
Lysis dynamics and membrane oligomerization pathways for Cytolysin A (ClyA) pore-forming toxin
Abstract
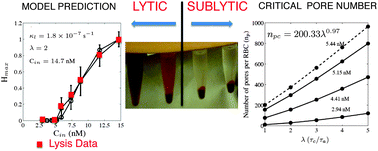
Pore-forming toxins are known for their ability to efficiently form transmembrane pores which eventually leads to cell lysis. The dynamics of lysis and underlying self-assembly or oligomerization pathways leading to pore formation are incompletely understood. In this manuscript the pore-forming kinetics and lysis dynamics of Cytolysin-A (ClyA) toxins on red blood cells (RBCs) are quantified and compared with experimental lysis data. Lysis experiments are carried out on a fixed mass of RBCs, under isotonic conditions in phosphate-buffered saline, for different initial toxin concentrations ranging from 2.94–14.7 nM. Kinetic models which account for monomer binding, conformation and oligomerization to form the dodecameric ClyA pore complex are developed and lysis is assumed to occur when the number of pores per RBC (np) exceeds a critical number, npc. By analysing the model in a sublytic regime (np < npc) the number of pores per RBC to initiate lysis is found to lie between 392 and 768 for the sequential oligomerization mechanism and between 5300 and 6300 for the non-sequential mechanism. Rupture rates which are first order in the number of RBCs are seen to provide the best agreement with the lysis experiments. The time constants for pore formation are estimated to lie between 1 and 20 s and monomer conformation time scales were found to be 2–4 times greater than the oligomerization times. Cell rupture takes places in 100s of seconds, and occurs predominantly with a steady number of pores ranging from 515 to 11 000 on the RBC surface for the sequential mechanism. Both the sequential irreversible and non-sequential kinetics provide similar predictions of the hemoglobin release dynamics, however the hemoglobin released as a function of the toxin concentration was accurately captured only with the sequential model. Each mechanism develops a distinct distribution of mers on the surface, providing a unique experimentally observable fingerprint to identify the underlying oligomerization pathways. Our study offers a method to quantify the extent and dynamics of lysis which is an important aspect of developing novel drug and gene delivery strategies based on pore-forming toxins.
 Please wait while we load your content...
Please wait while we load your content...

