 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modified nanofibrillated cellulose–polyvinyl alcohol films with improved mechanical performance
Sanna Virtanen*a,
Jari Vartianenb,
Harri Setäläb,
Tekla Tammelinb and
Sauli Vuotic
aVTT Technical Research Centre of Finland, Sinitaival 6, P.O. Box 1300, FI-33101 Tampere, Finland. E-mail: sanna.virtanen@vtt.fi; Fax: +358 20 722 3498; Tel: +358 40 180 3695
bVTT Technical Research Centre of Finland, Biologinkuja 7, P.O. Box 1000, FI-Espoo, Finland
cVTT Technical Research Centre of Finland, Valta-akseli, P.O. Box 21, FI-05201 Rajamäki, Finland
First published on 19th February 2014
Abstract
In this study chemically surface-modified nanofibrillated cellulose (NFC) was used at low levels (0.5 to 3 wt%) as reinforcement in a polyvinyl alcohol (PVA) matrix. The modified NFC–PVA films prepared by a solution casting technique showed improved mechanical performance and good optical properties. NFC was allylated and further epoxidised with hydrogen peroxide. The addition of 1 wt% epoxy-NFC enhanced the modulus and strength of the pure PVA film, 474% and 224%, respectively. This composite film exhibited visible light transmittance of 83%. The results also showed that 1 wt% epoxy-NFC loading was beneficial to improve the crystallinity of PVA. SEM characterization confirmed better dispersion of modified NFC within the PVA matrix compared to unmodified NFC. The result showed the favourable effect of chemically modified NFC on the mechanical properties of PVA compared to unmodified NFC as reinforcement.
Introduction
Cellulose is one of the most abundant biomaterials on earth, and it has been used successfully for the preparation of nanocomposites.1–10 New sources for nanocomposites are microfibrillated cellulose (MFC) and nanofibrillated cellulose (NFC). These nanoscale elements display high stiffness and strength, and cellulose hydroxyl groups offer reactive sites for chemical modification.11–15 There are currently several applications that serve as promising candidates for industrial scale use. Applications for filtering, modifying rheology, absorbing water and enforcing the strength of paper have been described in the literature.16–18 NFC can form complex networks and has good mechanical and barrier properties. MFC films have shown a potential for food packaging due to their high toughness and strength with the required oxygen and water vapour barrier properties. Such results have indeed been reported in the literature.19–21 High crystallinity of NFC could potentially decrease the permeability of the films. However, Fukuzumi et al.15 found good barrier properties for chemically modified MFC films, and similar studies were also reported by Syverud et al.16Biodegradable polymers (BDP) such as polyvinyl alcohol (PVA or PVOH) are alternatives, which could replace polyethylene or polypropylene to produce environmentally friendly and biodegradable composite materials. It has been reported that PVA degrades in a microbial active environment within 5–6 weeks.21 Due to biodegradability, water-solubility, excellent chemical resistance, gas barrier properties, biocompatibility, good thermo-stability, optical and physical properties,18,20,22,23 PVA and PVA-based composites are used in a wide range of applications including filtration materials,24 paper coatings,21 mats,25 films,26,27 and packaging materials.28 However, the low mechanical strength and integrity of PVA demand the use of reinforcing agents, for example, carbon nanotubes, cellulose nanofibres and chitin whiskers.29–32 Preparation of bio-composites based on cellulose fibres incorporated in PVA matrices has also been reported.18,20,23,26,27,33–36 However, to our knowledge the modification effect of NFC on PVA's mechanical, thermal and optical properties has not yet been widely studied. Using natural fibres to reinforce composite structures is a highly potential solution for reducing the dependency on petroleum-based and non-biodegradable materials. There has been an increasing interest in creating composite structures from bio-based materials with superior mechanical properties. Cellulose nanofibres have already been reported to improve the mechanical properties of polypropylene37 and polylactic acid.38–40
In this work we introduce an effective crosslinking method between NFC and PVA, which gives a significant improvement in mechanical properties of the PVA film. The chemically modified NFC has been used at low levels in combination with PVA in order to produce composite films with good optical transparency. Epoxy-NFC was prepared from allyl-NFC by using a similar method described in the literature.35 The morphology and thermal properties of the NFC- and epoxy-NFC–PVA composite films were characterized. The novel epoxy-NFC–PVA composites with enhanced mechanical performance could be utilized in a wide range of applications e.g. packaging, wash-away bags and coatings that require high strength properties. Other application areas for the epoxy-NFC–PVA films could be among functional engineering applications.
Experimental
Materials
Allyl glycidyl ether (99%, Sigma-Aldrich) was dried prior to use. Hydrogen peroxide (H2O2, 35 wt% in water), sodium carbonate (Na2CO3, 99.98%), tert-butanol (purum, ≥99.0%) and acetone (≥99.5%) were purchased from Sigma-Aldrich and used as received. Sodium bicarbonate (NaHCO3, Merck) and acetonitrile CH3CN (>99.9%, HPLC grade, Rathburn, Chemical limited, Scotland) were used as received. Pure water (18.2 M cm, Millipore) was used throughout the experiments. Polyvinyl alcohol (Mowiol 40-88, Mw 205![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 88% hydrolysed, Sigma-Aldrich) was used as a matrix polymer for the solution casting. 1,6-Diaminohexane (purity >99%, Fluka) was used as a catalyst to reactivate epoxy groups of NFC.
000 g mol−1, 88% hydrolysed, Sigma-Aldrich) was used as a matrix polymer for the solution casting. 1,6-Diaminohexane (purity >99%, Fluka) was used as a catalyst to reactivate epoxy groups of NFC.
Bleached birch kraft pulp was obtained from a Finnish pulp mill (UPM-Kymmene Oyj, Finland) and used as a starting material for production of the NFC. Nanofibrillation of the birch pulp was performed with a high pressure fluidizer (Microfluidics M-110EH, Microfluidics Int. Co., MA, USA) at VTT. Prior to fibrillation, the never-dried kraft pulp was pre-refined with a laboratory-scale refiner (Voith Labrefiner LR-1, Voith) reaching a Schopper-Riegler (SR) level over 90 SR. Then the pulp was diluted with water and passed through the fluidizer six times with an operating pressure of 1850 bars. The obtained aqueous gel-like NFC (dry matter content approximately 2%) was solvent-exchanged to acetone. After solvent-exchange, the dry-matter content was 3–4%. The carbohydrate composition of the birch pulp and NFC was very similar containing, 73% glucose, 26% xylose and 1% mannose.41 In addition 0.2% of residual lignin and 0.09% residual extractives were found from the pulp.42
Preparation of modified NFC–PVA composite films
Allylation and epoxidation of the solvent-exchanged NFC (dry matter content 3–4%) was performed by a similar procedure as reported previously.35 Aqueous solutions of PVA (7 wt%) were prepared by dissolving PVA in water at 90 °C for 2 h, and subsequently cooling the mixture to room temperature. Unmodified NFC and epoxy-NFC suspensions were dispersed with different loadings in these aqueous polyvinyl alcohol solutions (see Table 1). The mixtures were vigorously stirred overnight in order for the polymer to penetrate into the cellulose network. The epoxy groups of NFC were then reactivated with 1,6-diaminohexane (2 wt% of epoxy-NFC amount) and heating the suspension at 80 °C for 2 h under vigorous stirring, and then letting the mixture to cool to room temperature. Finally, the resulting suspensions were poured into Petri dishes with controlled levelling, and dried at ambient temperature for 7 days until the weight was constant. The final thicknesses of the films were approximately 100 μm ± 10 μm. Pure NFC and epoxy-NFC films were prepared from 2 wt% aqueous dispersions of NFC and dried at 55 °C until the weight was constant. The thickness of these films was approximately 20 μm.| Sample code | PVA (wt%) | Fibre | NFC content of PVA amount (wt%) |
|---|---|---|---|
| a * – a heat activated reference sample prepared without using a catalyst. | |||
| PVA | 100 | — | — |
| 1% NFC–PVA | 100 | NFC | 1 |
| 0.5% epoxy-NFC–PVA | 100 | Epoxy-NFC | 0.5 |
| 1% epoxy-NFC–PVA* | 100 | Epoxy-NFC | 1 |
| 1% epoxy-NFC–PVA | 100 | Epoxy-NFC | 1 |
| 1.5% epoxy-NFC–PVA | 100 | Epoxy-NFC | 1.5 |
| 3% epoxy-NFC–PVA | 100 | Epoxy-NFC | 3 |
Characterization of the NFC–PVA composite films
Scanning electron microscopy (SEM, JEOL JSM-6360 LV) was used to study the morphology of NFC, but also to observe the liquid nitrogen fractured surface of the NFC–PVA film and modified NFC–PVA composite films. For the SEM-imaging, NFC water dispersion was pipetted on the carbon tape and freeze-dried. All the samples were coated with a thin layer of gold to avoid charging during the SEM imaging process. The SEM images were taken at different magnifications.The thermal behaviour of pure PVA, NFC–PVA and modified NFC–PVA films was examined with a differential scanning calorimeter (NETZSH 204 F1 Phoenix) in the temperature range from 0 °C to 205 °C under nitrogen flow. The samples that weighed between 8 and 9 mg were tested at the same heating and cooling rates of 10 °C min−1. All the samples were held at 205 °C for 5 min to erase thermal history.
The tensile properties of the composite films were determined by using an Instron® 4505 Universal Testing machine at a cross-head speed of 5 mm min−1 at 25 °C. Prior to tensile testing the composite samples were cut into rectangle 1.5 cm × 7 cm film stripes. The film thickness average values were calculated from the measured thicknesses taken from five random positions on the film using a micrometre. The test specimens were equilibrated in an ambient environment for 7 days at a relative humidity of 50%. Five measurements were performed for each composite.
Light transmittance of the NFC and epoxy-NFC–PVA films were observed on UV-VIS spectrometer (Shimadzu, UV-2501 PC Series) in visible light wavelength range 190–800 nm at 25 °C. The transmittance spectra were acquired using air as background.
The water vapour permeability (WVP) of the composite films was determined gravimetrically using a modified ASTME-96 procedure. The sample test area was 25 cm2. Prior to testing the composite films were mounted on a circular aluminium dish (H.A. Buchel V/H, A.v.d. Korput, Baarn-Holland 45M-141), which contained water. Dishes were stored in test conditions of 23 °C and 50% relative humidity and weighed periodically until a constant rate of weight reduction was attained. Two measurements were made for each film and the mean value is reported.
The oxygen permeability (OP) measurements were performed with an Oxygen Permeation Analyser Model 8001 (Systech Instruments Ltd. UK). The tests were carried out at 23 °C and 50%, 70% and 90% relative humidity using circular samples with a test area of 5 cm2 and the partial pressure of oxygen was 1 atm. Two measurements were made for each film and the mean value is reported.
Results and discussion
Chemical modification of NFC
The NFC was modified by allylation and subsequent epoxidation as reported previously.35 The degree of substitution for the epoxy-NFC was 0.012 as determined by using a spectrophotometric assay adapted from the literature and also for comparison with 13CP/MAS NMR spectroscopy.35 PVA was used as a matrix material for the composite preparation and the reactivity of PVA's hydroxyl groups with the epoxy groups of the cellulose derivatives was exploited. The reaction scheme follows a standard epoxide ring-opening, where OH-group acts as a nucleophile. Nucleophilic substitution tends to happen to the least sterically hindered carbon as shown in Fig. 1.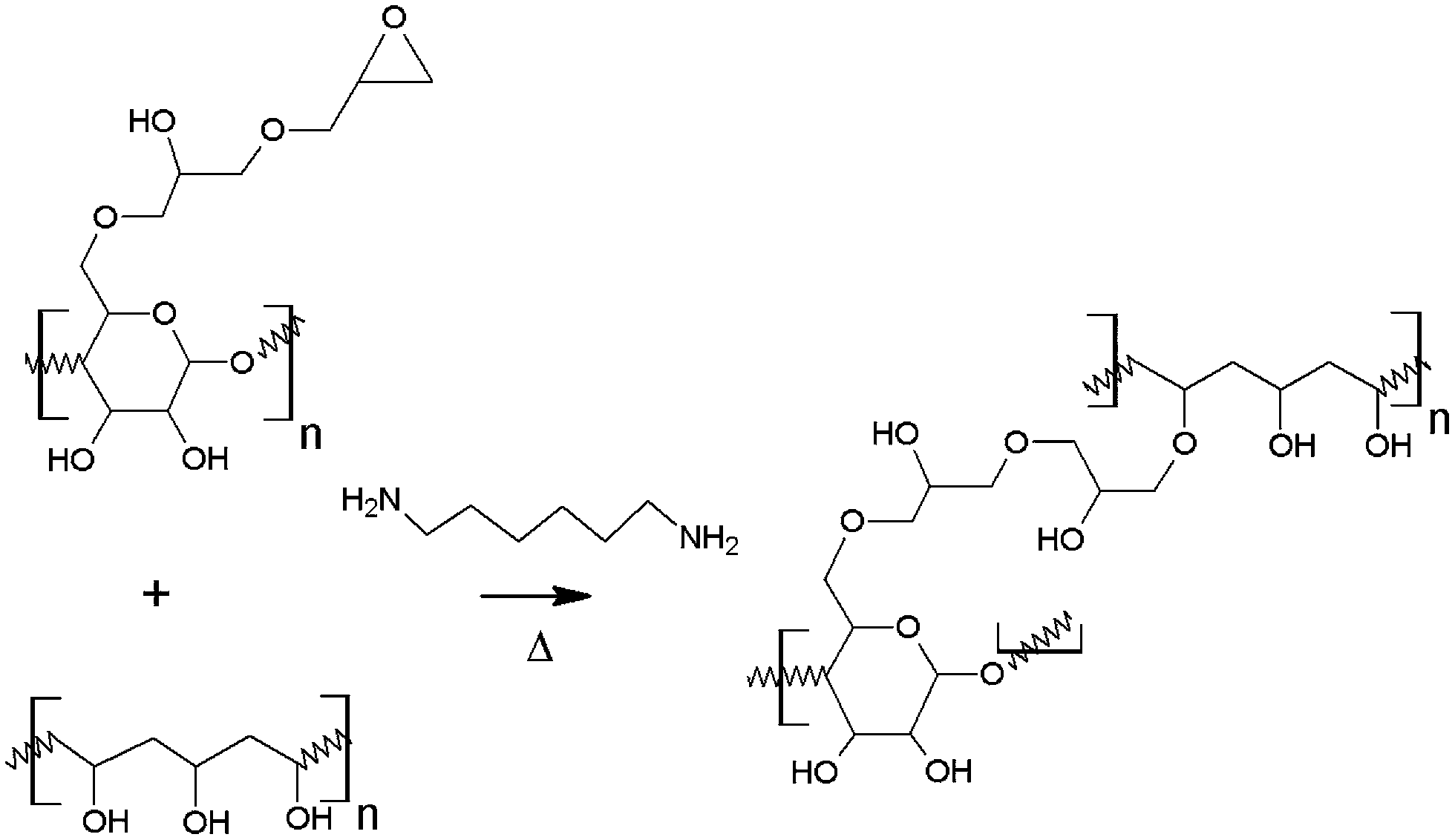 | ||
| Fig. 1 Schematic cross-linking mechanism of PVA with epoxy-NFC. | ||
Morphological characterization
Fig. 2 illustrates the SEM images of NFC and liquid nitrogen fractured surface of the NFC–PVA film and epoxy-NFC–PVA films. It can be seen that the small thread-like cellulose fibres having an average diameter of 67–133 nm, form web-like structures and bundles (Fig. 2a). The freeze-fractured composite film prepared with 1 wt% chemically unmodified NFC (Fig. 2b) showed agglomeration of the fibres, indicating the uneven dispersion in the matrix. The tensile strength of the composite made with 1 wt% unmodified NFC was 13% lower than that of pure PVA. The result may derive from the formation of agglomerates and non-uniform dispersion of NFC in the matrix as shown in Fig. 2b. In contrary, 1 wt% of surface modified NFC appeared as white dots and were firmly embedded in PVA (Fig. 2c). The enhanced modulus and tensile strength of the 1 wt% epoxy-NFC–PVA film could be explained by the lack of agglomerates and more homogenous dispersion of the chemically modified NFC in the matrix compared to the unmodified NFC. As shown in Fig. 2d, the modified NFC dispersed poorly in the PVA at higher loadings (1.5 wt%), which led to a decline in the mechanical properties of the composite.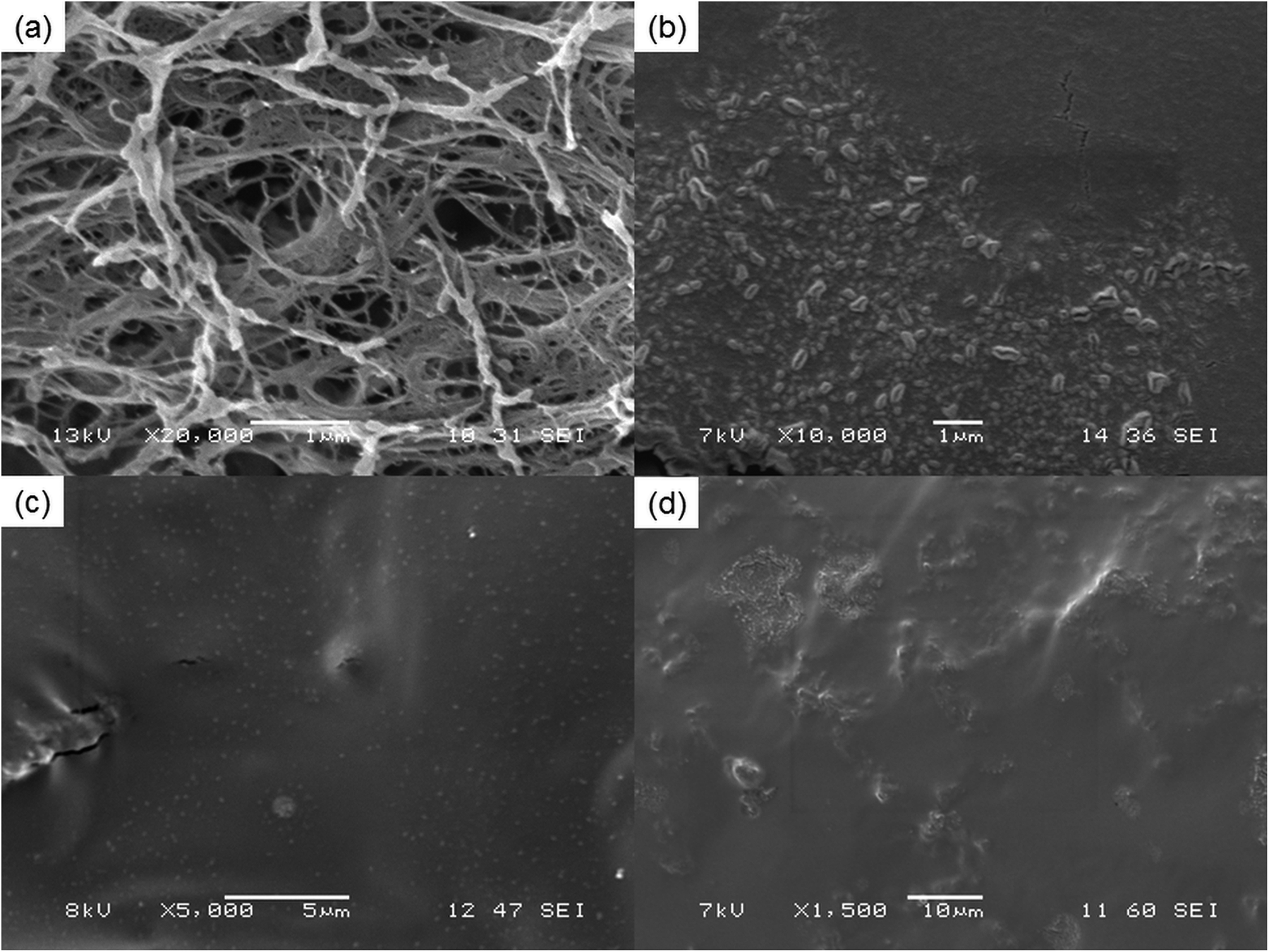 | ||
| Fig. 2 SEM images of pure NFC (a), and the freeze-fractured surface of films: 1 wt% NFC–PVA (b), 1 wt% epoxy-NFC–PVA (c) and 1.5 wt% epoxy-NFC–PVA (d), respectively at different magnification. | ||
Thermal properties
The differential scanning calorimetry (DSC) was used to investigate the effect of unmodified and modified NFC on the thermal and crystallization behaviour of PVA. PVA is a semicrystalline polymer that has strong physical inter-chain and intra-chain interactions such as hydrogen bonds in its structure. The functional groups of NFC could form bonds with PVA chains, and thus change the interactions between PVA chains. This may have an effect on the glass transition temperature and crystallization behaviour of PVA.31,43,44Glass transition temperature (Tg), melting temperature (Tm) and heat of fusion (ΔHm) of the samples were determined from the second heating scans. The first heating scans were excluded from the results because they contained latent heat of water absorbed in the samples. The crystallization temperatures (Tc) were determined from the first cooling scans. The first and second cooling scans were compared because PVA degrades near its melting temperature.45
The second and third heating curves of pure PVA and its composite films are plotted in Fig. 3. Fig. 4 represents the first and second cooling curve of all samples. An endothermic peak in the range of 177 °C to 185 °C was found to be the melting temperature for PVA (Fig. 3). Glass transition temperature of PVA and its composite films appeared as a shoulder around 65–66 °C in the DSC thermograms (Fig. 3). An exothermic peak, which appeared between 127 °C and 146 °C was detected for all samples and chosen as the crystallization temperature (Tc) of PVA (Fig. 4). The DSC thermograms showed that the additional thermal treatment cycles resulted in a decrement in the melting and crystallization temperatures. This phenomenon may result from sample degradation.46,47
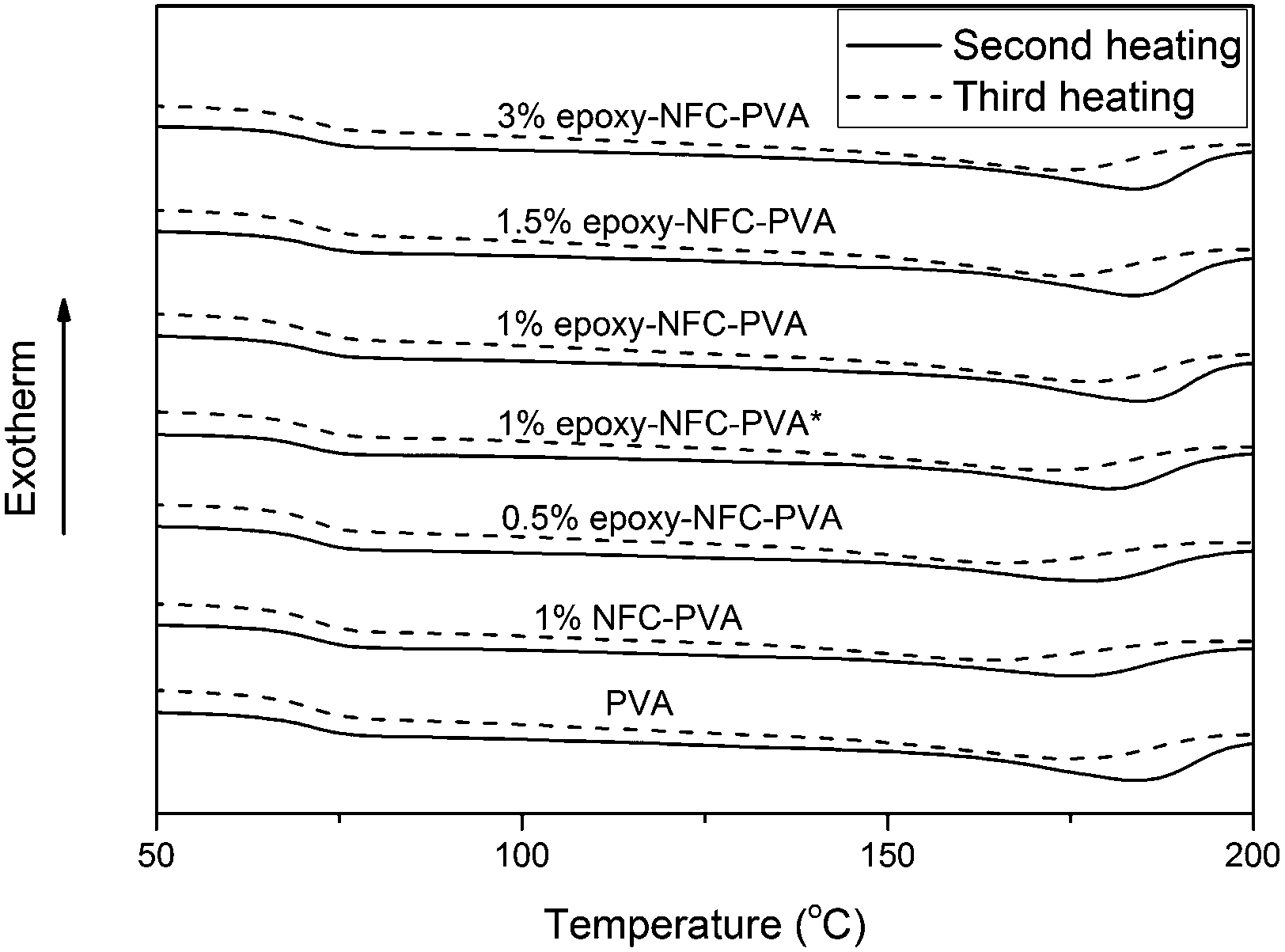 | ||
| Fig. 3 The DSC heating scans of PVA and its composite films. The solid line represents the second heating scan and the dashed line represents the third heating scan of the samples. | ||
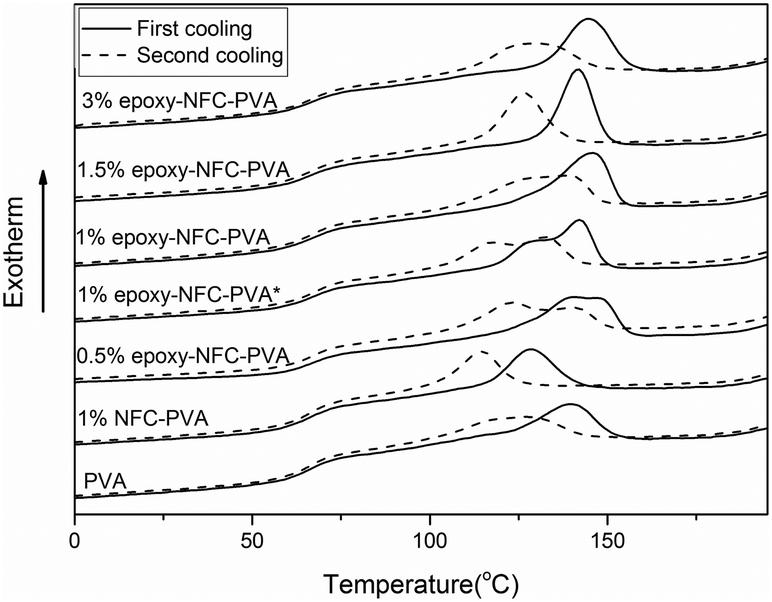 | ||
| Fig. 4 DSC cooling scans of PVA and its composite films. The solid line represents the first cooling scan and the dashed line represents the second cooling scan of the samples. | ||
Table 2 summarizes glass transition temperature, melting temperature, heat of fusion and crystallization temperature values determined from the DSC thermograms as well as the degree of crystallinity of all the samples. The degree of crystallinity (χc) of PVA and its composites was calculated using the following equation
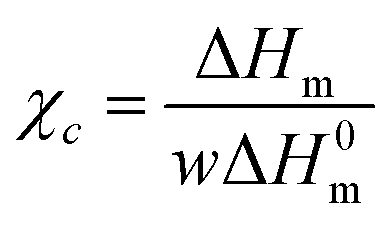 | (1) |
| Sample | Tg, mid-point (°C) | Tm (°C) | ΔHm (J g−1) | χc (%) | Tc (°C) |
|---|---|---|---|---|---|
| PVA | 70.6 | 184.2 | 17.9 | 12.9 | 139.6 |
| 1% NFC–PVA | 71.0 | 176.8 | 19.1 | 13.9 | 128.5 |
| 0.5% epoxy-NFC–PVA | 70.8 | 177.8 | 21.5 | 15.5 | 140.4 |
| 1% epoxy-NFC–PVA* | 71.0 | 180.6 | 24.0 | 17.4 | 142.0 |
| 1% epoxy-NFC–PVA | 72.0 | 184.8 | 26.3 | 19.1 | 145.6 |
| 1.5% epoxy-NFC–PVA | 71.4 | 184.0 | 25.2 | 18.4 | 141.7 |
| 3% epoxy-NFC–PVA | 71.0 | 184.0 | 25.9 | 19.0 | 144.5 |
Among the NFC and epoxy-NFC–PVA composite films, the 1 wt% epoxy-NFC–PVA sample showed the highest melting temperature, heat of fusion, degree of crystallinity and crystallization temperature. This might result from the chemical crosslinks between NFC and PVA. The glass transition temperature of PVA increased slightly with NFC and epoxy-NFC loadings. The largest increase of glass transition temperature of PVA was observed with the addition of 1 wt% epoxy-NFC. Since Tg describes the polymer chains flexibility, the increased Tg value of 1 wt% epoxy-NFC–PVA can be explained by the interactions between NFC and PVA that restrict the mobility of the PVA chains.
As presented in Table 2, the crystallization temperature of PVA decreased with the addition of NFC whereas 0.5, 1, 1.5, 3 wt% epoxy-NFC loadings increased the crystallization temperature of PVA. The decrease and increase of crystallization temperature of PVA might be a consequence of several factors such as the nucleating ability of NFC, sample degradation and physical interactions between PVA and NFC.43,46 Studies of the nucleating effect of nanosized materials on the crystallization behaviour of PVA have been reported.43,44,46
Optical properties
PVA is a visible light polarizer material, which allows its use as a raw material for polarizer film used in liquid crystal display (LCD) panels, lenses and optical filters.52 Fig. 5 shows the photographs of the pure PVA film and the PVA films with NFC and different epoxy-NFC contents. The flower-like patterns and letters can be clearly seen through the pure PVA film and its composite films, thus showing the films transparency. | ||
| Fig. 5 Transparency of pure PVA and its composite films with different NFC and epoxy-NFC contents: pure PVA (A), 1% NFC–PVA (B), 0.5% epoxy-NFC–PVA (C), 1% epoxy-NFC–PVA (D), 1.5% epoxy-NFC–PVA (E), 3% epoxy-NFC–PVA (F). For the imaging the rectangle shape film stripes were set on the flower-pattern substrate. | ||
Light transmittance of the composite films was observed in the visible light wavelength range as shown in Fig. 6. The PVA film was transparent with nearly 92% light transmittance at wavelength of 500 nm. The composite film with 0.5% epoxy-NFC showed nearly the same percentage of light transmittance (87%) as that of a pure PVA film. The light transmittance rate decreased from 85%, 82%, and 76% for the composite films containing 1, 1.5, and 3 wt% of epoxy-NFC, respectively. The light transmittance detected for the composite film filled with 1% NFC was 85%. This result shows that the addition of NFC and epoxy-NFC up to 1.5% into the PVA film produces films with high visible light transmittance, whereas pure NFC and epoxy-NFC films had a light transmittance values of 70% and 68%, respectively at wavelength of 500 nm (results not shown).
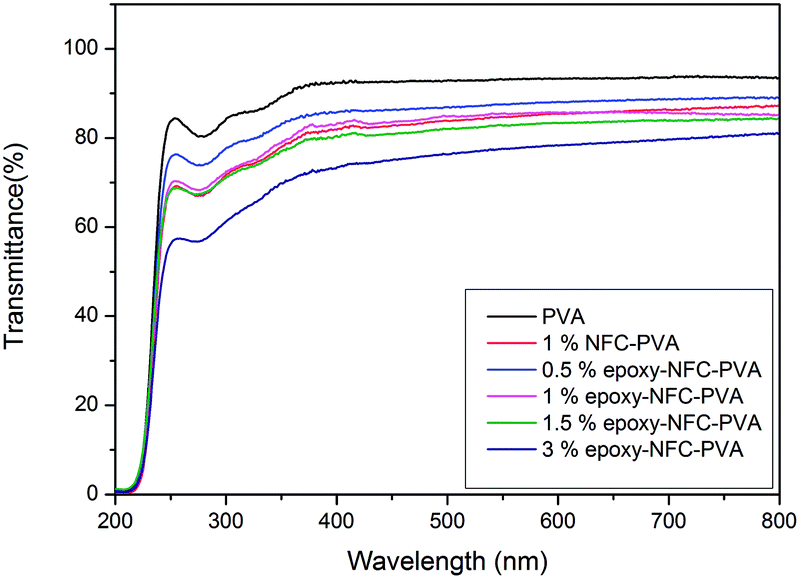 | ||
| Fig. 6 Optical properties of PVA film and composite films in the visible light wavelength range. | ||
Tensile properties
The mechanical properties of unmodified NFC, modified NFC films, and the composite films were performed by the tensile test. Fig. 7 shows the representative stress–strain curves of the pure NFC and epoxy-NFC films. Stress–strain curves for the pure PVA film and its composite films are plotted in Fig. 8. As seen from Fig. 7, the pure NFC film had a tensile strength of 122 MPa, while modified NFC formed more brittle film and broke at a lower strength of about 83 MPa. The tensile strain at break for these films was ∼5%. The result showed that pure NFC could exhibit good strength as such. The obtained tensile strength values for the pure NFC and epoxy-NFC films were higher compared to the reported value of NFC film.53 The pure PVA film showed lower strength (38 MPa), but had substantially higher strain than NFC and epoxy-NFC films (Fig. 8). The reinforcing effect of NFC in the soft chain polymer matrix, like PVA, has been reported.20 In this work, modified NFC was expected to increase the strength of PVA films through chemical cross-linking. Unmodified NFC was used as a reference filler in the polymer matrix.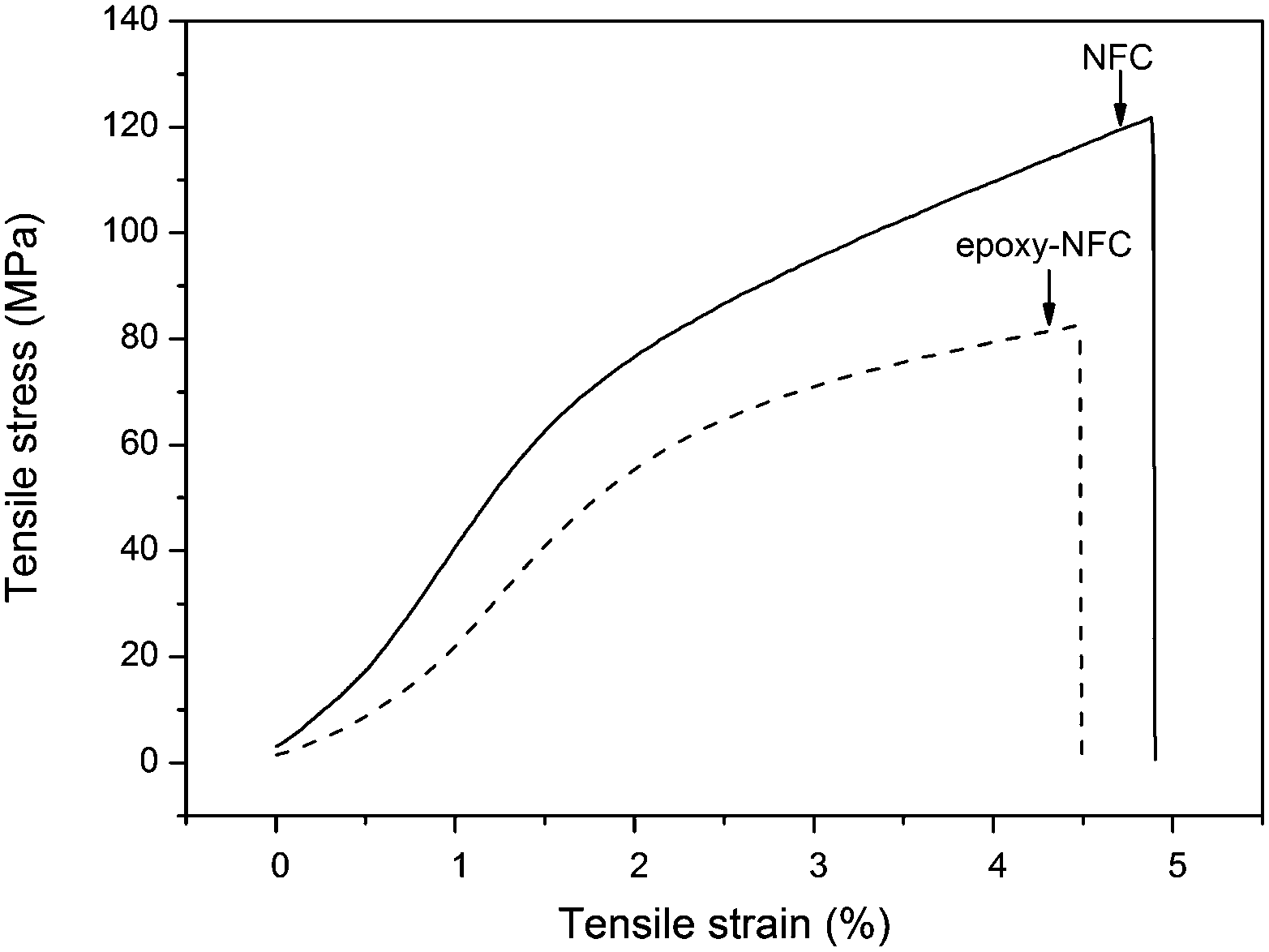 | ||
| Fig. 7 Stress–strain representative curves of pure NFC and modified NFC films. | ||
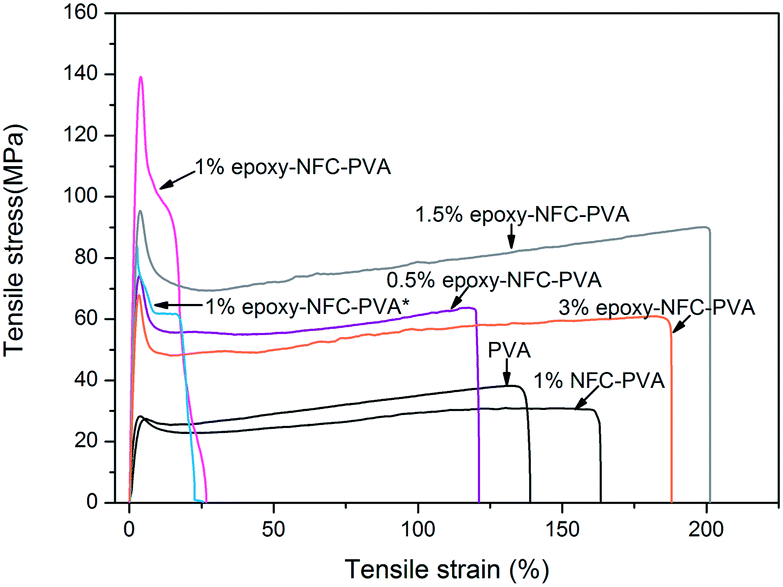 | ||
| Fig. 8 Stress–strain curves of PVA and the composite films. | ||
As shown in Fig. 8, an addition of 1 wt% of unmodified NFC decreased the tensile strength of PVA, while the strain of PVA improved. A 97% improvement in the tensile strength of PVA was obtained with only 0.5 wt% epoxy-NFC loading. This composite film was more rigid than the pure PVA film, and broke at strain of 121%. When comparing the reinforcing effect of 1 wt% epoxy-NFC to 1 wt% unmodified NFC, it seems that the addition of modified NFC yielded stronger and stiffer composite film, but the sample broke easily when compared to the pure PVA film. This stress–strain behaviour of the 1 wt% epoxy-NFC–PVA film could be attributed to the formation of crosslinks between the epoxy groups of NFC and the hydroxyl groups of PVA.
As the amount of NFC increased from 1 wt% to 3 wt%, the strength of the composite film decreased. However, even at the two highest loadings of epoxy-NFC, the strength of the composite films was 134% and 68% higher, respectively, than that of pure PVA. Epoxy-NFC at 1.5 wt% and 3 wt% loadings made the composites more flexible and the strain at break of PVA increased by 52% and 42%, respectively.
The favourable effect of chemically modified NFC on the mechanical properties of PVA compared to the unmodified NFC reinforced PVA composite are shown in Fig. 9. Among the NFC and epoxy-NFC–PVA composite films, the highest E-modulus (4.978 GPa) and tensile strength (123 MPa) were reached with 1 wt% of epoxy-NFC content. The result showed that the addition of 1 wt% epoxy-NFC could improve the E-modulus of PVA by 474% and the tensile strength by 224%. The improved modulus and strength of the 1 wt% epoxy-NFC–PVA sample can be explained by the increased crystallinity as determined by DSC analysis. A heat activated reference sample prepared without using a catalyst and denoted as 1 wt% epoxy-NFC–PVA* also showed a growing trend in mechanical properties. However, the increment in E-modulus and strength was not as significant as for the catalyst and heat activated 1 wt% epoxy-NFC–PVA sample. The result showed that the E-modulus and tensile strength tend to level off when the epoxy-NFC content was greater than 1 wt%. However, even at 1.5 wt% and 3 wt% epoxy-NFC loadings the tensile properties were much higher when compared to pure PVA or the 1 wt% unmodified NFC–PVA composite. At higher loadings, the reinforcing effect of NFC disappeared mainly because of the poor dispersion of cellulose fibrils in the matrix polymer (Fig. 2d). The tensile strength of the composite made with 1 wt% unmodified NFC was 13% lower than that of pure PVA, which might due to the non-uniform dispersion of NFC in PVA. The increment in the E-modulus of PVA was 56% following the addition of unmodified NFC, which shows that nanocellulose has ability to enhance modulus value of PVA.
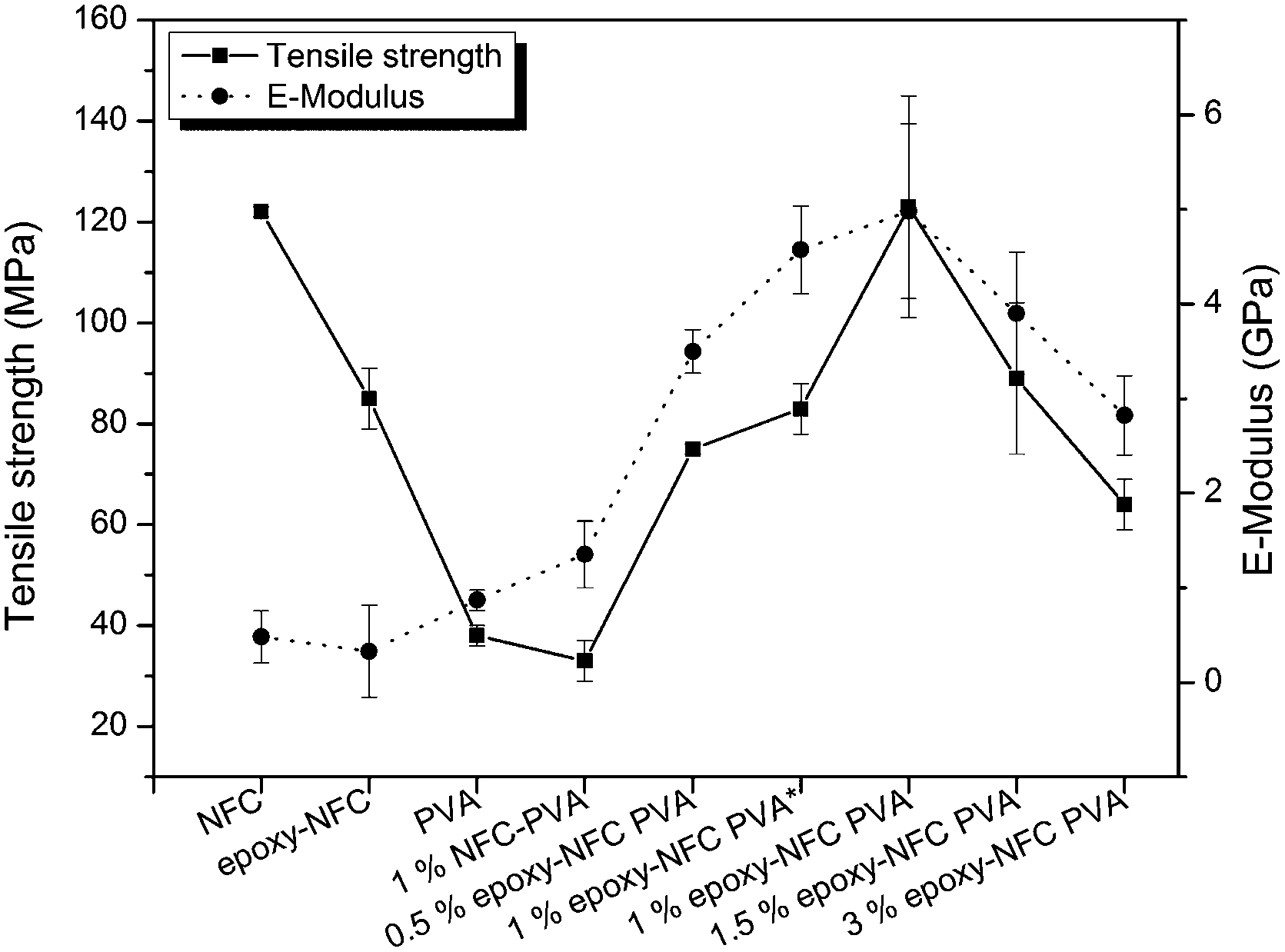 | ||
| Fig. 9 The effect of NFC and modified NFC addition on the mechanical properties of PVA. The error bars represent ±1 standard deviation of the data. | ||
The improved mechanical performance of the epoxy-NFC–PVA composites could be beneficial in the applications that require high strength properties including water-soluble applications such as agro-chemicals and detergents packaging, water-soluble laundry bags and coatings. Other potential application of the developed composites could be among electronic applications e.g. polarizer components with improved durability, stability during processing and thinner design.
The barrier performance
The barrier performance of the NFC and epoxy-NFC–PVA films were measured by the water vapour permeability (WVP) and oxygen permeability (OP) tests (Table 3). WVP measures the rate at which water vapour permeates through the composite film of a specific thickness at a specified temperature and relative humidity. Similarly, OP measures the amount of oxygen gas that passes through the specific size composite film over a given period. OP of all the films was below the detection limit (<0.1 cm3 mm per m2 per day) at 50% relative humidity. Low permeation of oxygen at dry conditions is typical for many biopolymers, but not so easily achieved with synthetic polymers. Obtained OP values are slightly lower as compared to polyamide, but a bit higher as compared to ethylene vinyl alcohol copolymer (EVOH). The permeation was strongly dependent on the humidity, and increased significantly at 90% relative humidity. This behaviour is very typical for nearly all biopolymers.54 WVP of all the films was relatively high due to the hydrophilic character of the raw materials. NFC and epoxy-NFC reinforcement did not change the barrier properties of PVA notably.| Sample code | OP 23 °C, 50% RH (cm3 mm per m2 per day) | OP 23 °C, 70% RH (cm3 mm per m2 per day) | OP 23 °C, 90% RH (cm3 mm per m2 per day) | WVP 23 °C, 100/50% RH (g mm per m2 per day) |
|---|---|---|---|---|
| PVA | <0.1 | 1.2 | 45 | 45 |
| 1% NFC–PVA | <0.1 | 1.4 | 50 | 50 |
| 0.5% epoxy-NFC–PVA | <0.1 | 1.6 | 60 | 55 |
| 1% epoxy-NFC–PVA | <0.1 | 1.5 | 57 | 52 |
| 1.5% epoxy-NFC–PVA | <0.1 | 1.8 | 65 | 60 |
| 2% epoxy-NFC–PVA | <0.1 | 1.4 | 52 | 56 |
| 3% epoxy-NFC–PVA | <0.1 | 1.3 | 48 | 49 |
Conclusions
The composite films of biodegradable PVA matrix reinforced with NFC and epoxy-NFC were successfully prepared by the solution casting method. The potential of using modified NFC as filler for PVA was evaluated based on the mechanical properties of the resulting composite films. The 1 wt% addition of surface-functionalized NFC to the PVA matrix increased the E-modulus and tensile strength of PVA by 474% and 224%, respectively. However, a 1 wt% loading of epoxy-NFC made the sample break more easily when compared to pure PVA.The addition of unmodified NFC to PVA did not improve the mechanical properties of PVA. Moreover, it was found that epoxy-NFC content affected on the mechanical performance of the composite film. Increasing the epoxy-NFC content from 1 wt% to 3 wt% in the PVA composite resulted in a 43% lower E-modulus value and a 48% lower tensile strength value, respectively. Results from SEM imaging showed that the 1 wt% epoxy-NFC formed a more uniform dispersion in the PVA matrix when compared to 1 wt% unmodified NFC. The better dispersion of the modified NFC in the PVA matrix led to improved mechanical performance. DSC analysis showed that the highest increment of melting temperature, heat of fusion, degree of crystallinity and crystallization temperature of PVA was achieved with the addition of 1 wt% epoxy-NFC. This behaviour could be explained by the interactions between the cellulose fibre surface and the adjacent PVA chains, and the nucleating effect of cellulose fibres as a result of surface functionalization of NFC. The DSC result correlated with the mechanical performance of the 1 wt% epoxy-NFC–PVA sample. As the amount of crystallinity increased, the PVA chains became more tightly packed and tensile strength and modulus of the sample increased.
Acknowledgements
The authors gratefully acknowledge financial support received from the Tekes project Naseva.Notes and references
- A. F. Turbak, F. W. Snyder and K. R. Sandberg, J. Appl. Polym. Sci.: Appl. Polym. Symp., 1983, 37, 815–827 CAS.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- A. Dufresne, Recent Res. Dev. Macromol. Res., 1998, 3, 455–474 CAS.
- L. Berglund, Cellulose-based nanocomposites, in Natural Fibres, Biopolymers and Biocomposites, ed. A. K. Mohanty, M. Misra and L. T. Drzal, Taylor & Francis, Boca Raton, 2005, pp. 807–832 Search PubMed.
- M. A. Azizi Samir, F. Alloin and A. Dufresne, Biomacromolecules, 2005, 6, 612–626 CrossRef PubMed.
- S. Kamel, eXPRESS Polym. Lett., 2007, 1, 546–575 CrossRef CAS.
- A. Dufresne, Can. J. Chem., 2008, 86, 484–494 CrossRef CAS.
- M. Hubbe, O. J. Rojas, L. A. Lucia and M. Sain, BioResources, 2008, 3, 929–980 Search PubMed.
- M. Nogi, S. Iwamoto, A. N. Nakagaito and H. Yano, Adv. Mater., 2009, 20, 1–4 Search PubMed.
- T. Ebeling, M. Paillet, R. Borsali, O. Diat, A. Dufresne, J. Y. Cavaille and H. Chanzy, Langmuir, 2009, 15, 6123–6126 CrossRef.
- A. N. Nakagaito, A. Fujimura, T. Sakai, Y. Hama and H. Yano, Compos. Sci. Technol., 2009, 69, 1293–1297 CrossRef CAS PubMed.
- S. Fakirov, D. Bhattacharyya and R. J. Shields, Colloids Surf., A, 2008, 313–314, 2–8 CrossRef PubMed.
- A. N. Nakagaito and H. Yano, Cellulose, 2008, 15, 555–559 CrossRef CAS.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS PubMed.
- K. Syverud and P. Stenius, Cellulose, 2009, 16, 75–85 CrossRef CAS.
- C. Aulin, S. Ahola, P. Josefsson, T. Nishino, Y. Hirose and M. Österberg, Langmuir, 2009, 25, 7675–7685 CrossRef CAS PubMed.
- J. Lu, T. Wang and L. T. Drzal, Composites, Part A, 2008, 39, 738–746 CrossRef PubMed.
- C. Aulin, G. Salazar-Alvarez and T. Lindström, Nanoscale, 2012, 4, 6622–6628 RSC.
- W. Zhang, X. Yang, C. Li, M. Liang, C. Lu and Y. Deng, Carbohydr. Polym., 2011, 83, 257–263 CrossRef CAS PubMed.
- M. Flieger, M. Kantorová, A. Prell, T. Řezanka and J. Votruba, Folia Microbiol., 2003, 48(1), 27–44 CrossRef CAS.
- O. W. Guirguis and M. T. H. Moselhey, Nat. Sci., 2012, 4(1), 57–67 CAS.
- I. M. Jipa, L. Dobre, M. Stroescu, A. Stoica-Guzun, S. Jinga and T. Dobre, Mater. Lett., 2012, 66, 125–127 CrossRef PubMed.
- Y. Shang and Y. Peng, Desalination, 2007, 204, 322–327 CrossRef CAS PubMed.
- J. Lu, T. Wang and L. T. Drzal, Composites, Part A, 2008, 39, 738–746 CrossRef PubMed.
- C. Tang and H. Liu, Composites, Part A, 2008, 39, 1638–1643 CrossRef PubMed.
- P. Alexy, D. Káchova, M. Kršiak, D. Bakoš and B. Šimkova, Polym. Degrad. Stab., 2002, 78, 413–421 CrossRef CAS.
- T. Zimmerman, E. Pöhler and T. Geiger, Adv. Eng. Mater., 2004, 6, 754–761 CrossRef CAS.
- B. H. Jeong, E. M. V. Hoek, Y. S. Yan, A. Subramani, X. F. Huang, G. Hurwitz, A. K. Ghosh and A. Jawor, J. Membr. Sci., 2007, 1–2, 1–7 CrossRef PubMed.
- K. K. H. Wong, J. L. Hutter, M. Zinke-Allmang and W. Wan, Eur. Polym. J., 2009, 45(5), 1349–1358 CrossRef CAS PubMed.
- D. Liu, X. Sun, H. Tian, S. Maiti and Z. Ma, Cellulose, 2013, 20, 2981–2989 CrossRef CAS PubMed.
- W. Li, X. Zhao, Z. Huang and S. Liu, J. Polym. Res., 2013, 20, 210 CrossRef PubMed.
- A. Bulota, A. S. Jääskeläinen, J. Paltakari and M. Hughes, J. Mater. Sci., 2011, 46, 3387–3398 CrossRef.
- M. Hrabalova, M. Schwanninger, R. Wimmer, A. Gregorova, T. Zimmerman and N. Mundigler, BioResources, 2011, 6(2), 1631–1647 CAS.
- S. Arola, T. Tammelin, H. Setälä, A. Tullila and M. B. Linder, Biomacromolecules, 2012, 13, 594–603 CrossRef CAS PubMed.
- W. Li, X. Zhao, Z. Huang and S. Liu, J. Polym. Res., 2013, 20(8), 1–7 CAS.
- D. M. Panaitescu, P. Nechita, H. Iovu, M. D. Iorga, M. Ghiurea and D. Serban, Mater. Plast., 2007, 44(3), 195–198 CAS.
- A. Iwatake, M. Nogi and H. Yano, Compos. Sci. Technol., 2008, 68, 2103–2106 CrossRef CAS PubMed.
- L. Suryanegara, A. N. Nakagaito and H. Yano, Compos. Sci. Technol., 2009, 69(7–8), 1187–1192 CrossRef CAS PubMed.
- M. Jonoobi, J. Harun, A. P. Mathew and K. Oksman, Compos. Sci. Technol., 2010, 70, 1742–1747 CrossRef CAS PubMed.
- P. Eronen, M. Österberg, S. Heikkinen, M. Tenkanen and J. Laine, Carbohydr. Polym., 2011, 86, 1281–1290 CrossRef CAS PubMed.
- S. Asikainen, A. Fuhrmann and F. Robertsen, Nord. Pulp Pap. Res. J., 2010, 25, 269–276 CAS.
- M. Liu, B. Guo, M. Du and D. Jia, Appl. Phys. A: Mater. Sci. Process., 2007, 88, 391–395 CrossRef CAS.
- Y. Srithep, L.-S. Turng, R. Sabo and C. Clemons, Cellulose, 2012, 19, 1209–1223 CrossRef CAS PubMed.
- X. Tang and S. Alavi, Carbohydr. Polym., 2011, 85, 7–16 CrossRef CAS PubMed.
- O. Probst, E. M. Moore, D. E. Resasco and B. P. Grady, Polymer, 2004, 45, 4437–4443 CrossRef CAS PubMed.
- B. Holland and J. Hay, Polymer, 2001, 42, 6775–6783 CrossRef CAS.
- M. S. Peresin, Y. Habibi, J. O. Zoppe, J. J. Pawlak and O. J. Rojas, Biomacromolecules, 2010, 11, 674–681 CrossRef CAS PubMed.
- A. P. Mathew, W. Thielemans and A. Dufresne, J. Appl. Polym. Sci., 2008, 109, 4065–4074 CrossRef CAS.
- J. N. Coleman, M. Cadek, R. Blake, V. Nicolosi, K. P. Ryan, C. Belton, A. Fonseca, J. B. Nagy, Y. K. Gun'ko and W. J. Blau, Adv. Funct. Mater., 2004, 14, 791–798 CrossRef CAS.
- M. Roohani, Y. Habibi, N. Belgacem, G. Ebrahim, A. N. Karimi and A. Dufresne, Eur. Polym. J., 2008, 44(8), 2489–2498 CrossRef CAS PubMed.
- Nitto Denko Corp., US Pat., 8 404 066, 2013.
- J. Y. Zhu, R. Sabo and X. Luo, Green Chem., 2011, 13, 1339–1344 RSC.
- C. Aulin, M. Gällstedt and T. Lindström, Cellulose, 2010, 17, 559–574 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |