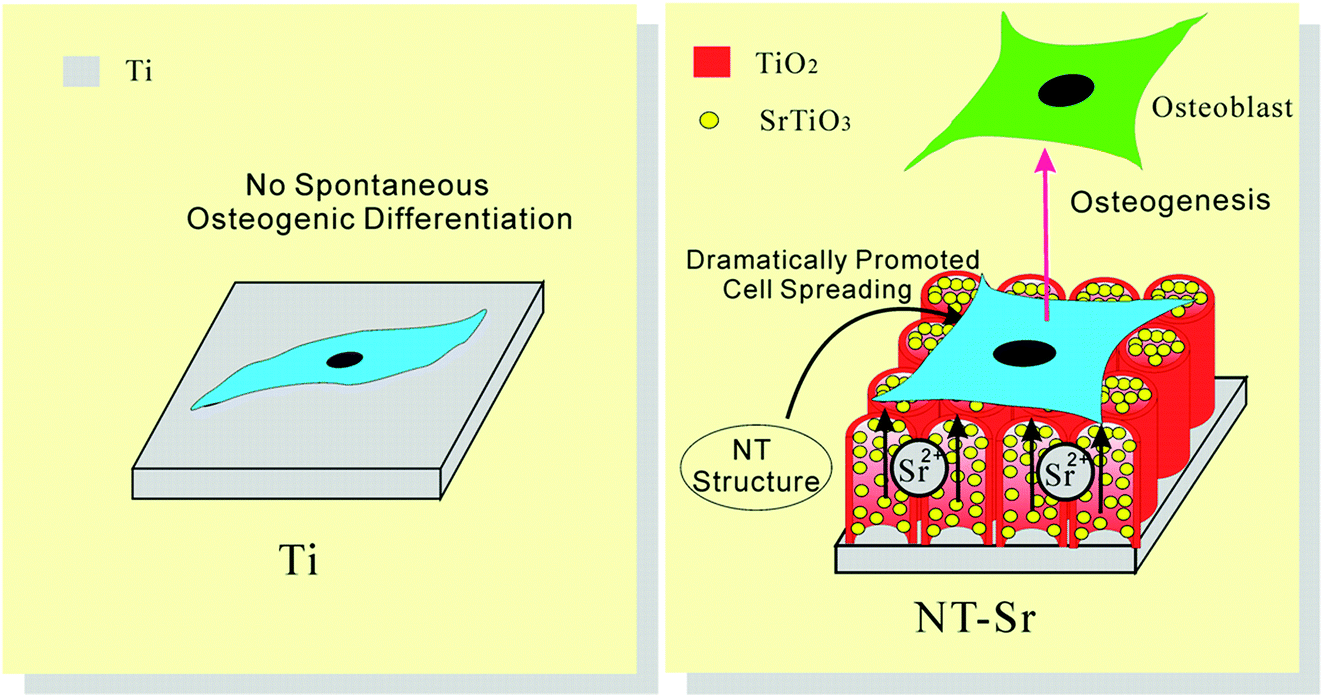 | ||
| Fig. 22 Schematic showing that the NT–Sr coating, combining the effect of Sr and the nanomorphology of NT, dramatically promotes MSC spreading and induces MSC selective differentiation towards osteoblasts. Reproduced from ref. 155, Copyright (2013), with permission from Elsevier. | ||
In addition to Sr, many agents have attractive properties in bone implant applications, for example, Zn and Mg. Besides the effects of positively regulating bone turnover and inhibiting bone resorption, Zn has good antibacterial and anti-inflammation ability. Zn has been incorporated into bioglasses and bioceramics. NT samples have been produced by anodization at 10 and 40 V (NT10 and NT40) were hydrothermally treated in 40 mL of 0.1 M zinc acetate at 200 °C for 1 and 3 h to produce the TiO2 NTs with different amounts of Zn designated as NT10–Zn1, NT10–Zn3, NT40–Zn1, and NT40–Zn3.157 The samples are annealed at 450 °C for 3 h in air to promote sample crystallization and strengthen the adhesion between the coating and Ti substrate. The amount of incorporated Zn can be adjusted by varying the structural parameters such as the NT diameter and length as well as hydrothermal treatment time. After the hydrothermal treatment, the nanotubular architecture is preserved. However, the diameter of the nanotubes on NT–Zn becomes smaller due to the transformation of TiO2 into ZnTiO3. The XPS depth profiles of Ti, O, and Zn of the NT–Zn samples demonstrate that a longer hydrothermal treatment incorporates more Zn and Zn is distributed along the entire length of the NTs on NT10–Zn3 and NT40–Zn3. The Zn concentrations are larger on the outer layers on NT10–Zn3 and NT40–Zn3 and decrease quickly with depth in the top 40 and 100 nm, respectively. To determine the total amount of Zn incorporated into the NT–Zn samples, the NT–Zn samples are dissolved in HF and HNO3 and the Zn contents are determined by ICP-AES. For the 1 cm2 coatings, the total Zn contents in NT10–Zn1, NT10–Zn3, NT40–Zn1, and NT40–Zn3 are 1.2, 11.4, 3.4 and 60.2 mg, respectively. The Zn release kinetics is assessed by immersing the samples in 5 mL PBS for a month. NT10–Zn3 delivers more Zn and NT40–Zn3 delivers the greatest quantity. The amounts of Zn released from NT40–Zn3 and NT10–Zn3 are 0.06 and 0.05 ppm in the first day, respectively and decrease quickly with time. After 20 days, about 0.01 ppm Zn is released. Zn incorporation is effective in preventing bacteria colonization on the specimens. The larger initial Zn release from NT40–Zn3 and NT10–Zn3 effectively kills adherent bacteria as well as surrounding planktonic bacteria in the early stage to prevent perioperative infection and foster normal wound healing as shown in Fig. 23. The long-term ability of NT40–Zn3 and NT10–Zn3 to prohibit bacteria colonization and decline in Zn release with time create the ideal scenario in clinical practice to harness the desirable defects while avoiding potential side effects associated with Zn overdose.
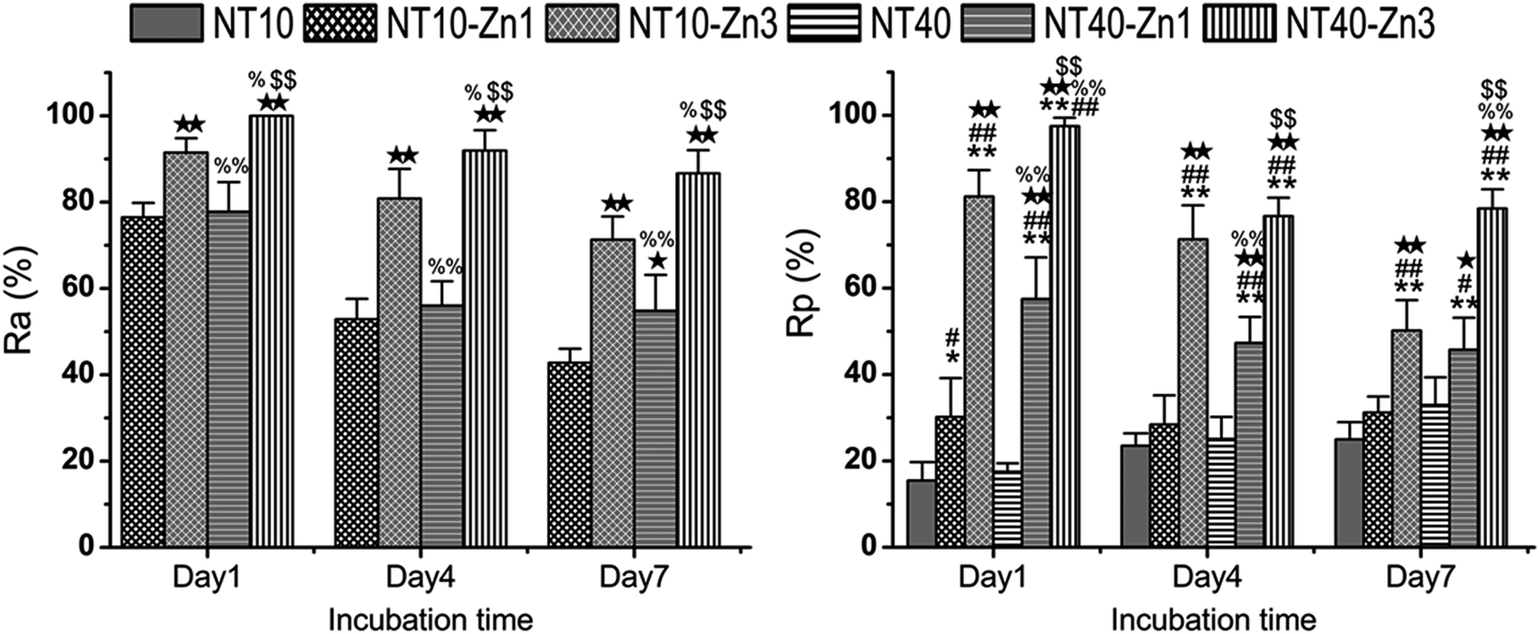 | ||
| Fig. 23 (A) Antibacterial rates versus adherent bacteria on the specimen (Ra) and against planktonic bacteria in the medium (Rp). *, ** p < 0.05 and 0.01 vs. NT10; #, ## p < 0.05 and 0.01 vs. NT40; ★, ★★ p < 0.05 and 0.01 vs. NT10–Zn1; %, %% p < 0.05 and 0.01 vs. NT10–Zn3; $, $$ p < 0.05 and 0.01 vs. NT40–Zn1. Reproduced from ref. 157, Copyright (2013), with permission from Elsevier. | ||
The effects of the NT–Zn coatings on the MSC functionalities and lineage commitment have been investigated. The NT–Zn coatings show no cytotoxicity as indicated by the LDH assay and are safe in long-term applications in vivo. The nontoxic nature of the NT–Zn samples is attributed to the small amounts of released Zn. The NT–Zn samples obviously enhance the initial adherent cell numbers, thus possibly initiating swift osseointegration in vivo. On NT10, NT10–Zn1, NT10–Zn3, and NT40–Zn3, the MSCs quickly assume a typical well spread osteoblast like cell shape and this effect is long lasting (Fig. 24). The better cell spreading on NT10–Zn1 and NT10–Zn3 than NT10 arises mainly from the effects of incorporated Zn since they have similar surface morphology.157 It is generally believed that well spread cells with bigger areas favor bone lineage and so the excellent ability to promote cell spreading is a positive indication of the osteogenesis inducing ability.
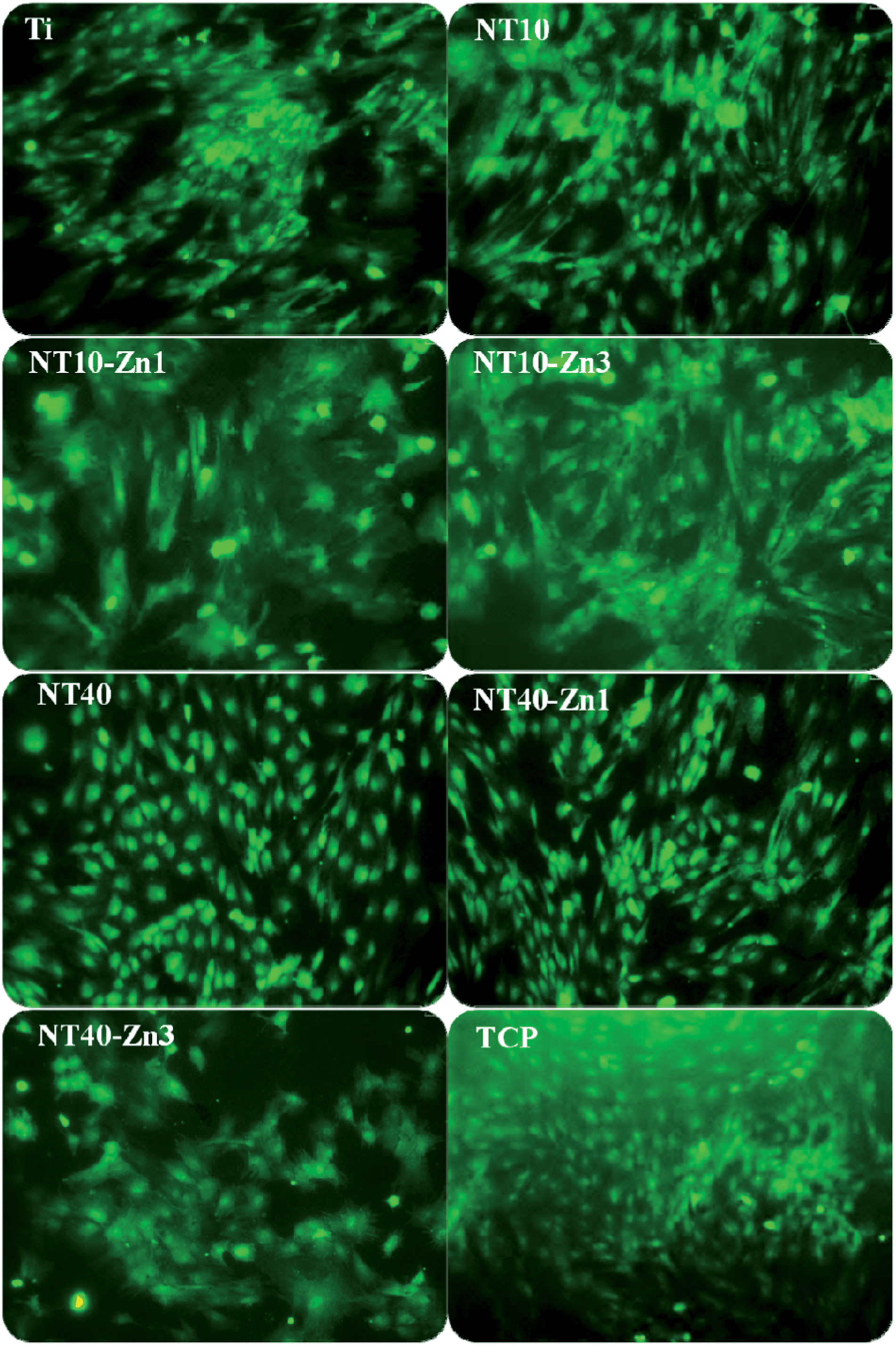 | ||
| Fig. 24 Fluorescence images of the green fluorescence protein (GFP) labeled MSCs showing the cell morphology on the samples (Ti, NT, NT–Zn) after culturing for 8 days. The samples were placed in 24 well plates and tissue culture plates (TCP) served as the control. Reproduced from ref. 157, Copyright (2013), with permission from Elsevier. | ||
Osteogenic differentiation of MSCs in the absence of exogenous OS has been observed. Samples NT10–Zn1, NT10–Zn3, and NT40–Zn3 induce dense nodular ALP areas after culturing for 10 d. MSC differentiation is attributed to signaling initiated by the surface topography and Zn. The ERK1/2 signaling is believed to play an important role in osteogenic differentiation of MSCs and found to be modulated by different biomaterials. NT40–Zn3 induces more even protein absorption and enhances cell spreading in turn leading to the highest ERK1/2 signaling activity (Fig. 25) and excellent cell osteogenic differentiation signaling. The biological effects on the NT–Zn coatings are ascribed to the combined effects of the topographical cues and Zn incorporation that lead to much higher activity of ERK1/2, osteoblast marker gene expression, and calcium deposition on the MSCs.
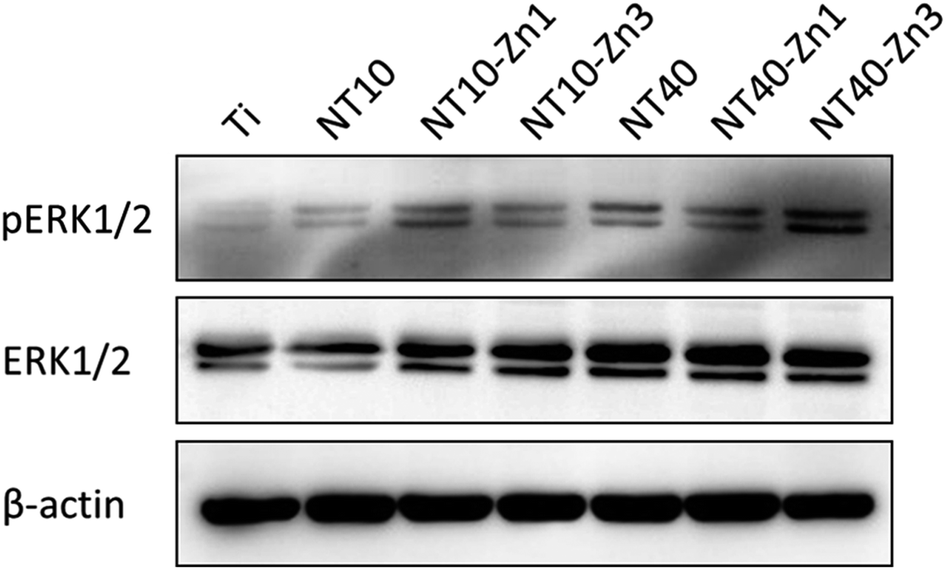 | ||
| Fig. 25 Western blot of pERK1/2 and ERK1/2 levels in the MSCs cultured on the different samples for 48 h. β-actin is used as a control for equal loading. Reproduced from ref. 157, Copyright (2013), with permission from Elsevier. | ||
The NT–Ag coating on Ti implants also show good long-term antibacterial ability but disappointingly they induce cytotoxicity.195 In comparison, the NT40–Zn3 structure has a larger Zn loading capacity and releases more Zn showing excellent osteogenesis inducing activity without cytotoxicity as well as good antibacterial effects. The NT–Zn coatings possess both osteogenesis inducing ability and antibacterial effects to prevent post-operation infection due to the release of Zn and have large potential in biomedical applications.
4.2 TiO2 NTAs-based electrode for biosensing
TiO2 NTAs fabricated by anodic oxidation of Ti have excellent biocompatibility, large surface area, good uniformity, and conformability over large areas and constitute a promising platform for protein and biomolecule immobilization and biosensing applications.196–199 It is crucial that the proteins can be immobilized onto the TiO2 NTs while retaining their native biological structure and functions.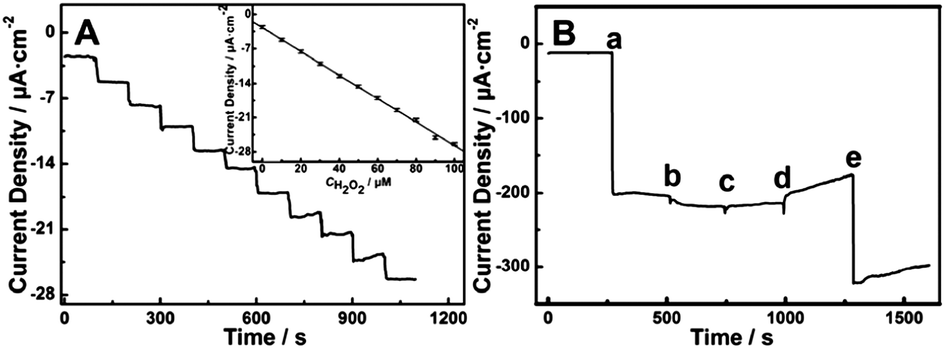 | ||
| Fig. 26 (A) Amperometric response obtained at the TiO2 NTA electrode in 0.1 M pH = 5.5 PBS upon successive injection of 10 mM H2O2 for each step at −0.25 V (vs. Ag/AgCl), the inset is the fitted current plot versus H2O2 concentration; (B) amperometric response at the TiO2 NTA electrode in 0.1 M pH = 5.5 PBS at −0.25 V with sequential injection of 2 mM H2O2 (a), 2 mM uric acid (UA) (b), 2 mM dopamine (DA) (c), 2 mM ascorbic acid (AA) (d) and 2 mM H2O2 (e). Adapted from ref. 204, Copyright (2013) The Royal Society of Chemistry. | ||
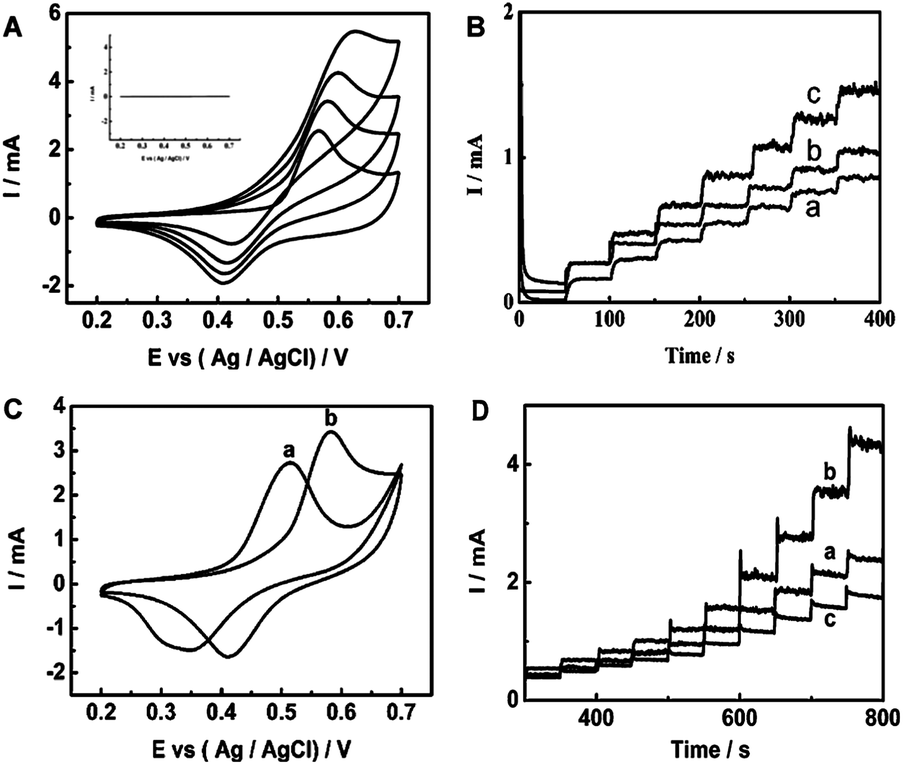 | ||
| Fig. 27 (A) CVs of the Ni–Cu/TiO2 electrode in 0.1 M NaOH solution with different concentrations of glucose: 0, 0.1, 0.2 and 0.4 mM (from inner to outer). Inset: TiO2 electrode in 0.1 M NaOH solution with 0.1 mM glucose. All the scan rates are 50 mV s−1. (B) Amperometric responses of Cu/TiO2NTs (a), Ni/TiO2 NTs (b) and Ni–Cu/TiO2 NTs (c) in 0.1 M NaOH with successive addition 0.1 mM glucose at +0.55 V. (C) CVs of Ni–Cu/Ti electrode (a) and Ni–Cu/TiO2 NTs electrode (b) in 0.1 M NaOH solution containing 0.1 mM glucose. All the scan rates are 50 mV s−1. (D) Amperometric responses for successive addition of a certain amount of glucose in 0.1 M NaOH solution at 0.6 V of activated Ni–Cu/TiO2 NTs deposited for different times: 11.2 s (a), 33.6 s (b) and 56 s (c). Adapted with permission from ref. 210 Copyright (2013) The Royal Society of Chemistry. | ||
Li et al. have reported photoactive TiO2 NTAs immobilized with HRP for visible-light-activated PEC detection of H2O2. The photocurrent spectra of HRP/TiO2 NTs show an obvious photocurrent response under visible-light irradiation (λ ≥ 400 nm), suggesting the possibility of PEC detection of H2O2 upon visible light irradiation. The photocurrent generated from the HRP/TiO2 NTs at 400 nm is significantly enhanced after addition of H2O2 into the solution and the photocurrent intensity goes up with H2O2 concentration. The HRP/TiO2 NTAs electrode displays a linear range of 5.0 × 10−7 to 3.5 × 10−5 M and low detection limit of 1.8 × 10−7 M in H2O2 determination.213 Zhang et al. describe the construction of a novel photoelectrochemical immunosensor for a-Synuclein (a-SYN) detection based on Au-doped TiO2 NTAs. The Au–TiO2 NTAs play an essential role in not only immobilization of the protein molecules, but also photocurrent generation during the process.214 The secondary antibody (Ab2) and GOx can be easily bound to the Au nanoparticles to yield {Ab2–Au–GOx} bioconjugates. The photocurrents are proportional to the a-SYN concentrations and the linear range of the immunosensor is from 50 pg mL−1 to 100 ng mL−1 with a detection limit of 34 pg mL−1. Cai et al. have developed a label-free tris(2,3-dibromopropyl)isocyanurate (TBC) immunosensor by immobilizing the anti-TBC polyclonal antibody on the CdTe/Au nanocrystals co-deposited TiO2 NTAs (CdTe/Au–TiO2 NTAs) substrate. The ternary hybrid CdTe/Au–TiO2 NTAs electrode shows enhanced photon absorption and photocurrent generation due to the synergistic effects of the CdTe NPs, Au NPs, and TiO2 NTAs. TBC is quantified by measuring the photocurrent intensity. Sensitive detection of TBC is achieved with a limit of detection (LOD) of 50 pM and linear range of 50 pM to 50 μM.215 WO3 NPs decorated core–shell TiC–C nanofiber arrays with high sensitivity have been reported for non-enzymatic photoelectrochemical biosensing.216 A high PEC sensitivity of 3388 mA mM−1 cm−2 in the range of 1–10 mM and 837 mA mM−1 cm−2 in the range of 10–100 mM together with a low detection limit of 11.2 nM (S/N = 3) for glucose are achieved under 50 mW cm−2 irradiation. The results provide insights into non-enzymatic PEC biological assays that are convenient, highly sensitive, and promising.216
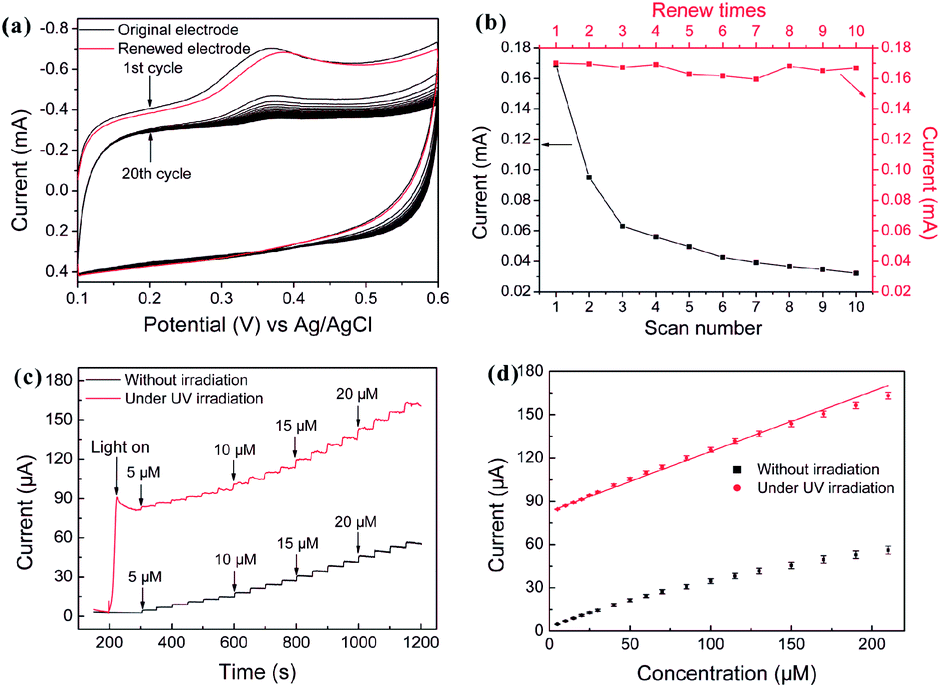 | ||
| Fig. 28 (a) CV profiles of 0.2 mM 5-HT in a 0.1 M pH = 7.4 PBS obtained from original (black) and renewed (red) C-doped TiO2-NTAs electrode. Scan rate: 100 mV s−1. (b) The peak current of different scan number and renew times. (c) Typical current–time response curves at the C-doped TiO2-NTAs electrode through successive addition of different concentrations of 5-HT (5–210 μM) into a stirred 0.1 M PBS (pH = 7.4) at an applied potential of 380 mV with and without UV irradiation, respectively. The electrodes are not removed between measurements at different concentrations without UV irradiation. (d) The calibration curve of the respond currents versus concentrations of 5-HT with and without UV irradiation. The error bars represent the respond current of three independent experiments. Adapted with permission from ref. 125 Copyright (2011) American Chemical Society. | ||
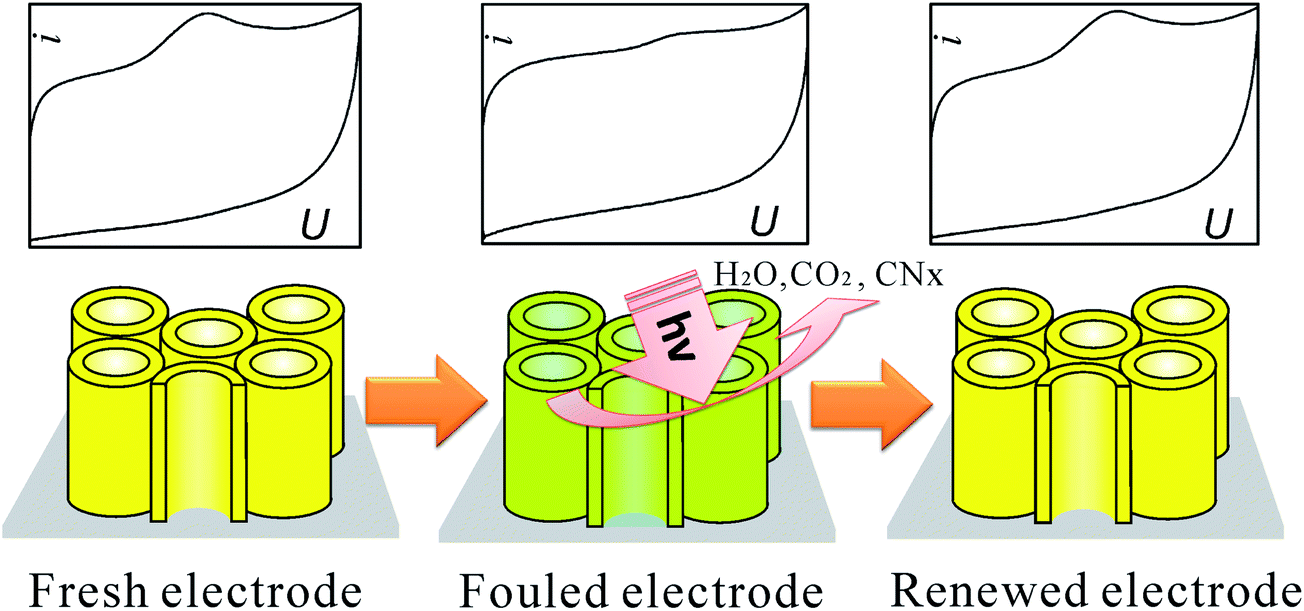 | ||
| Fig. 29 Photo-refreshing process of the C-doped TiO2 NTAs electrode for biosensor. Adapted with permission from ref. 125 Copyright (2011) American Chemical Society. | ||
TiO2 NTAs also possess excellent chemical sensing properties. For example, Ameen et al. have demonstrated that the TiO2 NTAs electrode has a high sensitivity of ∼40.9 μA mM−1 cm−2, detection limit of 0.22 μM, and short response time of 10 s in the detection of harmful phenyl hydrazine.198 The enhanced sensing properties are ascribed to the depleted oxygen layer on the TiO2 NT arrays, favorable electron transfer, and good electrocatalytic activity towards phenyl hydrazine.
5. Conclusions and outlook
Anodization of Ti to form TiO2 NTs is a promising technique to produce an ordered nanotopography on the surface to improve cellular response. The proper NTs promote the functions of osteoblasts, MSCs osteogenic differentiation in vitro, as well as implant osseointegration in vivo. TiO2 NTAs can be incorporated with Ag, Sr, and Zn to achieve both antibacterial and/or osteogenesis-inducing capability. It should be noted that the size and topography of the TiO2 NTA coatings must be optimized/modified in order to achieve the favorable cell response and osseointegration ability. Sample sterilization which is essential to experiments conducted in vitro and in vivo can be considered a post-treatment but common methods utilizing ultraviolet light, autoclaving, and ethanol treatment can change the surface properties of the samples and corresponding biological characteristics. Hence, the TiO2 NTs must be easily and effectively sterilized. Incorporation of Ag or antibiotics into TiO2 NTAs to enhance the antibacterial properties while preserving the osseointegrative properties are important to biomedical applications of Ti implants. Precise control of the NT length and diameter enables different loading and release capacities at different rates. Another topic that must be further investigated is the effect of wear debris generated from the TiO2 NT structures. The anodized TiO2 NTs are mechanically fragile and easily destroyed. Hence, the mechanical properties and adhesion strength between the TiO2 NTAs and underlying Ti substrate must be improved and the biological consequence of wear debris released from anodized nanotubular Ti must be better understood.Anodized TiO2 NTAs constitute a promising platform or vessel for protein or biomolecule immobilization in biosensing applications due to their excellent biocompatibility and large surface area. Although biosensors based on TiO2 NTAs have been reported, it is important that proteins or enzymes are immobilized on the TiO2 NTs so that direct electrochemistry of the redox proteins or enzymes can take place without chemical mediators. Optimization of the electrode properties by means of doping and modification lead to improved photo-electrochemical properties. Rapid and precise real-time detection of analytes requires that the electrochemical sensors can operate with a short response time, enhanced selectivity, sensitivity, and recoverability. A significant advantage of a biosensor composed of TiO2 NTAs is self-regeneration under light irradiation. As an example, the C-doped TiO2 NTAs electrode delivers excellent electrochemical performance in simultaneous determination of 5-HT and AA. Regeneration of the fouled surface can be readily accomplished by irradiation with UV or visible light and the high selectivity and sensitivity can be recovered without damaging the microstructure.
PEC detection based on TiO2 NTAs is a new and promising bioanalytical method. However, many biological systems are prone to UV radiation and the strong oxidizing power of photogenerated holes produced by TiO2 during UV illumination may induce destructive effects on some biomolecules. The low conductivity of TiO2 results in poor electron transport and poor detection sensitivity. Therefore, modification of the TiO2 NTAs to shift the threshold of the photoresponse into the visible light region and improve the conductivity is a viable approach to improve the PEC properties. By combining modern technologies such as nanofabrication and lab-on-chip designs, TiO2 NTAs can be made into efficient, low cost, and environmentally benign sensors suitable for biomedical diagnosis and environmental analysis.
Acknowledgements
This work was financially supported by National Science Foundation of China (50902104 and 21105077), Fundamental Research Funds for the Central Universities (HUST: 0118187030), National Science Foundation of Hubei Province (2013CFB327), Hong Kong Research Grants (RGC) General Research Funds (GRF) no. 112212, and City University of Hong Kong Applied Research Grants (ARG) no. 9667066 and 9667069.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., A, 2012, 13, 169–189 CrossRef CAS PubMed.
- X. B. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- X. Su, L. Wu, X. Zhan, J. Wu, Y. Wei and Z. Guo, J. Mater. Sci., 2012, 47, 2519–2534 CrossRef CAS.
- D. P. Macwan, P. N. Dave and S. Chaturvedi, J. Mater. Sci., 2011, 46, 3669–3686 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- R. Tenne1 and C. N. R. Rao, Philos. Trans. R. Soc., A, 2004, 362, 2099–2125 CrossRef PubMed.
- M. J. Green, N. Behabtu, M. Pasquali and W. W. Adams, Polymer, 2009, 50, 4979–4997 CrossRef CAS PubMed.
- R. S. Devan, R. A. Patil, J. H. Lin and Y. R. Ma, Adv. Funct. Mater., 2012, 22, 3326–3370 CrossRef CAS PubMed.
- P. M. Rørvik, T. Grande and M. A. Einarsrud, Adv. Mater., 2011, 23, 4007–4034 CrossRef PubMed.
- C. N. R. Rao and A. Govindaraj, Adv. Mater., 2009, 21, 4208–4233 CrossRef CAS PubMed.
- R. Tenne and G. Seifert, Annu. Rev. Mater. Res., 2009, 39, 387–413 CrossRef CAS.
- G. K. Mor, O. K. Varghese, M. Paulose, K. Shankar and C. A. Grimes, Sol. Energy Mater. Sol. Cells, 2006, 90, 2011–2075 CrossRef CAS PubMed.
- I. Paramasivam, H. Jha, N. Liu and P. Schmuki, Small, 2012, 8, 3073–3103 CrossRef CAS PubMed.
- J. Y. Huang, K. Q. Zhang and Y. K. Lai, Int. J. Photoenergy, 2013, 2013, 761971 Search PubMed.
- J. M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3–18 CrossRef CAS PubMed.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- Y. Jun, J. H. Park and M. G. Kang, Chem. Commun., 2012, 48, 6456–6471 RSC.
- W. Tan, B. Pingguan-Murphy, R. Ahmad and S. A. Akbar, Ceram. Int., 2012, 38, 4421–4435 CrossRef PubMed.
- S. Minagar, J. Wang, C. C. Berndt, E. P. Ivanova and C. Wen, J. Biomed. Mater. Res., Part A, 2013, 101, 2726–2739 CrossRef PubMed.
- H. Han, T. Song, E. K. Lee, A. Devadoss, Y. Jeon, J. Ha, Y. C. Chung, Y. M. Choi, Y. G. Jung and U. Paik, ACS Nano, 2012, 6, 8308–8315 CrossRef CAS PubMed.
- V. Galstyan, E. Comini, G. Faglia and G. Sberveglieri, Sensors, 2013, 13, 14161–14174 CrossRef PubMed.
- M. Salari, S. H. Aboutalebi, K. Konstantinov and H. K. Liu, Phys. Chem. Chem. Phys., 2011, 13, 5038–5041 RSC.
- F. Fabregat-Santiago, E. M. Barea, J. Bisquert, G. K. Mor, K. Shankar and C. A. Grimes, J. Am. Chem. Soc., 2008, 130, 11312–11316 CrossRef CAS PubMed.
- S. Bae, E. Shim, J. Yoon and H. Joo, Sol. Energy Mater. Sol. Cells, 2008, 92, 402–409 CrossRef CAS PubMed.
- A. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425–5438 RSC.
- K. C. Popat, M. Eltgroth, T. J. LaTempa, C. A. Grimes and T. A. Desai, Small, 2007, 3, 1878–1881 CrossRef CAS PubMed.
- P. Hoyer, Langmuir, 1996, 12, 1411–1413 CrossRef CAS.
- H. Imai, Y. Takei, K. Shimizu, M. Matsuda and H. Hirashim, J. Mater. Chem., 1999, 9, 2971–2972 RSC.
- S. Kobayashi, N. Hamasaki, M. Suzuki, M. Kimura, H. Shirai and K. Hanabusa, J. Am. Chem. Soc., 2002, 124, 6550–6551 CrossRef CAS PubMed.
- C. H. Lee, S. W. Rhee and H. W. Choi, Nanoscale Res. Lett., 2012, 7, 48–52 CrossRef PubMed.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS.
- Z. R. Tian, J. A. Voigt, J. Liu, B. Mckenzie and H. F. Xu, J. Am. Chem. Soc., 2003, 125, 12384–12385 CrossRef CAS PubMed.
- A. Nakahira, T. Kubo and C. Numako, Inorg. Chem., 2010, 49, 5845–5852 CrossRef CAS PubMed.
- V. Zwilling, E. Darque-Ceretti, A. Boutry-Forveille, D. David, M. Y. Perrin and M. Aucouturier, Surf. Interface Anal., 1999, 27, 629–637 CrossRef CAS.
- D. Gong, C. A. Grimes, O. K. Varghese, W. C. Hua, R. S. Singh, Z. Chen and E. C. Dickey, J. Mater. Res., 2001, 16, 3331–3334 CrossRef CAS.
- M. Paulose, H. E. Prakasam, O. K. Varghese, L. Peng, K. C. Popat, G. K. Mor, T. A. Desai and C. A. Grimes, J. Phys. Chem. C, 2007, 111, 14992–14997 CAS.
- D. A. Wang and L. F. Liu, Chem. Mater., 2010, 22, 6656–6664 CrossRef CAS.
- D. Regonini, C. R. Bowen, A. Jaroenworaluck and R. Stevens, Mater. Sci. Eng., R, 2013, 74, 377–406 CrossRef PubMed.
- S. P. Albu, P. Roy, S. Virtanen and P. Schmuki, Isr. J. Chem., 2010, 50, 453–467 CrossRef CAS PubMed.
- Y. Yang, S. P. Albu, D. Kim and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 9071–9075 CrossRef CAS PubMed.
- S. Berger, F. Jakubka and P. Schmuki, Electrochem. Commun., 2008, 10, 1916–1919 CrossRef CAS PubMed.
- Y. Shin and S. Lee, Nanotechnology, 2009, 20, 105301–105305 CrossRef PubMed.
- S. Berger, F. Jakubka and P. Schmuki, Electrochem. Solid-State Lett., 2009, 12, K45–K48 CrossRef CAS PubMed.
- I. Sieber, H. Hildebrand, A. Friedrich and P. Schmuki, Electrochem. Commun., 2005, 7, 97–100 CrossRef CAS PubMed.
- R. L. Karlinsey, Electrochem. Commun., 2005, 7, 1190–1194 CrossRef CAS PubMed.
- W. Wei, J. M. Macak and P. Schmuki, Electrochem. Commun., 2008, 10, 428–432 CrossRef CAS PubMed.
- W. Wei, J. M. Macak, N. K. Shrestha and P. Schmuki, J. Electrochem. Soc., 2009, 156, K104–K109 CrossRef CAS PubMed.
- H. A. El-Sayed and V. I. Birss, Nano Lett., 2009, 9, 1350–1355 CrossRef CAS PubMed.
- M. Yang, N. K. Shrestha and P. Schmuki, Electrochem. Commun., 2009, 11, 1908–1911 CrossRef CAS PubMed.
- A. Watcharenwong, W. Chanmanee, N. R. de Tacconi, C. R. Chenthamarakshan, P. Kajitvichyanukul and K. Rajeshwar, J. Electroanal. Chem., 2008, 612, 112–120 CrossRef CAS PubMed.
- X. M. Zhang, K. F. Huo, L. S. Hu and P. K. Chu, Nanosci. Nanotechnol. Lett., 2010, 2, 51–57 CAS.
- S. P. Albu, A. Ghicov and P. Schmuki, Phys. Status Solidi RRL, 2009, 3, 64–66 CrossRef CAS PubMed.
- S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048–3055 CrossRef CAS.
- S. Berger, H. Tsuchiya and P. Schmuki, Chem. Mater., 2008, 20, 3245–3247 CrossRef CAS.
- K. Yasuda and P. Schmuki, Electrochem. Commun., 2007, 9, 615–619 CrossRef CAS PubMed.
- K. Yasuda and P. Schmuki, Adv. Mater., 2007, 19, 1757–1760 CrossRef CAS PubMed.
- A. Ghicov, S. Aldabergenova, H. Tsuchiya and P. Schmuki, Angew. Chem., Int. Ed, 2006, 45, 6993–6996 CrossRef CAS PubMed.
- A. Ghicov, M. Yamamoto and P. Schmuki, Angew. Chem., Int. Ed., 2008, 41, 8052–8055 CrossRef PubMed.
- H. Tsuchiya, J. Nakata, S. Fujimoto, S. Berger and P. Schmuki, ECS Trans., 2008, 16, 359–367 CrossRef CAS.
- Y. C. Nah, A. Ghicov, D. Kim, S. Berger and P. Schmuki, J. Am. Chem. Soc., 2008, 130, 16154–16155 CrossRef CAS PubMed.
- X. J. Feng, J. M. Macak, S. P. Albu and P. Schmuki, Acta Biomater., 2008, 4, 318–323 CrossRef CAS PubMed.
- J. M. Macak, H. Tsuchiya, L. Taveira, A. Ghicov and P. Schmuki, J. Biomed. Mater. Res., Part A, 2005, 75, 928–933 CrossRef PubMed.
- C. A. Grimes and G. K. Mor, TiO2 Nanotube Arrays – Synthesis, Properties, and Applications, Springer, London, 2009 Search PubMed.
- L. L. Zhao, P. K. Chu, Y. Zhang and Z. Wu, J. Biomed. Mater. Res., Part B, 2009, 91, 470–480 CrossRef PubMed.
- C. N. Elias, J. H. C. Lima, R. Valiev and M. A. Meyers, JOM, 2008, 60, 46–49 CrossRef CAS PubMed.
- K. C. Popat, L. Leoni, C. A. Grimes and T. A. Desai, Biomaterials, 2007, 28, 3188–3197 CrossRef CAS PubMed.
- K. S. Brammer, S. H. Oh, J. O. allagher and S. H. Jin, Nano Lett., 2008, 8, 786–793 CrossRef CAS PubMed.
- J. Park, S. Bauer, K. v. d. Mark and P. Schmuki, Nano Lett., 2007, 7, 1686–1691 CrossRef CAS PubMed.
- J. Park, S. Bauer, K. A. Schlegel, F. W. Neukam, K. V. D. Mark and P. Schmuki, Small, 2009, 5, 666–671 CrossRef CAS PubMed.
- S. Oh and S. Jin, Mater. Sci. Eng., C, 2006, 26, 1301–1306 CrossRef CAS PubMed.
- K. C. Popat, M. Eltgroth, T. J. LaTempa, C. A. Grimes and T. A. Desai, Biomaterials, 2007, 28, 4880–4888 CrossRef CAS PubMed.
- K. Masuda and H. Fukuda, Science, 1995, 268, 1466–1468 Search PubMed.
- Q. Y. Lu, F. Gao, S. Komarneni and T. E. Mallouk, J. Am. Chem. Soc., 2004, 126, 8650–8651 CrossRef CAS PubMed.
- S. Shingubara, J. Nanopart. Res., 2003, 5, 17–30 CrossRef CAS.
- D. Losic, J. G. Shapter, J. G. Mitchell and N. H. Voelcker, Nanotechnology, 2005, 16, 2275–2281 CrossRef CAS PubMed.
- M. Assefpour-Dezfuly, C. Vlachos and E. H. Andrews, J. Mater. Sci., 1984, 19, 3626–3639 CrossRef CAS.
- Q. Y. Zeng, M. Xi, W. Xu and X. J. Li, Mater. Corros., 2013, 64, 1001–1006 CrossRef CAS PubMed.
- K. Yasuda and P. Schmuki, Electrochim. Acta, 2007, 52, 4053–4361 CrossRef CAS PubMed.
- J. M. MacAk, K. Sirotna and P. Schmuki, Electrochim. Acta, 2005, 50, 3679–3684 CrossRef CAS PubMed.
- H. Liu, L. Tao and W. Z. Shen, Nanotechnology, 2011, 22, 155603 CrossRef PubMed.
- K. Shankar, G. K. Prakasam, H. E. Yoriya, S. Yoriya, M. Paulose, O. K. Grimes and C. A. Grimes, Nanotechnology, 2007, 18, 065707 CrossRef.
- S. Yoriya and C. A. Grimes, Langmuir, 2010, 26, 417–420 CrossRef CAS PubMed.
- Q. Cai, M. Paulose, O. K. Varghese and C. A. Grimes, J. Mater. Res., 2005, 20, 230–236 CrossRef CAS.
- N. K. Allam and C. A. Grimes, J. Phys. Chem. C, 2007, 111, 13028–13032 CAS.
- K. Nakayama, T. Kubo, A. Tsubokura, Y. Nishikitani and H. Masuda, ECS Meeting Abstracts, 2005, 502, 819 Search PubMed.
- C. Richter, Z. Wu, E. Panaitescu, R. Willey and L. Menon, Adv. Mater., 2007, 19, 946–948 CrossRef CAS PubMed.
- R. Hahn, J. M. Macak and P. Schmuki, Electrochem. Commun., 2007, 9, 947–952 CrossRef CAS PubMed.
- Z. X. Su and W. Z. Zhou, Adv. Mater., 2008, 20, 3663–3667 CrossRef CAS PubMed.
- S. Berger, J. Kunze, P. Schmuki, D. LeClere, A. Valota, P. Skeldon and G. E. Thompson, Electrochim. Acta, 2009, 54, 5942–5948 CrossRef CAS PubMed.
- S. P. Albu, A. Ghicov, S. Aldabergenova, P. Drechsel, D. LeClere, G. E. Thompson, J. M. Macak and P. Schmuki, Adv. Mater., 2008, 20, 4135–4139 CAS.
- Y. Smith, R. Ray, K. Carlson, B. Sarma and M. Misra, Materials, 2013, 6, 2892–2957 CrossRef CAS.
- N. K. Allam and C. A. Grimes, Langmuir, 2009, 25, 7234–7240 CrossRef CAS PubMed.
- M. X. Zhang, K. F. Huo, H. R. Wang, W. Zhang and P. K. Chu, J. Nanosci. Nanotechnol., 2011, 11, 11200–11205 CrossRef PubMed.
- O. K. Varghese, D. W. Gong, M. Paulose, C. A. Grimes and E. C. Dickey, J. Mater. Res., 2003, 18, 156–165 CrossRef CAS.
- A. Roguska, M. Pisarek, M. Andrzejczuk, M. Dolata, M. Lewandowska and M. Janik-Czachor, Mater. Sci. Eng., C, 2011, 31, 906–914 CrossRef CAS PubMed.
- A. Ghicov, S. P. Albu, R. Hahn, D. Kim, T. Stergiopoulos, J. Kunze, C. A. Schiller, P. Falaras and P. Schmuki, Chem.–Asian. J., 2009, 4, 520–525 CrossRef CAS PubMed.
- Y. Yang, X. H. Wang and L. T. Li, J. Am. Ceram. Soc., 2008, 91, 632–635 CrossRef CAS.
- A. Tighineanu, T. Ruff, S. Albu, R. Hahn and P. Schmuki, Chem. Phys. Lett., 2010, 494, 260–263 CrossRef CAS PubMed.
- H. Zhou and Y. R. Zhang, J. Power Sources, 2013, 239, 128–131 CrossRef CAS PubMed.
- X. H. Lu, G. M. Wang, T. Zhai, M. H. Yu, J. Y. Gan, Y. X. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS PubMed.
- H. Wu, C. Xu, J. Xu, L. Lu, Z. Fan and X. Chen, Nanotechnology, 2013, 24, 455401–455407 CrossRef PubMed.
- M. Salari, K. Konstantinov and H. K. Liu, J. Mater. Chem., 2011, 21, 5128–5133 RSC.
- P. Xiao, Y. Zhang, B. B. Garcia, S. Sepehri, D. W. Liu and G. Cao, J. Nanosci. Nanotechnol., 2009, 9, 2426–2436 CrossRef CAS PubMed.
- G. Liu, F. Li, D. W. Wang, D. M. Tang, C. Liu, X. L. Ma, G. Q. Lu and H. M. Cheng, Nanotechnology, 2008, 19, 025606–025611 CrossRef PubMed.
- Y. H. Zhang, P. Xiao, X. Y. Zhou, D. W. Liu, B. B. Garcia and G. Z. Cao, J. Mater. Chem., 2009, 19, 948–953 RSC.
- Z. G. Lu, C. T. Yip, L. P. Wang, H. T. Huang and L. M. Zhou, ChemPlusChem, 2012, 77, 991–1000 CrossRef CAS PubMed.
- Y. K. Lai, J. Y. Huang, H. F. Zhang, V. P. Subramaniam, Y. X. Tang, D. G. Gong, L. Sundar, L. Sun, Z. Chen and C. J. Lin, J. Hazard. Mater., 2010, 184, 855–863 CrossRef CAS PubMed.
- J. M. Macak, A. Ghicov, R. Hahn, H. Tsuchiya and P. Schmuki, J. Mater. Res., 2006, 21, 2824–2828 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- J. H. Park, S. W. Kim and A. J. Bard, Nano Lett., 2006, 6, 24–28 CrossRef CAS PubMed.
- J. Y. Li, N. Lu, X. Quan, S. Chen and H. M. Zhao, Ind. Eng. Chem. Res., 2008, 47, 3804–3808 CrossRef CAS.
- H. Tang and D. Y. Li, J. Phys. Chem. C, 2008, 112, 5405–5409 Search PubMed.
- Y. Y. Zhang, W. Y. Fu, H. B. Yang, S. K. Liu, P. Sun, M. X. Yuan, D. Ma, W. Y. Zhao, Y. M. Sui, M. H. Li and Y. X. Li, Thin Solid Films, 2009, 518, 99–103 CrossRef CAS PubMed.
- W. F. Zhou, Q. J. Liu, Z. Q. Zhu and J. Zhang, J. Phys. D: Appl. Phys., 2010, 43, 035301 CrossRef.
- S. M. Zhang, Y. Y. Chen, Y. Yu, H. H. Wu, S. R. Wang, B. L. Zhu, W. P. Huang and S. H. Wu, J. Nanopart. Res., 2008, 10, 871–875 CrossRef CAS.
- Y. F. Tu, S. Y. Huang, J. P. Sang and X. W. Zou, Mater. Res. Bull., 2010, 45, 224–229 CrossRef CAS PubMed.
- O. Teruhisa, M. Takahiro and M. Michio, Chem. Lett., 2003, 32, 364–365 CrossRef.
- R. Hahn, F. Schmidt-Stein, J. Salonen, S. Thiemann, Y. Y. Song, J. L. Kunze, V. P. Lehto and P. Schmuki, Angew. Chem., Int. Ed., 2009, 48, 7236–7239 CrossRef CAS PubMed.
- A. Ghicov, J. M. Macak, H. Tsuchiya, J. Kunze, V. Haeublein, L. Frey and P. Schmuki, Nano Lett., 2006, 6, 1080–1082 CrossRef CAS.
- R. Hahn, A. Ghicov, J. Salonen, V. P. Lehto and P. Schmuki, Nanotechnology, 2007, 18, 105604 CrossRef.
- S. U. M. Kahn, M. Al-Shahry and W. B. J. Ingler, Science, 2002, 297, 2243–2245 CrossRef PubMed.
- L. S. Hu, K. F. Huo, R. S. Chen, B. Gao, J. J. Fu and P. K. Chu, Anal. Chem., 2011, 83, 8138–8144 CrossRef CAS PubMed.
- D. H. Kim, S. Fujimoto, P. Schmuki and H. Tsuchiya, Electrochem. Commun., 2008, 10, 910–913 CrossRef CAS PubMed.
- N. K. Shrestha, Y. C. Nah, H. Tsuchiya and P. Schmuki, Chem. Commun., 2009, 2008–2010 RSC.
- P. Agarwal, I. Paramasivam, N. K. Shrestha and P. Schmuki, Chem.–Asian J., 2010, 5, 66–69 CrossRef CAS PubMed.
- J. M. Macak, P. J. Barczuk, H. Tsuchiya, M. Z. Nowakowska, A. Ghicov, M. Chojak, S. Bauer, S. Virtanen, P. J. Kulesza and P. Schmuki, Electrochem. Commun., 2005, 7, 1417–1422 CrossRef CAS PubMed.
- I. Paramasivam, J. M. Macak and P. Schmuki, Electrochem. Commun., 2008, 10, 71–75 CrossRef CAS PubMed.
- I. Paramasivam, J. M. Macak, A. Ghicov and P. Schmuki, Chem. Phys. Lett., 2007, 445, 233–237 CrossRef CAS PubMed.
- S. H. Kang, Y. E. Sung and W. H. Smyrlb, J. Electrochem. Soc., 2008, 155, 1128–1135 CrossRef PubMed.
- W. T. Sun, Y. Yu, H. Y. Pan, X. F. Gao, Q. Chen and L. M. Peng, J. Am. Chem. Soc., 2008, 130, 1124–1125 CrossRef CAS PubMed.
- L. X. Yang, S. L. Luo, R. H. Liu, Q. Y. Cai, Y. Xiao, S. H. Liu, F. Su and L. F. Wen, J. Phys. Chem. C, 2010, 114, 4783–4789 CAS.
- Q. Kang, S. H. Liu, L. X. Yang, Q. Y. Cai and C. A. Grimes, ACS Appl. Mater. Interfaces, 2011, 3, 746–749 CAS.
- K. Y. Xie, J. Li, Y. Q. Lai, Z. A. Zhang, Y. X. Liu, G. G. Zhang and H. T. Huang, Nanoscale, 2011, 3, 2202–2207 RSC.
- K. Shankar, G. K. Mor, H. E. Prakasam, O. K. Varghese and C. A. Grimes, Langmuir, 2007, 23, 12445–12449 CrossRef CAS PubMed.
- F. X. Xiao, J. Phys. Chem. C, 2012, 116, 16487–16498 CAS.
- F. X. Xiao, J. Mater. Chem., 2012, 22, 7819–7830 RSC.
- Y. C. Liang, C. C. Wang, C. C. Kei, Y. C. Hsueh, W. H. Cho and T. P. Perng, J. Phys. Chem. C, 2011, 115, 9498–9502 CAS.
- J. G. Yu, G. P. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 16394–16401 CAS.
- J. F. Qi, X. N. Dang, P. T. Hammond and A. M. Belcher, ACS Nano, 2011, 5, 7108–7116 CrossRef CAS PubMed.
- A. Takai and P. V. Kamat, ACS Nano, 2011, 5, 7369–7376 CrossRef CAS PubMed.
- L. Z. Zhao, H. R. Wang, K. F. Huo, L. Y. Cui, W. R. Zhang, H. W. Ni, Y. M. Zhang, Z. F. Wu and P. K. Chu, Biomaterials, 2011, 32, 5706–5716 CrossRef CAS PubMed.
- Y. Y. Song, Z. Gao, K. Lee and P. Schmuki, Electrochem. Commun., 2011, 13, 1217–1220 CrossRef CAS PubMed.
- Y. K. Lai, J. J. Gong and C. J. Lin, Int. J. Hydrogen Energy, 2012, 37, 6438–6446 CrossRef CAS PubMed.
- J. M. Macak, B. G. Gong, M. Hueppe and P. Schmuki, Adv. Mater., 2007, 19, 3027–3031 CrossRef CAS PubMed.
- S. Oh, K. S. Moon and S. H. Lee, J. Nanomater., 2013, 2013, 9 Search PubMed.
- M. S. Aw, G. Karan and L. Dusan, J. Biomater. Nanobiotechnol., 2011, 2, 477–484 CrossRef CAS.
- C. Y. Tian, J. J. Xu and H. Y. Chen, Chem. Commun., 2012, 48, 8234–8236 RSC.
- Y. Y. Song, F. Schmidt-Stein, S. Bauer and P. Schmuki, J. Am. Chem. Soc., 2009, 131, 4230–4232 CrossRef CAS PubMed.
- Q. Y. Li, R. Li, L. L. Zong, J. H. He, X. D. Wang and H. J. Yang, Int. J. Hydrogen Energy, 2013, 38, 12977–12983 CrossRef CAS PubMed.
- X. M. Zhang, K. F. Huo, L. S. Hu, Z. W. Wu and P. K Chu, J. Am. Ceram. Soc., 2010, 93, 2771–2778 CrossRef CAS.
- Y. C. Xin, J. Jiang, K. H. Huo, T. Hu and P. K. Chu, ACS nano, 2009, 3, 3228–3234 CrossRef CAS PubMed.
- L. Z. Zhao, H. R. Wang, K. F. Huo, X. M. Zhang, Z. F. Wang, Y. M. Zhang, Z. Wu and P. K. Chu, Biomaterials, 2013, 34, 19–29 CrossRef PubMed.
- J. M. Macak, C. Zollfrank, B. J. Rodriguez, H. Tsuchiya, M. Alexe, P. Greil and P. Schmuki, Adv. Mater., 2009, 21, 3121–3125 CrossRef CAS PubMed.
- K. F. Huo, X. M. Zhang, H. R. Wang, L. Z. Zhao, X. Y. Liu and P. K. Chu, Biomaterials, 2013, 34, 3467–3478 CrossRef CAS PubMed.
- X. H. Zhu, Z. G. Liu and N. B. Ming, J. Mater. Chem., 2010, 20, 4015–4030 RSC.
- K. F. Huo, H. R. Wang, X. M. Zhang, Y. Cao and P. K. Chu, ChemPlusChem, 2012, 77, 323–329 CrossRef CAS PubMed.
- Y. L. Liao, W. X. Que, P. Zhong, J. Zhang and Y. C. He, ACS Appl. Mater. Interfaces, 2011, 3, 2800–2804 CAS.
- E. Conforto, D. Caillard, B. O. Aronsson and P. Descouts, Eur. Cells Mater., 2002, 3, 9–10 Search PubMed.
- W. Daecke, K. Veyel, P. r. Wieloch, M. Jung, H. Lorenz and A. K. Martini, J. Hand Surg., 2006, 31, 90–97 CrossRef PubMed.
- R. Kane and P. X. Ma, Mater. Today, 2013, 16, 418–423 CrossRef CAS PubMed.
- M. Tzaphlidou, Micron, 2005, 36, 593–601 CrossRef CAS PubMed.
- G. A. Crawford, N. Chawla, K. Das, S. Bose and A. Bandyopadhyay, Acta. Biomaterials, 2007, 3, 359–367 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- S. H. Oh, R. R. Finones, C. Daraio, L. H. Chen and S. H. Jin, Biomaterials, 2005, 26, 4938–4943 CrossRef CAS PubMed.
- A. Pittrof, S. Bauer and P. Schmuki, Acta Biomater., 2011, 7, 424–431 CrossRef CAS PubMed.
- Y. Bai, I. S. Park, H. H. Park, M. H. Lee, T. S. Bae, W. Duncan and M. Swain, Surf. Interface Anal., 2011, 43, 998–1005 CrossRef CAS PubMed.
- L. Z. Zhao, L. Liu, Z. F. Wu, Y. M. Zhang and P. K. Chu, Biomaterials, 2012, 33, 2629–2641 CrossRef CAS PubMed.
- L. Z. Zhao, S. L. Mei, P. K. Chu, Y. M. Zhang and Z. F. Wu, Biomaterials, 2010, 31, 5072–5082 CrossRef CAS PubMed.
- K. S. Brammer, S. Oh, C. J. Cobb, L. M. Bjursten, H. V. Heyde and S. Jin, Acta Biomater., 2009, 5, 3215–3223 CrossRef CAS PubMed.
- K. Das, S. Bose and A. Bandyopadhyay, J. Biomed. Mater. Res., Part A, 2009, 90, 225–237 CrossRef PubMed.
- S. Oh, C. Daraio, L. H. Chen, T. R. Pisanic, R. R. Finones and S. Jin, J. Biomed. Mater. Res., Part A, 2006, 78, 97–103 CrossRef PubMed.
- B. S. Smith, S. Yoriya, T. Johnson and K. C. Popat, Acta Biomater., 2011, 7, 2686–2696 CrossRef CAS PubMed.
- L. Peng, M. L. Eltgroth, T. J. LaTempa, C. A. Grimes and T. A. Desai, Biomaterials, 2009, 30, 1268–1272 CrossRef CAS PubMed.
- E. Gongadze, D. Kabaso, S. Bauer, J. Park, P. Schmuki and A. Iglič, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 47–69 CrossRef PubMed.
- J. Park, S. Bauer, A. Pittrof, M. S. Killian, P. Schmuki and K. v. d. Mark, Small, 2012, 8, 98–107 CrossRef CAS PubMed.
- M. S. Laranjeira, M. H. Fernandes and F. J. Monteiro, J. Biomed. Nanotechnol., 2013, 9, 1594–1606 CrossRef CAS PubMed.
- L. Z. Zhao, S. L. Mei, W. Wang, P. K. Chu, Y. M. Zhang and Z. F. Wu, Biomaterials, 2010, 31, 2055–2063 CrossRef CAS PubMed.
- L. Z. Zhao, S. L. Mei, W. Wang, P. K. Chu, Y. M. Zhang and Z. F. Wu, J. Biomed. Mater. Res., Part A, 2011, 96, 100–107 CrossRef PubMed.
- L. M. Bjursten, L. Rasmusson, S. Oh, G. C. Smith, K. S. Brammer and S. Jin, J. Biomed. Mater. Res., Part A, 2010, 92, 1218–1224 Search PubMed.
- W. Q. Yu, Y. L. Zhang, X. Q. Jiang and F. Q. Zhang, Oral Dis., 2010, 16, 624–630 CrossRef CAS PubMed.
- M. P. Neupane, I. S. Park, T. S. Bae, H. K. Yi, M. Uo, F. Watari and M. H. Lee, J. Mater. Chem., 2011, 21, 12078–12082 RSC.
- K. S. Brammer, C. J. Frandsen and S. Jin, Trends Biotechnol., 2012, 30, 315–332 CrossRef CAS PubMed.
- A. J. Salgado, O. P. Coutinho and R. L. Reis, Macromol. Biosci., 2004, 4, 743–765 CrossRef CAS PubMed.
- V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474–5491 CrossRef CAS PubMed.
- S. Oh, K. S. Brammer, Y. S. Li, D. Teng, A. J. Engler, S. Chien and S. Jin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2130–2135 CrossRef CAS PubMed.
- K. S. Moon, S. H. Yu, J. M. Bae and S. Oh, J. Nanomater., 2012, 2012, 252481 Search PubMed.
- J. Park, S. Bauer, P. Schmuki and K. v. d. Mark, Nano Lett., 2009, 9, 3157–3164 CrossRef CAS PubMed.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- N. Wang, H. Y. Li, W. L. Lü, J. H. Li, J. H. Wang, Z. T. Zhang and Y. R. Liu, Biomaterials, 2011, 32, 6900–6911 CrossRef CAS PubMed.
- L. Peng, A. D. Mendelsohn, T. J. LaTempa, S. Yoriya, C. A. Grimes and T. A. Desai, Nano Lett., 2009, 9, 1932–1936 CrossRef CAS PubMed.
- H. Cheng, Y. Li, K. F. Huo, B. Gao and W. Xiong, J. Biomed. Mater. Res., Part A, 2013 DOI:10.1002/jbm.a.35019.
- V. Alt, T. Bechert, P. Steinrucke, M. Wagener, P. Seidel, E. Dingeldein, E. Domann and R. Schnettler, Biomaterials, 2004, 25, 4383–4391 CrossRef CAS PubMed.
- Y. B. Xie, L. M. Zhou and H. T. Huang, Biosens. Bioelectron., 2007, 22, 2812–2818 CrossRef CAS PubMed.
- P. Xiao, B. B. Garcia, Q. Guo, D. W. Liu and G. Z. Cao, Electrochem. Commun., 2007, 9, 2441–2447 CrossRef CAS PubMed.
- S. Q. Liu and A. C. Chen, Langmuir, 2005, 21, 8409–8413 CrossRef CAS PubMed.
- S. Ameen, A. M. Shaheer, H. K. Seo and H. S. Shin, Appl. Phys. Lett., 2013, 103, 061602 CrossRef PubMed.
- M. C. Liu, G. H. Zhao, K. J. Zhao, X. L. Tong and Y. T. Tang, Electrochem. Commun., 2009, 11, 1397–1400 CrossRef CAS PubMed.
- F. H. Wu, J. J. Xu, Y. Tian, Z. C. Hu, L. W. Wang, Y. Z. Xian and L. T. Jin, Biosens. Bioelectron., 2008, 24, 198–203 CrossRef CAS PubMed.
- G. H. Zhao, Y. Z. Lei, Y. G. Zhang, H. X. Li and M. C. Liu, J. Phys. Chem. C, 2008, 112, 14786–14795 CAS.
- X. L. Cui, Z. Z. Li, Y. C. Yang, W. Zhang and Q. F. Wang, Electroanalysis, 2008, 20, 970–975 CrossRef CAS PubMed.
- L. Q. Zhang, L. Han, P. Hu, L. Wang and S. J. Dong, Chem. Commun., 2013, 49, 10480–10482 RSC.
- D. W. Li, W. L. Zhai, Y. T. Li and Y. T. Long, Microchim. Acta, 2014, 181, 381–387 CrossRef.
- Q. Kang, L. X. Yang and Q. Y. Cai, Bioelectrochemistry, 2008, 74, 62–65 CrossRef CAS PubMed.
- X. Pang, D. M. He, S. L. Luo and Q. Y. Cai, Sens. Actuators, B, 2009, 137, 134–138 CrossRef PubMed.
- Y. Wang, J. Chen, C. P. Zhou, L. Zhou, Y. Y. Kong, H. Y. Long and S. A. Zhong, Electrochim. Acta, 2014, 115, 269–276 CrossRef CAS PubMed.
- C. X. Wang, L. W. Yin, L. Y. Zhang and R. Ga, J. Phys. Chem. C, 2010, 114, 4408–4413 CAS.
- X. L. Li, J. Y. Yao, F. L. Liu, H. C. He, M. Zhou, N. Mao, P. Xiao and Y. H. Zhang, Sens. Actuators, B, 2013, 181, 501–508 CrossRef CAS PubMed.
- Z. D. Gao, J. Guo, N. K. Shrestha, R. Hahn, Y. Y. Song and P. Schmuki, Chem.–Eur. J., 2013, 19, 15530–15534 CrossRef CAS PubMed.
- S. L. Luo, F. Su, C. B. Liu, J. X. Li, R. H. Liu, Y. Xiao, Y. Li, X. N. Liu and Q. Y. Cai, Talanta, 2011, 86, 157–163 CrossRef CAS PubMed.
- D. Chen, H. Zhang, X. Li and J. H. Li, Anal. Chem., 2010, 82, 2253–2261 CrossRef CAS PubMed.
- Y. R. An, L. L. Tang, X. L. Jiang, H. Chen, M. C. Yang, L. T. Jin, S. P. Zhang, C. G. Wang and W. Zhang, Chem.–Eur. J., 2010, 16, 14439–14446 CrossRef CAS PubMed.
- H. Feng, L. P. Zhou, J. Z. Li, T. N. Y. Wang, L. J. Yuan, Z. H. Yan and Q. Y. Cai, Analyst, 2013, 138, 5726–5733 RSC.
- X. M. Zhang, K. H. Huo, X. Peng, R. Z. Xu, P. H. Li, R. S. Chen, G. Zheng, Z. W. Wu and P. K. Chu, Chem. Commun., 2013, 49, 7091–7093 RSC.
- M. Liu, G. Zhao, Y. Tang, Z. Yu, Y. Lei, M. Li, Y. Zhang and D. Li, Environ. Sci. Technol., 2010, 44, 4241–4246 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |