
Solar photo-thermochemical syntheses of 4-bromo-2,5-substituted oxazoles from N-arylethylamides†
Abstract
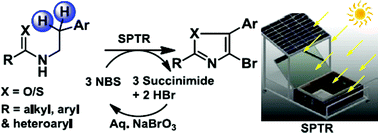
Solar photo-thermochemical C(sp3)–H bromination, conducted efficiently in a specially designed reactor, was reported recently. In the present study, the more complex formation of 4-bromo-2,5-substituted oxazoles from N-arylethylamides using this approach was achieved. The one-pot syntheses were carried out with N-bromosuccinimide–dichloroethane over 6 h (10.00 am to 4.00 pm) on sunny days. The isolated yields were in the range 42–82%. Benzylic bromination, followed by O–C bond formation through intramolecular nucleophilic substitution, and a second benzylic bromination followed by HBr elimination, gave the oxazole ring. A third bromination of the ring yielded the final product. The feasibility of synthesizing thiazole derivatives using a similar approach was also demonstrated. During the course of the reactions, succinimide and HBr were co-generated in the aqueous phase. Treatment with NaBrO3 and additional acid returned the reagent in 57% isolated yield upon chilling. The overall methodology was greener as a result.
 Please wait while we load your content...
Please wait while we load your content...

