 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Pure shift” 1H NMR, a robust method for revealing heteronuclear couplings in complex spectra†
Juan A. Aguilara,
Gareth A. Morrisb and
Alan M. Kenwright*a
aDepartment of Chemistry, University of Durham, South Road, Durham DH1 3LE, UK. E-mail: j.a.aguilar@durham.ac.uk; a.m.kenwright@durham.ac.uk
bSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: g.a.morris@manchester.ac.uk
First published on 23rd January 2014
Abstract
We investigate the utility of “pure shift” techniques in revealing heteronuclear couplings in complex 1H NMR spectra. The results show the technique to be a robust and valuable complement to the standard 1H spectrum, and an attractive alternative to heteronuclear decoupling since the technique is independent of the size of the heteronuclear couplings and the chemical shift range(s) of the heteronuclei involved. We highlight some possible artefacts, and the subtle effects due to the presence of 13C nuclei in otherwise symmetric molecules when bilinear rotational decoupling (BIRD) elements are present in the pulse sequence.
Introduction
Since the seminal discovery by Fried and Sabo in 1953 that the replacement of a single hydrogen atom in cortisol by a fluorine atom increased the pharmaceutical activity by an order of magnitude,1 the use of fluorine in medicinal chemistry has become widespread, to the point where around 25% of human and veterinary pharmaceuticals and agrochemicals currently in use contain at least one fluorine atom. At first sight this statistic may appear surprising given that there are very few naturally occurring organofluorine compounds2 and the majority of the few that are known have high toxicity, but it is a result of the ability of fluorine to modify the pKa of neighbouring groups, affect lipophilicity, hydrogen bonding and other binding interactions and, in many cases, block pathways leading to rapid metabolisation of the drug thereby increasing its bioavailability.Many of these effects can be traced to the particular steric and electronic properties of the C–F bond.3 The importance of fluorine in the modern pharmaceutical industry has been the subject of a number of reviews,4,5 and the same unique properties have seen its use spread to the wider arena of synthetic chemical biology.6 These developments have enjoyed a symbiotic relationship with the wider developments in synthetic organofluorine chemistry over the same period from a specialist niche area for a few intrepid workers 50 years ago to a mainstream discipline in modern synthetic chemistry.7,8
In view of this, the ability to characterise fluorinated organic molecules routinely has become much more important in recent years and NMR plays a key role in this, particularly, for example, in distinguishing between possible constitutional isomers in multiply substituted aromatic and hetero-aromatic rings with one or more fluorine substituents. There are a considerable number of well-established NMR experiments that are potentially useful here including two-dimensional homonuclear 1H experiments (such as COSY and NOESY) and two-dimensional heteronuclear 1H–13C and 1H–19F experiments (such as HSQC, HMBC and HOESY). The experiments involving manipulation of 1H and 19F simultaneously impose particular NMR hardware requirements (the ability to generate two “high-band” frequencies simultaneously and preferably a probe that can be tuned to 1H and 19F simultaneously). While spectrometer systems that meet these requirements have become much more common in recent years, they are by no means ubiquitous. Furthermore, routine two-dimensional experiments involving fluorine are often rendered problematic, both by the wide range of fluorine chemical shifts9 and by the variability of fluorine coupling constants.10
Regardless of whether the experimenter has the hardware and the knowledge to run the aforementioned experiments, the reality is that the experiment that really plays a pivotal role in the day-to-day decision making process is the simple one-dimensional 1H spectrum. For better or for worse, this is the experiment that non-specialists use right away to check reaction outcomes and to decide whether to continue with a synthetic procedure or to ask for technical assistance in running more sophisticated NMR experiments. In theory, the presence in 1H spectra of splittings due to 1H–19F coupling should improve the decision-making process, but in reality these are often hidden under a mass of homonuclear (1H–1H) splittings. Very early on it was realised11 that a possible solution to this problem was to eliminate the latter from 1H spectra, as this would reveal the 1H–19F coupling, while obviating both the need for a fluorine channel and the need for a skilled operator. The alternative approach, decoupling fluorine while observing proton, is technically challenging, both because of the dispersion of the fluorine signals and the variability of the fluorine couplings. Although often ignored, these should be taken into account when setting the decoupler.13 Unfortunately, suppressing homonuclear couplings in a broadband and efficient manner has proven to be one of the most intractable problems in NMR history. Only recently have practical techniques able to produce good quality results in reasonable amounts of time been developed. These techniques have been produced primarily to address the problem of limited spectral dispersion (peak overlap) in 1H-NMR but, as we shall see, some can be used equally well to solve the problem at hand. These techniques are the ideal addition to the simple 1H spectrum and even provide a useful complement to the 19F one, if that is available, as they can be run in a few minutes by non-specialists, even under automation, without the need for time consuming calibrations and without the need for fluorine pulsing capabilities. Furthermore, they are not limited to the case of fluorine; they can also be used to reveal couplings to elements such as phosphorous, platinum, rhodium, tin, thallium, mercury and silver.
Several schemes have been proposed to collapse homonuclear multiplets into singlets. These have been variously described as “broadband homonuclear decoupling” and “pure shift” techniques, as well as by other names. The question is which technique or techniques to use for the task at hand. The ideal technique should produce phase sensitive results in reasonable amounts of time and deal well with strong coupling. The old 2D J-resolved experiment used to reveal heteronuclear splittings fails in both areas, although attempts have been made to address both problems.14,15 One practical experiment that deals well with strong coupling and produces good quality data in a matter of minutes is a hybrid experiment16 that uses the BIRD rotation proposed by Pines et al.17 and the chemical shift sampling scheme of Zangger and Sterk18 extended by Morris and Nilsson.19 The pulse sequence used is shown in Fig. 1 and is discussed more extensively in ref. 16. The combination of BIRD and hard proton 180° rotations refocuses the evolution under the homonuclear coupling but allows the chemical shift to be sampled. A chunk of data lasting 1/sw1 is acquired, where sw1 is an integer submultiple of sw, and t1 is incremented in larger steps 1/sw1, typically of several tens of ms. In principle a classic Zangger–Sterk pulse sequence can be also used, but the presence of second-order, strongly coupled signals, very common in aromatic systems, made us favour the former. In reality they complement one another.
 | ||
| Fig. 1 Pulse sequence for the measurement of pure shift 1H spectra by the modified BIRD method, narrow RF pulses are 90° and wide 180°; 180° Broadband Inversion Pulses (BIP)12 are indicated by a diagonal line; the dotted line indicates a pulse omitted on alternate transients. The pulse sequence can be found in the ESI.† | ||
In order to analyse the adequacy of the technique, several aspects need consideration: firstly, the ability of the technique to eliminate homonuclear splittings leaving only the heteronuclear ones; secondly, its robustness; thirdly, its potential limitations.
Experimental
All spectra were recorded in CDCl3 at 298 K using a Varian VNMRS-600 spectrometer. The pulse sequence used to obtain the pure shift spectra and the macro needed to assemble the data can be found in the ESI.† The carbon pulses used were 125 μs long Broadband Inversion Pulses (BIP).12 The experiment was optimised for a typical 1JCH = 160 Hz. Deviations from this value lead to signal attenuation. Between 32 and 64 increments were used, depending on the desired resolution, while the number of transients varied from 2 to 8. sw1 was set to 50 Hz and experimental times varied from 3 to 15 minutes. Typical concentration ranged from 10 to 100 mM. Spin-system simulations were performed using the spectral simulation software package in the Varian VNMR 6.1c software,20 which is an implementation of the LAOCOON program.21Results and discussion
The ability of the technique to eliminate homonuclear splittings leaving only heteronuclear ones is exemplified in Fig. 2. The 1-fluorobenzene 1H spectrum (Fig. 2C) is moderately complex, yet there is no difficulty in identifying the heteronuclear splittings in the pure shift spectrum (Fig. 2B), so resolution and clarity have been gained. The observed couplings of 9.1 and 5.7 Hz are consistent with the relevant protons being ortho and meta to the fluorine. The third signal does not show any resolved coupling, as should be the case for a signal located in a para position with respect to the fluorine. In the second example, Fig. 3, a similar situation arises, but this time the presence of the proton–fluorine coupling helps identification of the fluorinated ring and the position of the fluorine atom (para) with respect to the second ring. The proton–fluorine coupling constants measured independently in the 1H pure shift and 19F spectra in Fig. 2 agree to within experimental error (±0.1 Hz) but it has to be borne in mind that the BIRD pure shift experiment deals with protons attached to 13C while the fluorine one mainly deals with protons attached to 12C, so minor differences may be due to the fact that the two spectra are predominantly of different isotopologues (see discussion below). | ||
| Fig. 2 19F (A), 1H pure shift BIRD (B) and 1H (C) spectra of 1-fluorobenzene in CDCl3. As can be seen in (B), all homonuclear couplings have been suppressed, revealing splittings due to fluorine. Strong coupling sidebands are labelled with *. See text for details. | ||
 | ||
| Fig. 3 1H pure shift BIRD spectrum (A) of 4-fluoro-4′-nitro-1,1-biphenyl in CDCl3 and its corresponding 1H spectrum (B). In this case the experiment distinguishes between the rings and indicates that the fluorine is located para with respect to the non-fluorinated ring. Strong coupling sidebands are marked with *. See text for details. | ||
The two examples presented so far do not pose particularly challenging problems, and indeed their structures were both known in advance and were chosen for illustrative purposes. The next two samples, however, were real unknown cases submitted by industrial users for structure confirmation. The first unknown sample, Fig. 4, exemplifies how the technique complements the 19F spectrum. In this case, the proton–fluorine couplings cannot easily be determined from the fluorine spectrum but they can be easily found from the pure shift spectrum. In other cases where fluorine multiplets are well resolved, these couplings could, in principle, be determined directly from the fluorine spectrum but because couplings to multiple protons or to other fluorine nuclei may be present, the analysis is often far from trivial. In such cases, the pure shift spectrum would greatly facilitate analysis.
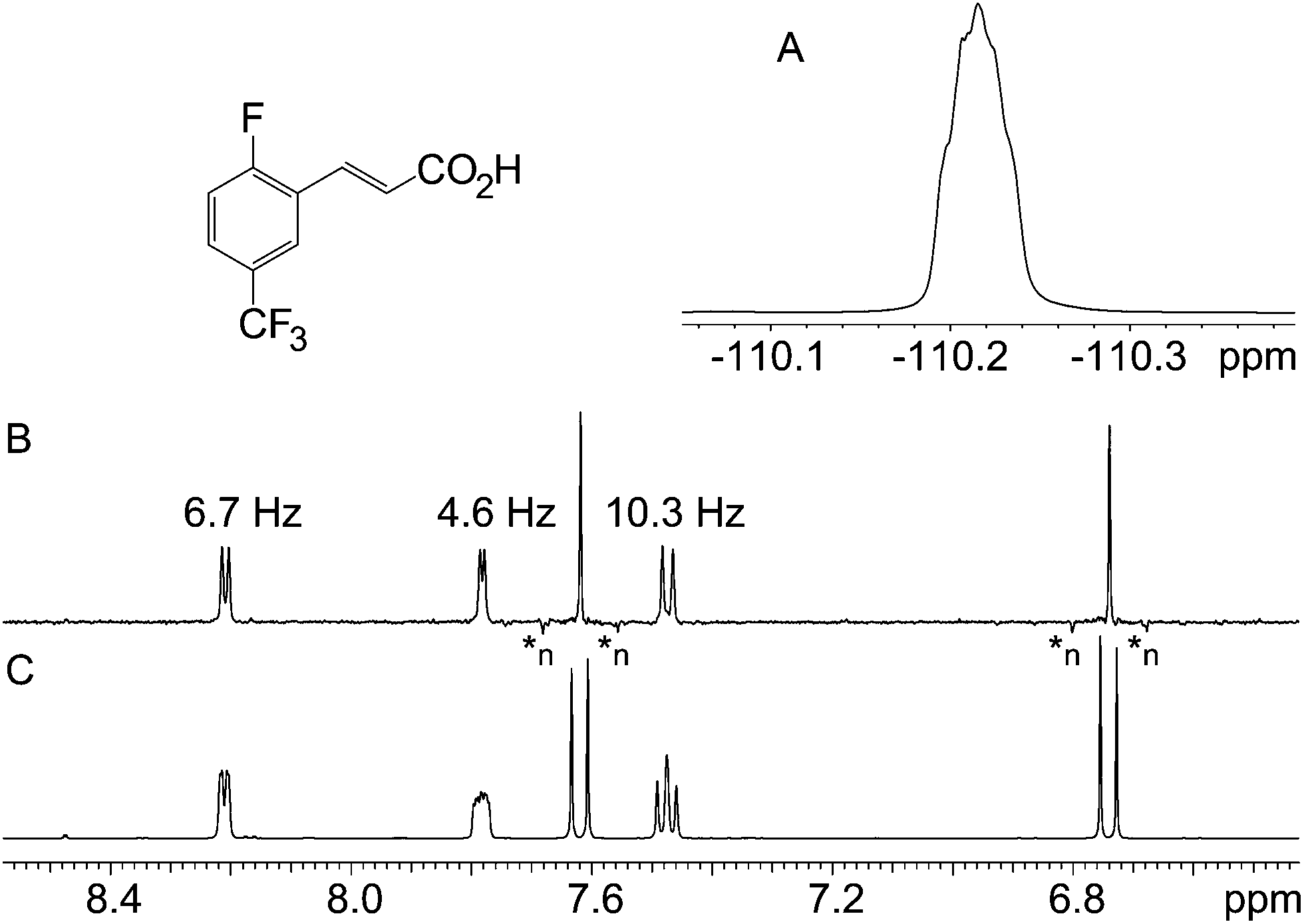 | ||
| Fig. 4 19F (A), 1H pure shift BIRD (B) and regular 1H (C) spectra of 2-fluoro-5-(trifluoromethyl)cinnamic acid. Decoupling artefacts characterised by negative intensity are labelled as *n. | ||
The second of the “unknown” samples, Fig. 5, is a fluoronitrobenzene, but its substitution pattern was unknown. The fact that there are four distinct proton signals shows that it must be either 2-fluoronitrobenzene or 3-fluoronitrobenzene. In this case, the significant feature is the fact that the apparent doublet signal at about 7.9 ppm in the normal 1H spectrum is essentially unchanged in the pure shift spectrum, showing that the proton involved has little 1H–1H coupling. This immediately identifies the sample as 3-fluoronitrobenzene, since the isolated proton must be in the 2 position.
 | ||
| Fig. 5 1H pure shift (A) and regular 1H (B) spectra of 3-fluoronitrobenzene. | ||
To assess the robustness of the experiment, it was run under automation over a few months using samples submitted to Durham University's NMR service. The vast majority of the samples analysed contained aromatic units displaying second order spectra. In all cases the same parameters were used, illustrating the fact that the experiment can be run by non-experts. The results show that the pulse sequence generally performs well, even in the presence of strong coupling. The most common artefacts are sidebands, marked with an asterisk in the examples presented. Such artefacts have not been reported previously, but seem to be as common as second order spectra. Although undesirable, their importance is small once experimenters become familiar with them. This type of artefact only becomes problematic when the experiment is used to reveal heteronuclear splittings of spins with low natural abundance, such as 29Si, or when several species of very dissimilar concentrations are present. The origin of the artefacts is still under investigation but is almost certainly a consequence of strong coupling. Because the wavefunctions of strongly coupled spins mix, the BIRD element partly inverts the coupled partner bound to 12C, not fully refocusing its proton–proton coupling interaction with the 13C-bound partner. As a result, there is additional evolution during t1 that translates into these sidebands. The effect seems to be related to similar problems reported for HETCOR and related experiments.22
A second class of artefact, previously reported in ref. 18, is a negative sideband caused by the decoupling method. Such artefacts are typically a few percent of the main signal and are often too weak to see. They can be recognised by their sign and because they are separated by multiples of sw1 Hz. An example can be seen in Fig. 4B.
The relative resilience of BIRD-based pulse sequences where spin systems showing strong coupling are concerned arises from the lifting of 1H near-degeneracy caused by the one-bond 1H–13C coupling present in the minority of molecules observed when using BIRD. Spin systems that are strongly coupled in per-12C species will often be weakly coupled in a mono-13C isotopologue, and even where a mono-13C system is still strongly coupled this will usually apply only to one of the two 13C spin states (i.e. in general at most one of the two 13C satellites in the 1H spectrum will be strongly coupled). Conversely, in some otherwise symmetric systems, the presence of a 13C spin will break the symmetry, introducing strong coupling that would not otherwise intrude. This is exemplified by the case of 1,4-difluorobenzene. The reason for this effect (previously unreported in the literature relating to homonuclear broadband decoupling but commonplace elsewhere) lies in the fact that the spin system of interest is not that of Fig. 6B, but rather that of Fig. 6A, where the presence of 13C breaks the symmetry, making all of the nuclei inequivalent and introducing strong coupling. This results in unwanted features, as shown in Fig. 6A. To emphasise the point, the sample was subjected to another pure shift technique (Zangger–Sterk) that does not require the selection of protons coupled to 13C. The results are basically identical to the parent 1H spectrum, as expected.
 | ||
| Fig. 6 1H pure shift BIRD (A), pure shift ZS (B) and regular 1H (C) spectra of 1,4-difluorobenzene. The presence of the 13C breaks the symmetry of the spin system, making all nuclei inequivalent, introducing strong coupling and leading to extra features such as those marked *. Pulse sequences such as pure shift BIRD that select protons coupled to 13C are susceptible (A) to such artefacts; pulse sequences that do not select protons coupled to 13C, such as ZS (B), can be used to avoid the problem. | ||
The effects of 13C can be illustrated by simulating spectra with all homonuclear couplings set to zero. Simulation of the spectrum expected on the basis of a single chemical shift for the four hydrogen atoms, the 1H–1H coupling constants and the 1H–19F coupling constants set to the values determined by Wray et al.23 (Fig. SI-1†) shows reasonably good agreement with the normal 1H spectrum, confirming that some other factor is affecting the pure shift spectrum. We need only concern ourselves here with 13C nuclei that are directly bonded to 1H, and the symmetry of the molecule means that we need only consider a single 13C site. We do not need to consider heteronuclear 13C–1H coupling, since its effects are largely eliminated by the heteronuclear decoupling during acquisition, but we do need to consider the isotope shifts introduced by the presence of a single 13C nucleus in the molecule. These effectively render the 4 1H environments chemically inequivalent, and the same is true for the two 19F environments. The isotope shifts involved are small (typically 1 to 10 ppb), but have been determined previously both for fluorine24 and for proton,25 and were included in the simulation. The fact that the isotope shifts are small means that both the 1H–1H and 19F–19F couplings now give rise to strong second order effects in the spectra observed. A simulation of the conventional 1H spectrum of the spin system including the isotope shifts due to the presence of a single 13C in the molecule and all the 1H–1H, 19F–19F, and 1H–19F couplings is shown in the ESI.†
It should be noted that signals such as OH and NH are eliminated, as the pulse sequence selects only those protons coupled to 13C. In addition, non-equivalent geminal protons are not decoupled from one another, as they are both bonded to the same carbon. They typically appear as pairs of doublets since couplings to third partners are eliminated. In cases where strong coupling is not a pressing problem or when methylene signals are important, Zangger–Sterk or Pell–Keeler15 sequences can be used instead. In addition, some recently described ultra-fast pure shift BIRD pulse sequences should be considered.26,27
Conclusions
The pure shift BIRD experiment is a robust, simple to run pulse sequence that both simplifies 1H spectra and reveals heteronuclear couplings. Minor artefacts are produced that may be obtrusive in cases where coupling to isotopes of low natural abundance is of interest, or where mixtures with very different concentrations are concerned, and complications can arise where the presence of 13C breaks symmetry.Acknowledgements
The authors are grateful to JRD Fluorochemicals and Mr Antal Harsanyi for their generous donation of samples. This work was supported by the Engineering and Physical Sciences Research Council (grant number EP/I007989/1).Notes and references
- J. Fried and E. Sabo, J. Am. Chem. Soc., 1953, 75, 2273–2274 CrossRef CAS.
- D. O'Hagan and D. B. Harper, J. Fluorine Chem., 1999, 100, 127–133 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS PubMed.
- S. L. Cobb and C. D. Murphy, J. Fluorine Chem., 2009, 130, 132–143 CrossRef CAS PubMed.
- G. Sandford, Philos. Trans. R. Soc., A, 2000, 358, 455–471 CrossRef CAS.
- W. R. Dolbier Jr, J. Fluorine Chem., 2005, 126, 157–163 CrossRef PubMed.
- W. R. Dolbier Jr, Guide to Fluorine NMR for Organic Chemists, John Wiley & Sons, Inc., Hoboken, 2009 Search PubMed.
- J. W. Emsley, Progress in NMR Spectroscopy, 1976, 10, 83–756 CrossRef.
- L. D. Hall and S. Sukumar, J. Am. Chem. Soc., 1979, 101, 3120–3121 CrossRef CAS.
- M. A. Smith, H. Hu and A. J. Shaka, J. Magn. Reson., 2001, 151, 269–283 CrossRef CAS.
- E. Kupce and R. Freeman, J. Magn. Reson., 1995, 177, 246–256 Search PubMed.
- M. J. Thrippleton, R. A. E. Edden and J. Keeler, J. Magn. Reson., 2005, 174, 97–109 CrossRef CAS PubMed.
- A. J. Pell and J. Keeler, J. Magn. Reson., 2007, 189, 293–299 CrossRef CAS PubMed.
- J. A. Aguilar, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed. Engl., 2011, 50, 9716–9717 CrossRef CAS PubMed.
- J. R. Garbow, D. P. Weitekamp and A. Pines, Chem. Phys. Lett., 1982, 93, 504–509 CrossRef CAS.
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486–489 CrossRef CAS.
- M. Nilsson and G. A. Morris, Chem. Commun., 2007, 933–935 RSC.
- Varian Associates, Palo Alto, California, USA.
- S. Castellano and A. A. Bothner-By, J. Chem. Phys., 1964, 41, 3863–3869 CrossRef CAS PubMed.
- G. A. Morris and K. I. Smith, Mol. Phys., 1987, 61, 467–483 CrossRef CAS.
- V. Wray, L. Ernst and E. Lustig, J. Magn. Reson., 1977, 21, 1–21 Search PubMed.
- F. J. Weigert and J. D. Roberts, J. Am. Chem. Soc., 1971, 93, 2361–2369 CrossRef.
- V. A. Chertkov and N. M. Sergeyev, J. Magn. Reson., 1976, 21, 159–163 CAS.
- A. Lupulescu, G. L. Olsen and L. Frydman, J. Magn. Reson., 2012, 218, 141–146 CrossRef CAS PubMed.
- L. Paudel, R. W. Adams, P. Király, J. A. Aguilar, M. Foroozandeh, M. J. Cliff, M. Nilsson, P. Sándor, J. P. Waltho and G. A. Morris, Angew. Chem., Int. Ed. Engl., 2013, 52, 11616–11619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The pure shift BIRD pulse sequence, the phase cycle table and the macro necessary to assemble the pure shift 1D data. Simulations presented in the paper are also included. See DOI: 10.1039/c3ra46745g |
| This journal is © The Royal Society of Chemistry 2014 |