PVDF film tethered with RGD-click-poly(glycidyl methacrylate) brushes by combination of direct surface-initiated ATRP and click chemistry for improved cytocompatibility†
Fei
He‡
a,
Baiwen
Luo‡
b,
Shaojun
Yuan
*a,
Bin
Liang
a,
Cleo
Choong
*b and
Simo Olavi
Pehkonen
c
aMultiphase Mass Transfer & Reaction Engineering Lab, College of Chemical Engineering, Sichuan University, Chengdu, 610065, China. E-mail: yuanshaojun@gmail.com; Fax: +86 28 85460557; Tel: +86 28 85999978
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798. E-mail: cleochoong@ntu.edu.sg
cChemical Engineering Program, Masdar Institute of Science and Technology, PO Box 54224, Abu Dhabi, United Arab Emirates
First published on 5th November 2013
Abstract
To enhance the cytocompatibility of polyvinylidene fluoride (PVDF) films, arginine–glycine–aspartic acid (RGD) peptide-click-poly(glycidyl methacrylate) (PGMA) polymer brushes were grafted onto the PVDF surface by the combination of surface-initiated atom transfer radical polymerization (ATRP) and click reaction. The direct initiation of the secondary fluorine atoms of PVDF backbone allowed for grafting of the PGMA brushes containing reactive epoxy groups. Subsequent introduction of the azide groups onto the side chain of PGMA brushes was achieved by the ring-open reaction of the epoxy groups with sodium azide. The cell adhesive RGD peptide was finally conjugated onto the PGMA brushes via alkyne-azide click reaction. Kinetic studies revealed that the PGMA chain growth from the PVDF surface was consistent with a “controlled” process, and that the amount of immobilized RGD peptide on the PGMA brushes increased with the concentration of the pendant azide groups. The specificity of cellular interactions of adipose tissue-derived stem cells (ASCs) on the functionalized PVDF films was investigated. Results demonstrated that cell adhesion and proliferation of ASCs were significantly improved on the RGD-immobilized PVDF substrates, and this improvement was positively correlated with the surface concentration of covalently-bonded RGD peptide. With the inherent superior chemical and mechanical properties of PVDF films and the biocompatible nature of cell-adhesive peptides, surface functionalized PVDF films are potentially useful for biomedical and tissue engineering applications.
1. Introduction
In recent years, a great deal of effort has been devoted to developing novel polymeric materials with improved functionality for tissue engineering applications. Such synthetic templates possess mechanical and physicochemical properties that allow them to replace defective functions of a tissue or part of an organ (such as heart valves, blood vessels and tendons etc.). Upon interfacing with biological systems, these biomaterials need to avoid triggering an adverse reaction of the organism (i.e. non-cytotoxic) and perform with an appropriate host response in their specific applications.1 Considering the potential risk in long-term applications and the costly and time-consuming certification process of newly-developed polymers for implants, prostheses and biomedical devices, some established polymeric materials, such as polypropylene (PP),2–4 polyethylene terephthalate (PET)5 and polytetrafluorethylene (PTFE),6–8 are currently being used in clinical long-term applications as hernia meshes, artificial ligaments and vascular grafts. As a particular inert polymer of superior mechanical strength and durability, polyvinylidene fluoride (PVDF) has received much attention and generated considerable interest for use in implant components.9,10 As compared to PET, the PVDF material is more resistant to hydrolysis and degradation, and preserves the mechanical stability even after some years of implantation.11 Moreover, aging does not result in the increase in bending stiffness as described for PP.11 However, the lack of biocompatibility due to the inherent hydrophobicity on the PVDF surface may limit its usage in the biomedical applications. Materials for biomedical or tissue engineering purpose should provide physic-chemical properties and cellular motifs matching the surroundings to support the desired cell response, e.g. adhesion, proliferation, or differentiation.12 It is therefore desired to modulate the surface properties of PVDF for promoting its interactions with biomolecules to improve the biocompatibility and hydrophilicity.Different approaches have been developed to modify the PVDF for the improvement of hydrophilicity and physiological properties. One is material matrix modification including blending and copolymerization,13–15 which usually needs to incorporate the compatibility and hydrophilic copolymers into the PVDF matrix. The other is surface modification or surface coating. This method presents a way to preserve the mechanical properties of established materials and to improve the surface biocompatibility.16 Since PVDF is lacking in reactive sites to allow for surface modification, surface activation pretreatments, such as ozone treatment,17 plasma irradiation,18,19 electron beam,20 γ-ray radiation,21 and ultraviolet (UV) treatment,22,23 have been widely used to produce radicals or radical-generating moieties on the PVDF surfaces. Subsequent thermally-induced polymerization can provide specific functional groups onto the PVDF surfaces for desirable functionality, such as improved hydrophilicity, biocompatibility, fouling-resistance, and conductivity. Among them, ozone or plasma-induced copolymerization offers an effective approach to introduce hydrophilicity by grafting of functional copolymers, such as poly(acrylic acid) (PAA),24 poly(methacrylic acid) (PMAA),25 poly(ethylene glycol) (PEG),18,26N-vinyl pyrrolidone (PVP),27 and poly(sulfobetaine methacrylate) (PSBMA).28,29 However, these free radical polymerization techniques suffer from lacking appropriate control over the graft yield of polymer brushes, the undesired homopolymerization of monomers, and the unfavorable reactions between reactive groups of polymer brushes and the surface.30 Moreover, backbone degradation and gelation can occur as a result of the high energy of ion bombardment or UV radiation.19 As a consequence, an alternative approach that allows for controlling over the polymer brush density, structure and composition is highly expected for the modification of PVDF.
Progress in polymerization has made it possible to produce polymer chains or brushes on a solid surface with a controlled length, composition and structure.30,31 As a “living/controlled” radical polymerization method, atom transfer radical polymerization (ATRP) has attracted considerable attention for its good controllability over the molecular weight, polydispersity, and chain-end functionality of the grafted polymers, as well as its capability of affording a high graft yield with the robustness and versatility of living radical polymerization retained.31 This approach provides an alternative strategy for tethering the PVDF surface with a specific functionality. To proceed with ATRP reactions, the pendant chemical groups containing radically transferable halogen atoms on the polymer substrates is indispensable to serve as the initiation sites. One common way is to introduce the alkyl halides (bromides or chlorides) to the pretreated PVDF surfaces described above.17,32 The other way is proposed by Mayes et al. to direct graft copolymerization of methacrylates from PVDF by using pendant secondary fluorine atoms as ATRP macroinitiators for the preparation of functionalized derivatives.33 However, the initiating efficiency has been often very low owing to the low reactivity of secondary fluorine atoms, and thus leading to low graft density of copolymers.34 For example, only about 0.1% of the secondary fluorine in PVDF can initiate the graft copolymerization of tert-butyl methacrylates.33 Despite these advantages, the direct surface-initiated ATRP offers a simple one-step process for ATRP, and has been extensively used to prepare hydrophilic and amphiphilic polymer brushes on the PVDF surface.35–39 The pendant reactive functional groups, such as hydroxyl, carboxyl, amino and epoxide groups, on the hydrophilic polymer brushes are able to act as highly active binding sites for biomolecules.37 Thereby, it would be feasible to promote the biocompatibility of PVDF by the conjugation of biologically active molecules to the grafted hydrophilic polymer brushes.
Post-polymerization modification allow for the implementation of small-molecule organic chemistry reactions to homopolymers in a controlled manner and under mild conditions.40 This approach circumvents a great many problems associated with direct polymer synthesis and enables the creation of biomolcules-polymeric systems for novel applications. In particular, an azide-alkyne click reaction is a highly efficient and orthogonal organic reaction that offers high selectivity, excellent tolerance for functional groups and no extraneous side products.41 This reaction is especially appealing for biological labeling and coupling reactions due to their facile incorporation into biomolecules through post-synthetic modification, enzymatic transfer, and azide-modified nutrients for metabolic functionalization.42 Thereby, the combination of surface-initiated ATRP and click reaction provides a novel, well-controlled and efficient process of the synthesis of a wide variety of new biomaterials with potential uses as drug delivery agents, tissue engineering scaffolds, and dispersible nanomaterials.43–45 As compared to the conventional methods for bioconjugation, the combination process of surface-initiated ATRP and click reaction allows to covalently immobilize biomolecules, such as fibronectin, laminin, and arginine–glycine–aspartic acid (RGD) peptide, on the substrates with desired densities and structures.46 Furthermore, this combination process also provides two major advantages. One is that ATRP enables the construction of polymer chains on the substrates, and the amount of reactive functional groups for biomolecule conjugation can be readily controlled by varying the reaction conditions.47 The other is that the click conjugation process can be well-controlled and carried out efficiently.48 However, to the best of our knowledge, only a few studies have been devoted to modifying PVDF by the combination process of ATRP and click reaction to trigger specific functionality.49
In the present study, the main purpose was to synthesize the RGD-click-poly(glycidyl methacrylate) (PGMA) polymer brushes on the PVDF film surface by the combination of direct surface-initiated ATRP and click reaction for the improvement of cytocompatibility. The PVDF surfaces were first grafted with PGMA polymer brushes via direct surface-initiated ATRP of GMA using the secondary fluorine atoms as the initiation sites, followed by introducing the azide groups on the PGMA side chains by the ring-open reaction of epoxy groups with sodium azide. Subsequent conjugation of RGD peptide to the azide-terminated side chains of PGMA brushes was accomplished by Cu(I)-catalyzed azide-alkyne click reaction. RGD peptide was chosen as the model cell-adhesive biomolecule to functionalize fluoropolymer substrates for the improvement of cellular function and biocompatibility,1,50–52 due to its widespread distribution and use throughout the organism, its effectiveness in binding integrin to stimulate cell adhesion and its biological impact on cell survival and growth.53 Success in each surface modification step was ascertained by attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR), X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM) and the measurement of static water contact angles. Due to the abundance and accessibility of fat tissue in the average person and the availability of reliable methods to isolate and expand the adult stem cells, adipose-derived stem cells (ASCs) have been widely recognized as an ideal and attractive source of autologous cells for tissue engineering.54 As such, in vitro cellular studies of ASCs were performed on the functionalized PVDF films to evaluate the cell–material interactions, as well as to demonstrate the improvement of the biocompatibility.
2. Experimental
2.1 Materials
PVDF films with a thickness of 0.5 mm were purchased from Goodfellow Cambridge, Ltd. (Huntingdon, UK), and were used as received. The monomer glycidyl methacrylate (GMA, 97%) was obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA), and was passed through an inhibitor removal column prior to being stored at −4 °C. 4-Pentynoic acid (95%), N,N′-diisopropyl carbodiimide (DIC, 99%), sodium azide (NaN3, 99%), ammonium chloride (NH4Cl, 99.5%), copper(I) bromide (CuBr, 99%), copper(II) bromide (CuBr2, 98%), 2,2′-bipyridine (bpy, 99%), copper(II) sulfate (CuSO4, 99%) and sodium ascorbate (98%) were obtained from Sigma-Aldrich Chemical Co. and were used as received. Solvents, such as methanol, toluene, acetone, dimethyl sulfoxide (DMSO), dichloromethane (DCA), N,N-dimethylformamide (DMF) and tetrahydrofuran (THF) were of reagent grade and used without further purification. RGD (H-Arg-Gly-Asp-OH) was obtained from Bachem Bioscience (King of Prussia, PA, USA). Adipose tissue-derived stem cells (ASCs) and cell culture medium (DMEM/F-12) were purchased from Life Technologies™ Singapore. Culture medium supplements, such as foetal bovine serum (FBS), trypsin–EDTA (0.25%), bovine brain extract, amphotericin, penicillin, and streptomycin were obtained from Life Technologies™ Singapore. The AlamarBlue® (AB) assay reagent and the LIVE/DEAD® Cell Viability Kit were also purchased from Life Technologies™ Singapore. The alkyne-functionalized RGD was synthesized using a previously-reported protocol with slight modifications.552.2 Direct surface-initiated ATRP of GMA and azidation of PGMA side chains
The PVDF films were sliced into round-shaped specimens with a diameter of 2 cm, followed by washing with copious amount of acetone, methanol, and deionized water in that order prior to being dried in a vacuum oven at ambient temperature. A typical solution polymerization process with a molar ratio of [GMA (monomer)]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CuBr (catalyst)]:[CuBr2 (deactivator)]:[bpy (ligand)] at 150:1.0:0.2:2.0 in the mixture of methanol and deionized water (4:1, v/v) was carried out as follows. 3 ml of GMA (22 mmol) and 45.8 mg (0.29 mmol) bpy were added to 5 ml of the methanol–water mixture in a Pyrex® tube. The PVDF film specimens were introduced to the reaction mixture, followed by purging with argon for 20 min. Subsequently, about 21.5 mg of CuBr (0.15 mmol) and 6.6 mg of CuBr2 (0.029 mmol) were added to the reaction mixture. After purging with argon for another 10 min, the reaction tube was sealed under an argon atmosphere with a rubber stopper. The reaction was allowed to proceed at 80 °C under continuous stirring for a predetermined time. At the end of the reaction, the PGMA-grafted PVDF substrates were removed and washed thoroughly with excess amount of THF, ethanol, deionized water, in that order, to ensure the complete removal of the physical adsorbed reactants before being dried under reduced pressure. The resultant PGMA-grafted PVDF substrates from 2 h and 8 h of ATRP reaction were denoted as the PVDF-g-PGMA1 and PVDF-g-PGMA2 surfaces, respectively.
:[CuBr (catalyst)]:[CuBr2 (deactivator)]:[bpy (ligand)] at 150:1.0:0.2:2.0 in the mixture of methanol and deionized water (4:1, v/v) was carried out as follows. 3 ml of GMA (22 mmol) and 45.8 mg (0.29 mmol) bpy were added to 5 ml of the methanol–water mixture in a Pyrex® tube. The PVDF film specimens were introduced to the reaction mixture, followed by purging with argon for 20 min. Subsequently, about 21.5 mg of CuBr (0.15 mmol) and 6.6 mg of CuBr2 (0.029 mmol) were added to the reaction mixture. After purging with argon for another 10 min, the reaction tube was sealed under an argon atmosphere with a rubber stopper. The reaction was allowed to proceed at 80 °C under continuous stirring for a predetermined time. At the end of the reaction, the PGMA-grafted PVDF substrates were removed and washed thoroughly with excess amount of THF, ethanol, deionized water, in that order, to ensure the complete removal of the physical adsorbed reactants before being dried under reduced pressure. The resultant PGMA-grafted PVDF substrates from 2 h and 8 h of ATRP reaction were denoted as the PVDF-g-PGMA1 and PVDF-g-PGMA2 surfaces, respectively.
The azido groups on the surface of PVDF-g-PGMA films were generated through the ring-open reaction of the pendant oxirane rings in the GMA segment of the copolymer chain with sodium azide (NaN3). Briefly, the corresponding PVDF-g-PGMA surfaces were immersed in a 30 ml of DMF solution containing NaN3 (0.33 g, 5.1 mmol) and NH4Cl (0.27 g, 5.1 mmol). The transformation reaction was allowed to proceed at 50 °C for 12 h under vigorous stirring. After the reaction, the resulting azide-terminated PVDF (referred to as the PVDF-g-P(GMA)-N3) surfaces were washed thoroughly with copious amounts of deionized water to remove excess NaN3 prior to being dried under reduced pressure at ambient temperature.
2.3 Click grafting of RGD peptide onto the azide-terminated PGMA side chains
The RGD peptide-functionalized PVDF surfaces were synthesized via click reaction of PVDF-g-P(GMA)-N3 surfaces with alkyne-functionalized RGD peptide. Briefly, the PVDF-g-P(GMA)-N3 films were immersed in a Pyrex® tube containing the mixture solution of DMSO (5 ml) and deionized water (5 ml). Subsequently, about 3.2 mg CuSO4 (0.02 mmol), 8.0 mg of sodium ascorbate (0.04 mmol) and 8.5 mg of alkyne-functionalized RGD (0.02 mmol) (the molar ratio of 1:2:1) were added into the reaction mixture. The reaction solution was purged with argon for 30 min under stirring. The reaction tube was sealed with the rubber stopper and kept in a 50 °C oil bath for 24 h to initiate the click reaction. At the end of the reaction, the RGD-grafted PVDF (denoted as PVDF-g-P(GMA)-click-RGD) surfaces were immersed in a large volume of PBS solution with gentle stirring to remove the physically-adsorbed RGD peptide over 24 h at ambient temperature, followed by washing thoroughly with PBS and deionized water prior to being dried under reduced pressure. The resultant RGD peptide-grafted PVDF surfaces were referred to as the PVDF-g-P(GMA)1-click-RGD and PVDF-g-P(GMA)2-click-RGD surfaces, respectively.
2.4 Determination of grafting density of PGMA and immobilized RGD
The grafting density of the grafted P(GMA) brushes and immobilized RGD peptide on the PVDF surfaces was determined by measurement of the grafting yield as described in detail previously.56 The grafting yield (GY) can be calculated by the following equation:where Wa and Wb represent the weights of the dry PVDF films after and before graft polymerization (or immobilization of RGD peptide), respectively, and A is the film area (3.2 cm2). At least three pieces of PVDF films were measured and the averaged GY values were obtained. Furthermore, X-ray photoelectron spectroscopy (XPS) measurement was also carried out to qualitatively determine the relative amount of immobilized RGD peptide on the PVDF surfaces. The N 1s core-level signal was used as an indicator of the immobilized RGD peptide. The [N]/[C] ratio, as determined from the sensitivity-factor-corrected N 1s and C 1s core-level spectral area, indicated the relative abundance of RGD peptide on the PVDF surface.
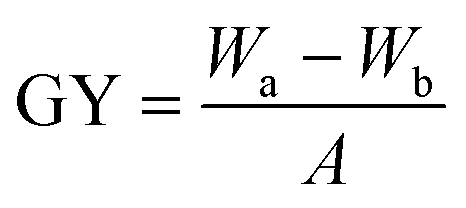
2.5 Surface characterization
Attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) was used to obtain infrared absorption spectra of the functionalized PVDF surface. The measurements were performed on a Spectrum GX FTIR spectrometer (Perkin-Elmer Inc., Waltham, MA) equipped with a smart performer accessory by using a germanium (Ge) crystal with an incident angle of 45° and a sampling area of 2 mm2. The chemical compositions of the functionalized PVDF film surfaces were also characterized by XPS on a Kratos AXIS His spectrometer using a monochromatised Al Kα X-ray source (1486.6 eV photons) using procedures described in detail previously.57 The static water contact angles of the pristine and functionalized PVDF films were measured at 25 °C and 60% relative humidity using a sessile drop method with a 3 μl water droplet on a FTÅ 200 contact angle goniometer (First Ten Angstroms Inc., Portsmouth, VA). The contact angles reported were the mean values from three substrates, with the value of each substrate obtained by averaging the contact angles for at least three surface locations. The surface topography of the functionalized PVDF surfaces was investigated by atomic force microscopy (AFM). A multimode scanning probe microscope equipped with a Nanoscope IIIa controller (Digital Instrument, Santa Barbara, CA) was used to capture the AFM images with a scan size of 10 mm × 10 mm in tapping mode in air. Nanoscope software version 4.43r8 was used to determine the root-mean-square (RMS) roughness.2.6 Cell culture
Adipose tissue-derived stem cells (ASCs) were cultured in a DMEM/F-12 culture medium, supplemented with HEPES, 10% fetal bovine serum, 1% L-glutamine, 1% MEM non-essential amino acids solution 10 mM (100×), and 1% penicillin-streptomycin in a 5% CO2 environment at 37 °C. The DMEM/F-12 medium was changed every other day. Upon 90% confluency, cells were harvested by trypsinization by 0.25% trypsin–EDTA solution. ASCs between passages 4–6 were used for subsequent experiments.2.7 Cell adhesion and proliferation
To assess ASCs adhesion and proliferation on the functionalized PVDF films, tissue culture polystyrene (TCPS) was used as positive controls. Prior to cell seeding, the PVDF films were sterilized by immersing in a 70% (v/v) ethanol solution for 60 min, and followed by rinsing with sterile PBS twice and equilibrated in the culture medium for 2 h. The ASCs suspension was resuspended in a fresh medium to a concentration of 1 × 104 cells per ml (measured by a hemocytometer). For cell adhesion assay, PVDF films were first placed at the bottom of each well of a 24-well culture plate. 0.5 ml aliquots of cell suspension (5000 cells per well) were subsequently seeded onto the surface of prewetted PVDF films in each well. Cell-seeded PVDF films were incubated in the 5% CO2 atmosphere at 37 °C for 24 h to allow for cell attachment. At the end of the incubation period, the PVDF films were washed twice with a PBS solution (pH 7.4) to remove any loosely-attached cells, and then were fixed with paraformaldehyde (4%, v/v in PBS solution) for 2 h, followed by dehydration with serial ethanol solutions (25%, 50%, 75%, 90% and 100%) prior to being dried under reduced pressure at ambient temperature. Scanning electron microscopy (SEM) images were finally captured on different PVDF film surfaces to observe cell adhesion and spreading behavior. Cell nuclei and F-actin were counter-stained with DAPI and rhodamine–phalloidine (1:200), respectively. For relative cell numbers and spreading area of ASCs on the surface-functionalized PVDF, nuclei and F-actin filament were stained and the representative images were visualized using a Nikon ECLIPSE Ti–S fluorescence microscope (emission at 515 nm and 635 nm, Nikon Instruments, Tokyo, Japan) at 100× magnification. The DAPI-stained fluorescence images were also captured to evaluate the adherent cell numbers.
Cell viability and proliferation were determined using the AlamarBlue® (AB) assay. Briefly, 0.5 ml of ASCs cell suspension (1 × 104 cells per ml) was seeded into each well of a 24-well plate containing the pristine and functionalized PVDF films, and incubated in a 5% CO2 environment at 37 °C for 1, 3, 5 and 7 days according to previously established protocols.56 Briefly, at each time points, the culture medium was removed and 0.5 ml of the AB solution (10% AB solution in culture medium without FBS) was added to the wells. The plates were incubated in a 5% CO2 atmosphere at 37 °C for 4 h. Fluorescence analysis using an excitation wavelength of 570 nm and an emission wavelength of 585 nm was carried out with a SpectralMax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Cell numbers were calculated using a standard curve correlating with the fluorescence emission.
2.8 Cell imaging
At the end of 7 days of incubation, cell viability and coverage on different PVDF film surfaces were determined by using the LIVE/DEAD® Viability/Cytotoxicity Kit following the manufacturer's instructions. Briefly, the culture medium was pipetted out and each specimen was washed thrice with the PBS solution. The working solution containing 2 mM calcein AM and 4 mM EthD-1 was subsequently added to each specimen. After incubation in a 5% CO2 atmosphere at 37 °C for 45 min, the surfaces were visualized using Nikon fluorescence microscopy. Fluorescence images of adhered cells were captured using NIS-Elements Br software at five locations on the specimen surfaces.2.9 Statistical analysis
Each experiment was carried out in triplicate (n = 3), and the data presented as mean ± standard deviation (SD) unless otherwise stated. Statistical analysis was carried out by means of one-way analysis of variance (ANOVA) with Tukey's post hoc test. A confidence level of 95% (p < 0.05) was considered statistically significant.3. Results and discussion
The PVDF film surface tethered with RGD peptide-click-poly(glycidyl methacrylate) (PGMA) polymer brushes is prepared according to the reaction sequence schematically shown in Fig. 1: (i) PGMA polymer brushes are directly grafted onto PVDF film surface by using the secondary fluorine atoms of PVDF backbone as initiation sites to provide reaction epoxy groups, (ii) subsequent introduction of the azide groups onto the PGMA side chains is achieved by the ring-open reaction of epoxy groups with sodium azide (NaN3), (iii) RGD peptide is immobilized onto the azide-terminated PGMA brushes through the Cu(I)-catalyzed azide-alkyne click reaction. Details of each reaction step are discussed below.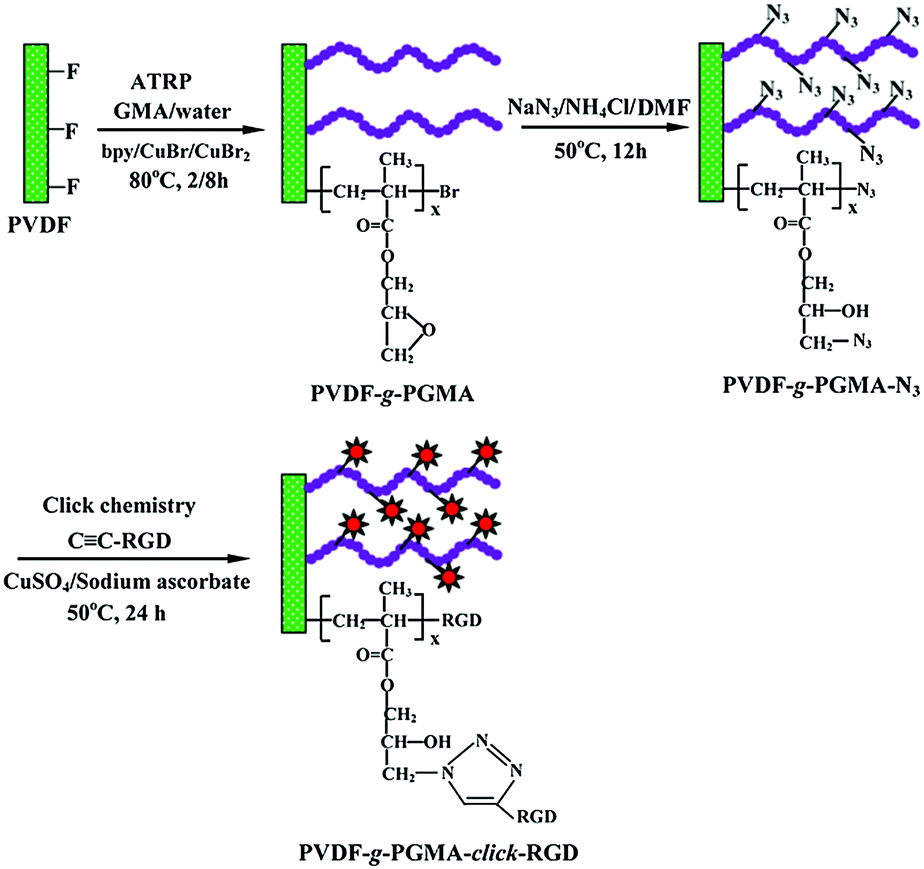 | ||
| Fig. 1 Schematic illustration of the process of direct surface-initiated ATRP of GMA from the PVDF film surface to give the PVDF-g-PGMA surface, subsequent introduction of azide groups via ring-open reaction of the epoxy groups on the PGMA brushes with sodium azide to produce the PVDF-g-PGMA-N3 surface, and the conjugation of RGD peptide to the PGMA brushes via alkyne-azide click reaction. | ||
3.1 Direct surface-initiated ATRP of GMA from the PVDF surfaces
Atom transfer radical polymerization (ATRP) has been employed to design and synthesize PVDF-based graft copolymers using secondary fluorinated groups of vinylidene fluoride (VDF) units in PVDF backbone as the initiation sites,35,38,58 although the directly-initiated ATRP reaction suffers from low initiation efficiency and requires high reaction temperature to activate the strong C–F bond (a bond energy of 486 kJ mol−1).37 In this study, direct surface-initiated ATRP of GMA was conducted on the PVDF film surfaces to produce the PVDF-g-PGMA surfaces. The chemical composition of the PVDF film surface was characterized by FTIR and XPS spectra.
Fig. 2 shows the ATR-FTIR spectra of the pristine PVDF and PVDF-g-PGMA surfaces from 2 h, 4 h and 8 h of ATRP reaction, respectively. For the pristine PVDF, the characteristic absorption band in the wavenumber region of 1120–1280 cm−1 is associated with the –CF2– group of the PVDF main chains.57 The band at the wavenumber of 1400 cm−1 is attributable to the –C–F– group of the PVDF (Fig. 2a). The successful grafting of PGMA brushes onto the PVDF surface can be deduced from the appearance of an additional peak at the wavenumber of 1724 cm−1, attributable to the characteristic stretching of ester carboxyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group of the grafted GMA segments (Fig. 2b–d).57 For the copolymers of PVDF-g-PGMA from different reaction time, the ratio of the intensity of the absorption band of the ester carboxyl groups to that of the –CF– at 1400 cm−1 ([–CO]/[–CF] ratio) reflects the relative graft amount of PGMA on the PVDF film surfaces. Both the intensity of the –CO absorption and the ratio of [–CO]/[–CF] undergo a noticeable increase with prolonging the reaction time, implying that the grafting density of PGMA brushes on the PVDF film surface is dependent on the ATRP time. This result is consistent with the controllable manner of the ATRP reaction.
O) group of the grafted GMA segments (Fig. 2b–d).57 For the copolymers of PVDF-g-PGMA from different reaction time, the ratio of the intensity of the absorption band of the ester carboxyl groups to that of the –CF– at 1400 cm−1 ([–CO]/[–CF] ratio) reflects the relative graft amount of PGMA on the PVDF film surfaces. Both the intensity of the –CO absorption and the ratio of [–CO]/[–CF] undergo a noticeable increase with prolonging the reaction time, implying that the grafting density of PGMA brushes on the PVDF film surface is dependent on the ATRP time. This result is consistent with the controllable manner of the ATRP reaction.
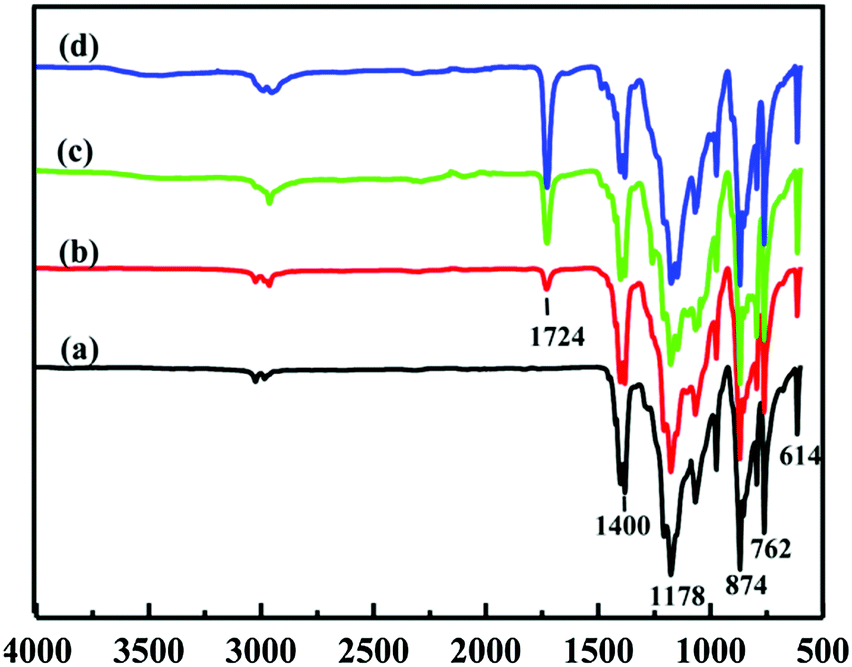 | ||
| Fig. 2 ATR-FTIR spectra of the (a) pristine PVDF film, (b) PVDF-g-PGMA surface from 2 h of ATRP time (PVDF-g-PGMA1), (c) PVDF-g-PGMA surface from 4 h of ATRP time, and (d) PVDF-g-PGMA surface from 8 h of ATRP time (PVDF-g-PGMA2). | ||
Fig. 3 shows the respective wide scan and C 1s core-level XPS spectra of the pristine PVDF, the PVDF-g-PGMA1 from 2 h of ATRP reaction and the PVDF-g-PGMA2 surfaces from 8 h of ATRP reaction. For the pristine PVDF film, only C 1s and F 1s signals are observed on the wide scan spectrum (Fig. 3a). The C 1s core-level spectrum can be curve-fitted with three peak components with binding energies (BEs) at 284.6, 285.8 and 290.5 eV, attributable to the neutral –CH, –CH2–CFx and –CF2 species,59 respectively (Fig. 3b). The [–CH2–CFx]:[–CF2] area ratio of about 1.0:1.0 is consistent with the chemical structure of VDF units. In the case of PVDF-g-PGMA1 surface from 2 h of ATRP time, the appearance of an additional O 1s signal and the clear increase in the relative intensity of C 1s signal in the wide scan spectrum (Fig. 3c), arising from the grafted PGMA chains, indicates the successful graft copolymerization of GMA from the PVDF main chains. The two additional C 1s peak components with BE at 286.2 eV for the –C–O species and at 288.5 eV for the O–CO species are also associated with the grafted PGMA chains (Fig. 3d).59 The [–C–O]:[O–CO] peak component area ratio of about 2.8:1 is comparable to the theoretical ratio of 3:1 for the chemical structure of the GMA repeat unit. The decrease in static water contact angles from 98 ± 3° (for the pristine PVDF surface) to about 78 ± 4° of the PVDF-g-PGMA1 surface is consistent with the presence of PGMA brushes on the PVDF surface (Table 1). Upon prolonging the ATRP time to 8 h, the only residual minor F 1s signal, together with the significant increase in O 1s and C 1s signals, in the wide scan spectrum of the PVDF-g-PGMA2 surface indicates a substantial increase in the thickness and surface density of the grafted PGMA brushes (Fig. 3e), albeit that the thickness of the PGMA brushes is somewhat less than the probing depth of the XPS technique (about 10 nm in an organic matrix59). This result is further ascertained by the significant decrease in the peak components of –C–CFx and –CF2 and the concomitantly marked increase in the peak components of –C–O and O–CO of the curve-fitted C 1s core-level spectrum for the PVDF-g-PGMA2 surface (Fig. 3f). It has been reported that the thickness of the poly(ethylene glycol)monomethacrylate (PEGMA) brushes grafted onto the PVDF surface was around 6.9 nm after 5 h of ATRP reaction under similar reaction conditions.35 The further decrease in the static water contact angle to about 67 ± 3° for the PVDF-g-PGMA2 surface also confirms that the thickness of the grafted PGMA brushes is gained gradually with an increase in the ATRP reaction time (Table 1). Thus, the PGMA brushes have been successfully grafted from the PVDF surface to cater for further functionalization.
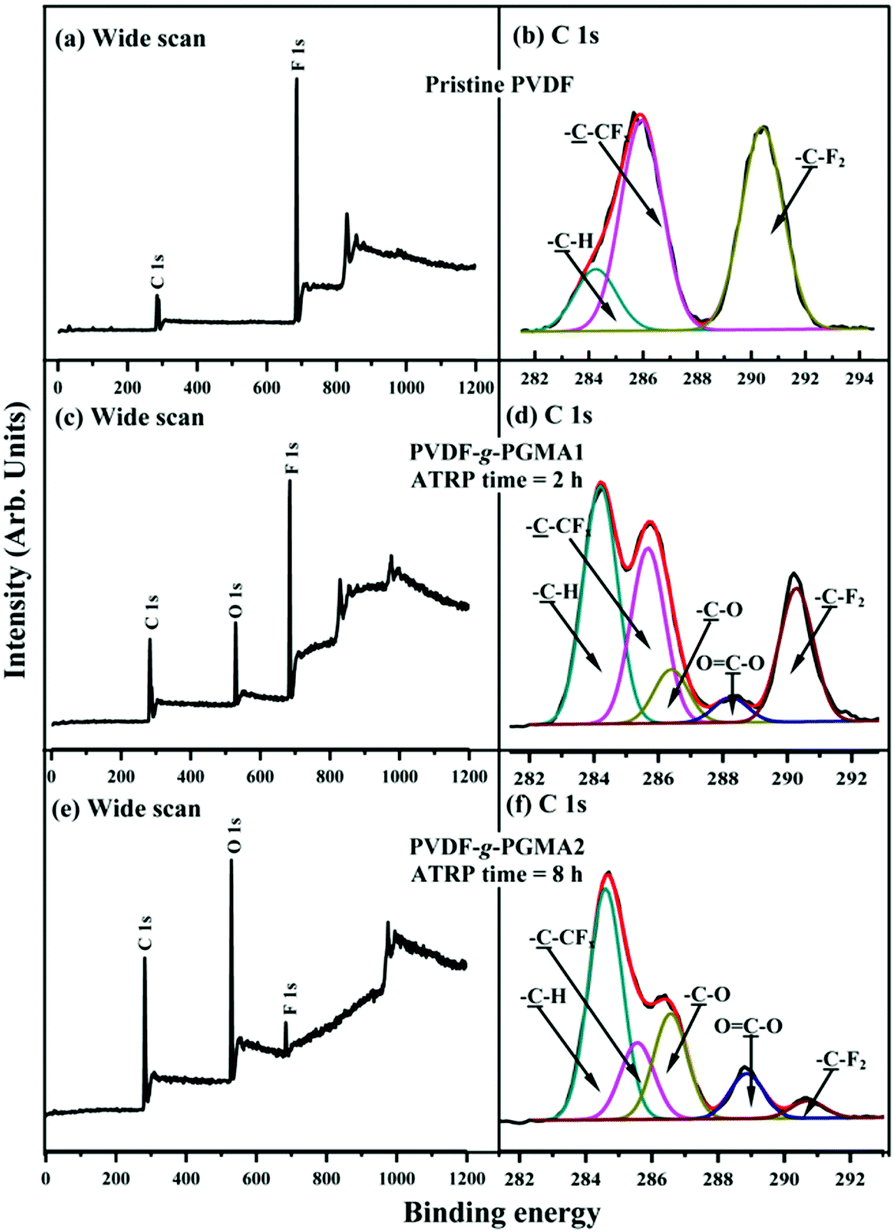 | ||
| Fig. 3 Wide scan and C 1s core-level XPS spectra of the (a and b) pristine PVDF film surface, (c and d) PVDF-g-PGMA1 surface from 2 h of ATRP time, and (e and f) PVDF-g-PGMA2 surfaces from 8 h of ATRP time. | ||
| Sample | GYd (μg cm−2) (mean ± SDe) | [N]/[C]f | WCAg (degree) | WCA images |
|---|---|---|---|---|
a
Reaction conditions: [GMA]:[CuBr]:[CuBr2]:[bpy] = 150:1:0.2:2 in the mixture of methanol and deionized water (4:1, v/v) at 80 °C.
b
Reaction conditions: the corresponding PVDF-g-P(GMA) surfaces incubated in a mixture DMF solution of NaN3 and NH4Cl (molar ratio 1:1) at 50° for 12 h.
c
Reaction conditions: [C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–RGD]:[CuSO4]:[sodium ascorbate] = 1:1:2 (molar ratio) in PBS solution at 50 °C for 24 h.
d Grafting yield (GY) is defined as GY = (Wa − Wb)/A, where Wa and Wb corresponds to the weight of the dry films before and after grafting of polymer brushes, respectively, and A is the film area (about 3.2 cm2).
e SD denotes standard deviation.
f Determined from the corresponding sensitivity factor-corrected element core-level spectral area ratios.
g WCA denotes static water contact angles. C–RGD]:[CuSO4]:[sodium ascorbate] = 1:1:2 (molar ratio) in PBS solution at 50 °C for 24 h.
d Grafting yield (GY) is defined as GY = (Wa − Wb)/A, where Wa and Wb corresponds to the weight of the dry films before and after grafting of polymer brushes, respectively, and A is the film area (about 3.2 cm2).
e SD denotes standard deviation.
f Determined from the corresponding sensitivity factor-corrected element core-level spectral area ratios.
g WCA denotes static water contact angles.
|
||||
| Pristine PVDF | — | — | 98 ± 3 |

|
| PVDF-g-PGMA1a | 3.63 ± 0.97 | — | 78 ± 4 |

|
| PVDF-g-PGMA2a | 9.79 ± 2.35 | — | 67 ± 3 |

|
| PVDF-g-PGMA1-N3b | 0.32 ± 0.13 | 0.023 | 61 ± 4 |

|
| PVDF-g-PGMA2-N3b | 0.86 ± 0.72 | 0.067 | 52 ± 3 |

|
| PVDF-g-PGMA1-click-RGDc | 0.97 ± 0.34 | 0.12 | 54 ± 3 |

|
| PVDF-g-PGMA2-click-RGDc | 2.19 ± 0.87 | 0.19 | 47 ± 4 |

|
In this study, the grafting yield (GY) was employed to evaluate the kinetics of PGMA chain growth directly from the PVDF surface. Fig. 4 shows the change in the GY values of the grafted PGMA brushes as a function of the ATRP reaction time. An approximate linear increase in the GY value of the grafted PGMA chains with polymerization time is clearly observed for the PVDF-g-PGMA surface, indicating that the growth of PGMA chains proceeds in a controlled manner by varying the reaction time. However, the fitted line deviates from zero due to the existence of an induction period at the beginning of the polymerization. The GY values of the PVDF-g-PGMA1 and PVDF-g-PGMA2 surfaces are about 3.63 ± 0.97 and 9.79 ± 2.35 μg cm−2 (Table 1), respectively. This is consistent well with the XPS results that the relative amount of the O–CO species in the curve-fitted C 1s spectrum and the [O–CO]/[–C–F2] area ratio increases with the increase in the graft polymerization time.
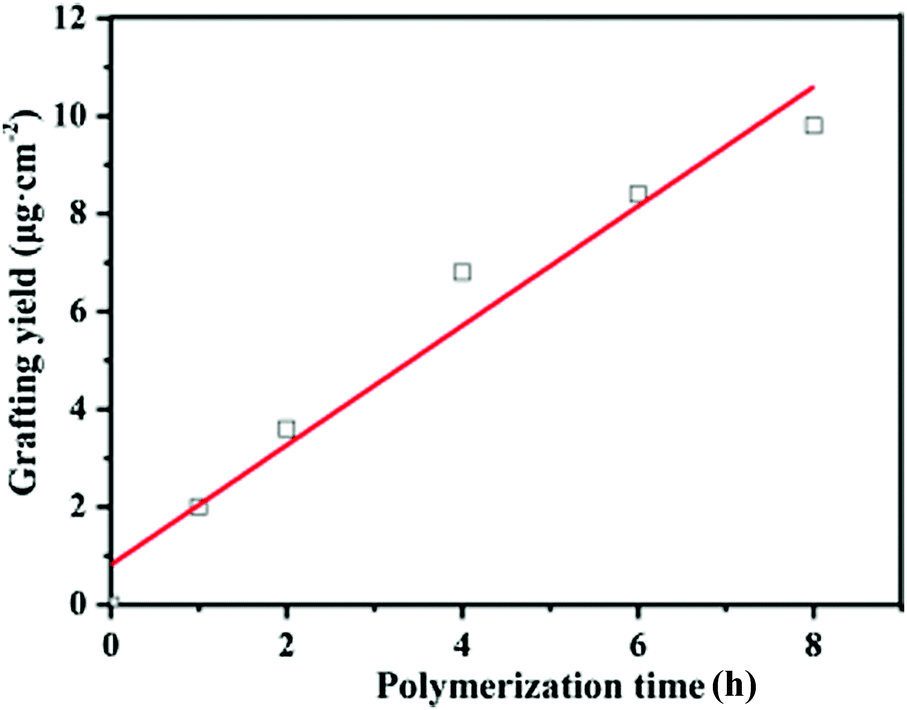 | ||
| Fig. 4 A linear relationship was observed for the grafting yield (GY) of the PGMA brushes formed on the PVDF-g-PGMA surface, as the result of direct surface-initiated ATRP of GMA from the PVDF surface. The polymer chain growth from the PVDF surface took place in a well-controlled manner by varying the reaction time. | ||
3.2 Azidation of the PGMA chains by the ring-open reaction with sodium azide
Due to bearing the pendant reactive epoxy groups in side chains, poly(glycidyl methacrylate) (PGMA) offers a very attractive platform to introduce complex functional moieties and/or to immobilize biomolecules via post-polymerization modification. To facilitate the direct functionalization of the PVDF-g-PGMA via azide-alkyne click reaction, the oxirane groups of the grafted PGMA chains take place the ring-open reaction by the presence of azide sodium, and thus resulting in the formation of reactive azido and hydroxyl groups. The former can be utilized for click reactions,60 while the latte for the improvement of hydrophilicity and the immobilization of biomolecules. The addition of a protic or Lewis acid to the azide-epoxide mixture prevents the hydroxyl functional groups from participating in side reactions when the reaction is carried out in an aqueous solutions.49 Parts (a) and (b) of Fig. 5 show the respective FTIR spectra of the PVDF-g-PGMA1-N3 and the PVDF-g-PGMA2-N3 surfaces. Upon opening of the oxirane rings of the GMA segments, an additional band with the wavenumber at 2130 cm−1, attributable to the stretching vibration of the azide groups in the PGMA-N3 side chains, is visible on the PVDF-g-PGMA1-N3 and the PVDF-g-PGMA2-N3 surfaces, respectively, as compared to the FTIR spectra of the PVDF-g-PGMA surface (Fig. 2b and d). The relatively stronger intensity of azide groups on the PVDF-g-PGMA2-N3 surface than that of the PVDF-g-PGMA1-N3 surface suggests a higher surface density of azide groups on the PVDF-g-PGMA2-N3 surface (inset of Fig. 5a and b). The appearance of another new absorption band at the wavenumber of 3300 cm−1, characteristics of the hydroxyl groups of PGMA-N3 side chains, also confirms the success in opening of the oxirane groups by sodium azide (Fig. 5a and b).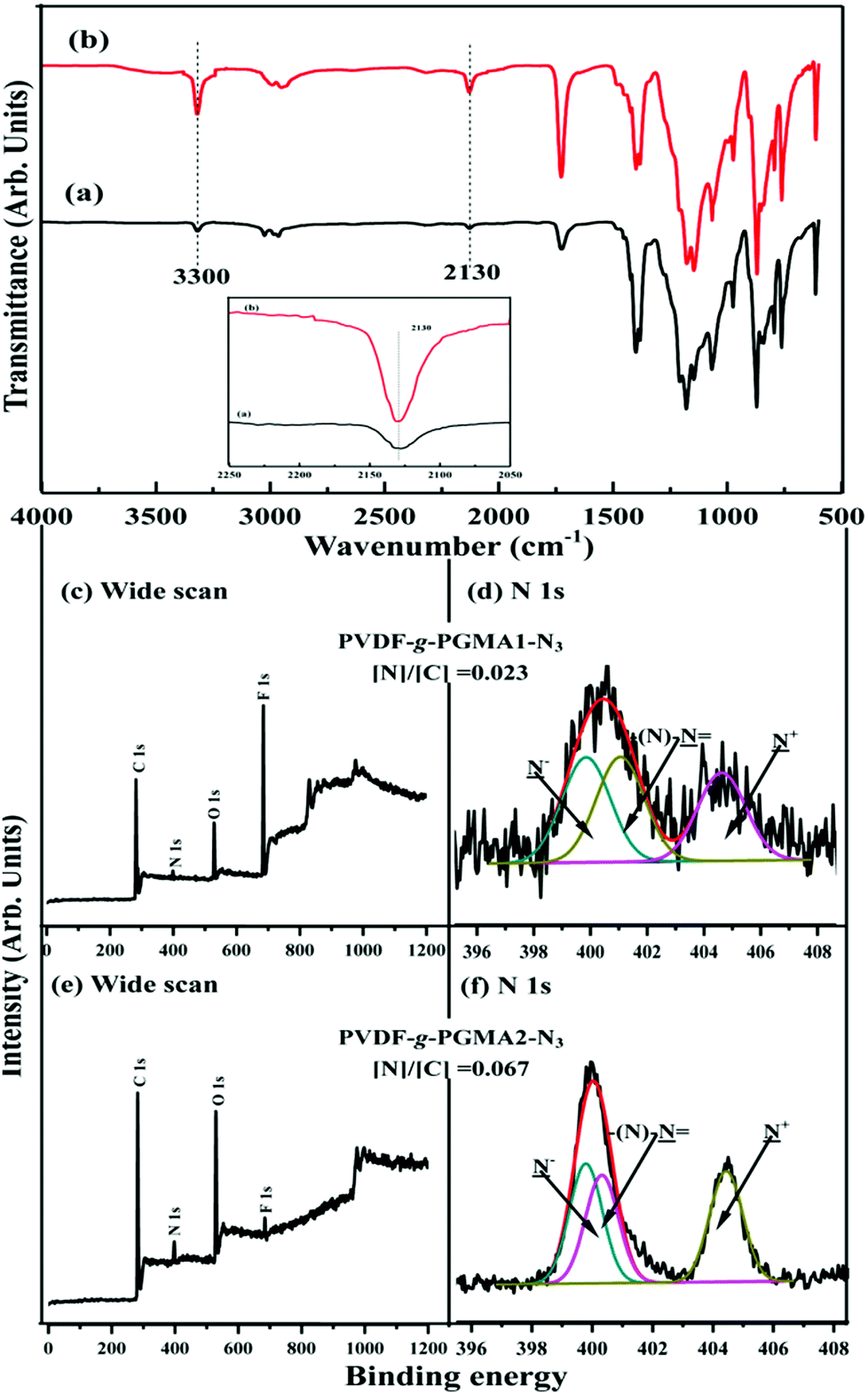 | ||
| Fig. 5 FTIR and XPS spectra of the (a, c and d) PVDF-g-PGMA1-N3 (from PVDF-g-PGMA1), (b, e and f) PVDF-g-PGMA2-N3 (from PVDF-g-PGMA2) surfaces. Inset shows an enlarged view of the 2250–2050 cm−1 region of the FTIR spectra. | ||
The chemical composition of the azide-functionalized PVDF surface was also ascertained by XPS characterization. Parts (c–f) of Fig. 5 show the respective wide scan and N 1s core-level XPS spectra of the PVDF-g-PGMA1-N3 and the PVDF-g-PGMA2-N3 surfaces. The appearance of an additional N 1s signal (at a BE of about 400 eV) in the wide scan spectra is associated with the introduced azide groups on the PGMA side chains, because of the absence of N signal in the PVDF and PGMA (Fig. 5c and e). The curve-fitted N 1s core-level spectra of the PVDF-g-PGMA-N3 surfaces consists of three components with BEs at 399.9, 401.2 and 404.6 eV, attributable to the negatively charged nitrogen (–N−), the imine nitrogen (–N), and the positively charged nitrogen (–N+), respectively (Fig. 5d and f). The molar ratio of the three peak components is about 1:1:1, as determined from the spectral area ratio of the three species, consistent with the theoretical ratio for the chemical structure of azide groups (–NN+N−). Furthermore, the [N]/[C] ratios for the PVDF-g-PGMA1-N3 and the PVDF-g-PGMA2-N3 surfaces, determined from the sensitivity-factor-corrected C 1s and N 1s spectral areas, are found to be about 0.023 and 0.067, respectively, which is further evidence that the surface density of the azide groups on the PVDF-g-PGMA2-N3 surface is higher than that of the PVDF-g-PGMA1-N3 surface. The static water contact angles of the PVDF-g-PGMA1-N3 and PVDF-g-PGMA2-N3 surfaces decrease to about 52 ± 3° and 61 ± 4° (Table 1), respectively, because of the surface concentration of hydroxyl groups generated in the ring-open reaction of the oxirane groups of the grafted PGMA chains. Thus, the azide groups are successfully introduced to the PGMA chains by the ring-open reactions with sodium azide, and their surface density is dependent on the surface concentration of the grafted PGMA brushes.
3.3 Immobilization of RGD peptide via click reaction
Arginine–glycine–aspartic acid (RGD), as a primary amino acid sequence in the extracellular matrix protein, is vital for integrin receptor mediated cell attachment.53 Integrin-mediated binding of cells is the foundation for cell growth and differentiation and is the dominant mechanism by which cells communicate with noncellular surroundings.61 The Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reactions are excellent candidates to functionalize polymeric materials due to their high yields and specificity.62 In this study, the alkyne-functionalized RGD peptide was immobilized onto the azide-termined PGMA brushes via the Cu(I)-catalyzed azide-alkyne click reactions to produce the PVDF-g-PGMA-click-RGD surfaces. Fig. 6a and b show the FTIR spectra of the PVDF-g-PGMA1-click-RGD and the PVDF-g-PGMA2-click-RGD surface, respectively. The successful immobilization of RGD peptide onto the azide-terminated PGMA brushes can be deduced from the appearance of three additional bands at 3398 cm−1 for the combined N–H and O–H stretching vibration, 1658 cm−1 for the amide I (CONH) and 1560 cm−1 for the amide II (N–H bending vibration),56 as well as the disappearance of the absorption bands of the azide groups. Moreover, the relative intensity of these three new bands for the PVDF-g-PGMA2-click-RGD surface is noticeably higher than that for the PVDF-g-PGMA1-click-RGD surfaces (inset of Fig. 6a and b), indicative of the high surface density of the immobilized RGD peptide on the former sample surface.
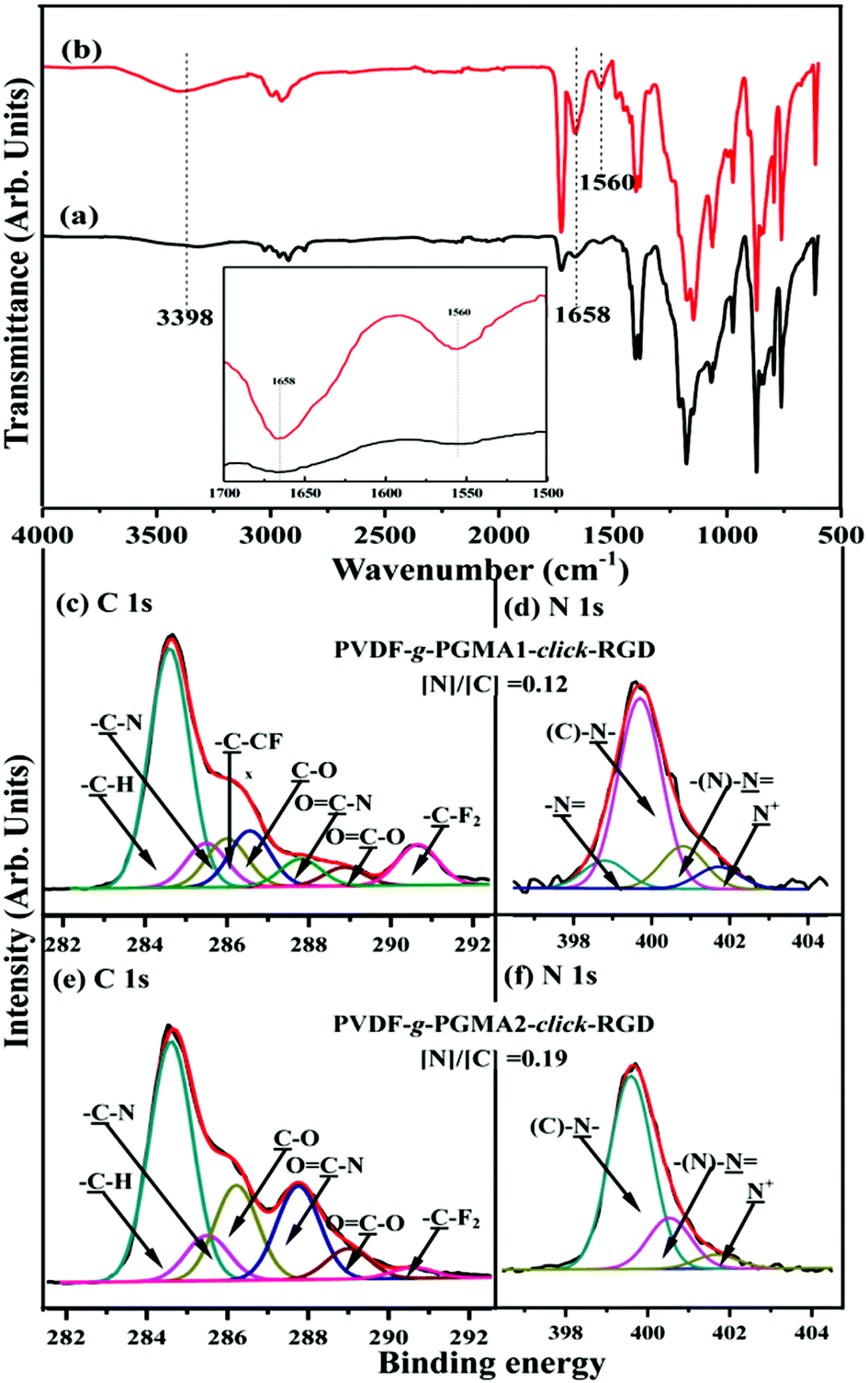 | ||
| Fig. 6 FTIR and XPS spectra of the (a, c and d) PVDF-g-PGMA1-click-RGD (from PVDF-g-PGMA1-N3), (b, e and f) P PVDF-g-PGMA2-click-RGD (from PVDF-g-PGMA2-N3) surfaces. Inset shows an enlarged view of the 2250–2050 cm−1 region of the FTIR spectra. | ||
The RGD-immobilized PVDF surfaces were also characterized by XPS, and the results are shown in Fig. 6c–f. For the PVDF-g-PGMA1-click-RGD surface, the curve-fitted C 1s core-level spectrum consists of seven peak components with BEs at 284.6, 285.5, 285.9, 286.6, 287.8, 288.8 and 290.6 eV, attributable to the –C–H, –C–N, –C–CFx, –C–O, OC–N, OC–O and –CF2 species, respectively (Fig. 6c). The C–N species is associated with the linkages in RGD peptide itself, as well the linkage between the PGMA chains and RGD peptide, while the OC–N and OC–O species are ascribed to the peptide bonds in RGD peptide.55 The N 1s core-level spectrum is curve-fitted into four peak components for the amine groups (–N) at 398.4 eV, the C–N species of the triazole rings at 399.7 eV, the imine moiety of the triazole rings at 400.6 eV and the residual N+ species at 401.8 eV (Fig. 6d).59 In the case of the PVDF-g-PGMA2-click-RGD surface, the disappearance of the –C–CFx and –CF2 species indicates the high surface density of the immobilized RGD peptide (Fig. 6e). This is consistent with the increase in the [N]/[C] ratio of 0.12 for the PVDF-g-PGMA1-click-RGD surface to 0.19 for the PVDF-g-PGMA2-click-RGD surface. Furthermore, the grafting densities of RGD peptide are calculated to be 0.94 ± 0.34 and 2.19 ± 0.87 μg cm−2, respectively, for the PVDF-g-PGMA1-click-RGD and PVDF-g-PGMA2-click-RGD surfaces (Table 1). The surface hydrophility of the RGD immobilized surface undergoes a further improvement, as the static water contact angles decrease to about 54 ± 3° for the PVDF-g-PGMA1-click-RGD surface and 47 ± 4° for the PVDF-g-PGMA2-click-RGD surface (Table 1). This is consistent with the presence of enriched carboxyl groups (-COOH) and amino groups (–NH2) in the chemical molecules of RGD peptide. The above results are consistent with the successful immobilization of RGD peptide on the PVDF surface, and the surface density of immobilized RGD is positively correlated with the surface grafting concentration of the PGMA brushes.
3.4 Surface topography
The changes in surface topography of the PVDF films after each modification step were characterized by atomic force microscopy (AFM). Fig. 7 shows the representative AFM height images of the pristine PVDF and functionalized PVDF film surfaces with scanned areas of 10 μm × 10 μm. The pristine PVDF is relatively uniform and smooth with the root-mean-square (RMS) surface roughness values of about 6.74 nm (Fig. 7a). Upon grafting of PGMA brushes, the surface roughness of the PVDF films shows an obvious increase with the increase in the graft polymerization reaction time, as the RMS values of the PGMA-grafted PVDF film increase from only 10.12 nm from 2 h of ATRP reaction (Fig. 7b) to about 31.24 nm from 8 h of ATRP reaction (Fig. 7c). This result is consistent with the previous findings that the grafting of the polymeric brushes can lead to a significant increase in surface roughness value, as well as surface morphology.32 A further slight increase in surface roughness is caused by the immobilization of RGD peptide onto the PGMA side chains, since the corresponding RMS value increases to 13.23 nm and 37.24 nm, respectively, for the PVDF-g-P(GMA)1-click-RGD and PVDF-g-P(GMA)2-click-RGD surfaces (Fig. 7d and e). The immobilization of RGD peptide can broaden the PGMA domains on the PVDF film surface, thus resulting in a further increase in the surface roughness. As a consequence, the surface topography measured by AFM is an alternative way to ascertain successful surface modification of the PVDF film.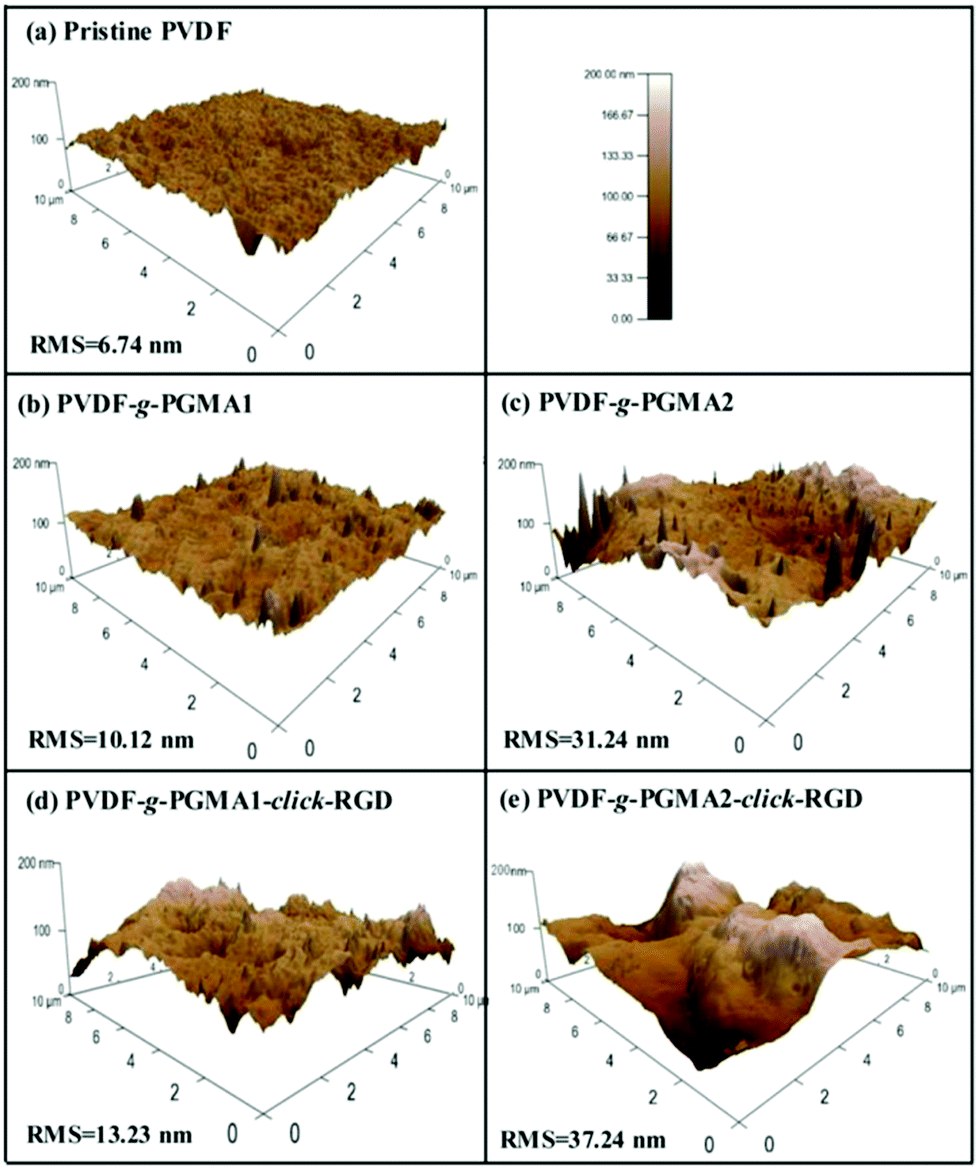 | ||
| Fig. 7 Surface topography determined by representative AFM images of the (a) pristine PVDF film surface, (b) PVDF-g-PGMA1 surface from 2 h of ATRP time, (c) PVDF-g-PGMA2 surface from 8 h of ATRP time, (d) PVDF-g-PGMA1-click-RGD surface, and (e) PVDF-g-PGMA2-click-RGD surface. The root-mean-squared (RMS) surface roughness of the different PVDF substrates was obtained over a scan size of 10 mm × 10 mm. | ||
SEM images of the pristine PVDF and functionalized PVDF film surfaces were also obtained to determine the change in surface microstructures after surface modification (ESI, Fig. S1†). Obviously, the pristine PVDF film shows a smooth surface (Fig. S1†), and no microporous structure can be observed from the cross-sectional view (Fig. S1b†). Upon grafting of PGMA polymer brushes, the surface morphology of PVDF films becomes rougher than that of the pristine PVDF surface (Fig. S1c and d†). The conjugation of RGD peptide to PGMA polymer brushes via click reaction can not only further increase the surface roughness, but also give rises to a porous surface microstructure (Fig. S1e and f†). However, the surface modification cannot alter the bulk structure of the PVDF film, as revealed by the cross-section images of the PVDF-g-P(GMA)2-click-RGD film (Fig. S1g†). As a result, these results are well consistent with those from the observation of AFM images.
3.5 Improved cell adhesion on the RGD-functionalized PVDF surfaces
Cell adhesion is the first event of cell–biomaterial interaction after cells are plated on the substrates, which is medicated by cell membrane receptors (mainly from the integrin superfamily). The cell attachment and spreading properties of various PVDF substrates were investigated by seeding adipose tissue-derived stem cells (ASCs) on the films for 24 h of incubation in a 5% CO2 atmosphere, and the representative SEM images are shown in Fig. 8. The pristine PVDF film surface shows the least conductive for supporting cellular interactions, since almost no ASCs are discernible on the film surface (Fig. 8a). This result is consistent with the previous findings that the PVDF substrate is unfavorable for cell adhesion and proliferation due to its intrinsic hydrophobic nature and the lack of cell-recognition motifs.39,63 In spite of the noticeable changes in surface hydrophilicity and roughness achieved after the grafting of PGMA brushes, no significant improvement in initial cell adhesion can be observed on the PVDF-g-PGMA2 surface relative to the pristine PVDF surfaces (Fig. 8b), indicating that other factors, such as biological cues, play a vital role in triggering positive cell interactions. Upon immobilization of the cell adhesive RGD peptide, a substantial improvement in initial cell adhesion is observed on the PVDF-g-PGMA1-click-RGD and the PVDF-g-PGMA2-click-RGD surfaces (Fig. 8c and e). Virtually, more ASCs are present on the PVDF-g-PGMA2-click-RGD surface than the PVDF-g-PGMA1-click-RGD surface, implying that the cell adhesion is positively correlated with the surface concentration of immobilized RGD peptides. Moreover, the attached ASCs exhibit good spreading behavior on the RGD-immobilized PVDF film surfaces (Fig. 8d and f), especially for the PVDF-g-PGMA2-click-RGD surface, on which ASCs show a more elongated ribbon shape than other substrates, and microfilaments are clearly observed around the cells (Fig. 8f). Generally, anchorage-dependent cells, such as ASCs, need to strongly adhere to substrates, in order to survive, spread, and transfer signals into the cytosol to maintain cell homeostasis. The extent of cell spreading on substrates plays a crucial role in cell proliferation. The above results demonstrates that cell adhesion and spreading of ASCs on the PVDF film surfaces are significantly improved as a result of the positive cell–material interactions from the presence of the bioactive RGD peptide motifs. | ||
| Fig. 8 SEM images of adipose tissue-derived stem cells (ASCs) adhered on the (a) pristine PVDF, (b) PVDF-g-PGMA2 surface from 8 h of ATRP time, (c and d) PVDF-g-PGMA1-click-RGD and (e and f) PVDF-g-PGMA2-click-RGD surfaces after 24 h of cell culture in a 5% CO2 atmosphere at a seeding density of 104 cells per cm2. | ||
Shape, adhesion, mobility and stability of cells are connected with the formation of the actin cytoskeleton.64 In this study, cell nuclei and F-actin were counter-stained to qualitatively determine relative adherent cell numbers and spreading areas of ASCs on the film surfaces. Fig. 9 shows the respective DAPI and F-action stained ASCs on the pristine PVDF, PVDF-g-PGMA2, PVDF-g-PGMA1-click-RGD, and PVDF-g-PGMA2-click-RGD surfaces. To accurately visualize the relative adherent cell numbers through the DAPI-labelled ASCs, the fluorescence images of DAPI-stained ASCs adhered on the pristine PVDF and surface functionalized PVDF surfaces were obtained (ESI, Fig. S2†). It is readily to conclude that the relative cell adhesion on the RGD-immobilized PVDF films is significantly greater than those on the pristine and PGMA-grafted film surfaces (Fig. S2†), and that the relative adherent cell numbers of ASCs on the RGD-immobilized PVDF surfaces are connected with the surface concentration of the immobilized RGD peptide, as a more pronounced cell adhesion is observed on the PVDF-g-PGMA2-click-RGD surface over the PVDF-g-PGMA1-click-RGD surface (Fig. S2c and d†). As shown by F-actin stained fluorescence images, the spreading areas of the attached ASCs on the RGD-immobilized PVDV surfaces are substantially enhanced relative to the pristine PVDF and PGMA-grafted PVDF surfaces (Fig. 9). In addition, the ASCs observed on the RGD-immobilized PVDF are spread out to a large extent and maintain the polygonal morphology with clear observation of actin filaments (Fig. 9c and d). Cells with pronounced actin stressed fibres are associated with the RGD peptide motifs immobilized on the film surface. These results are in good agreement with the above SEM results that the RGD peptides immobilized on the PVDF films can significantly enhance initial cell adhesion and spreading with the formation of the actin cytoskeleton.
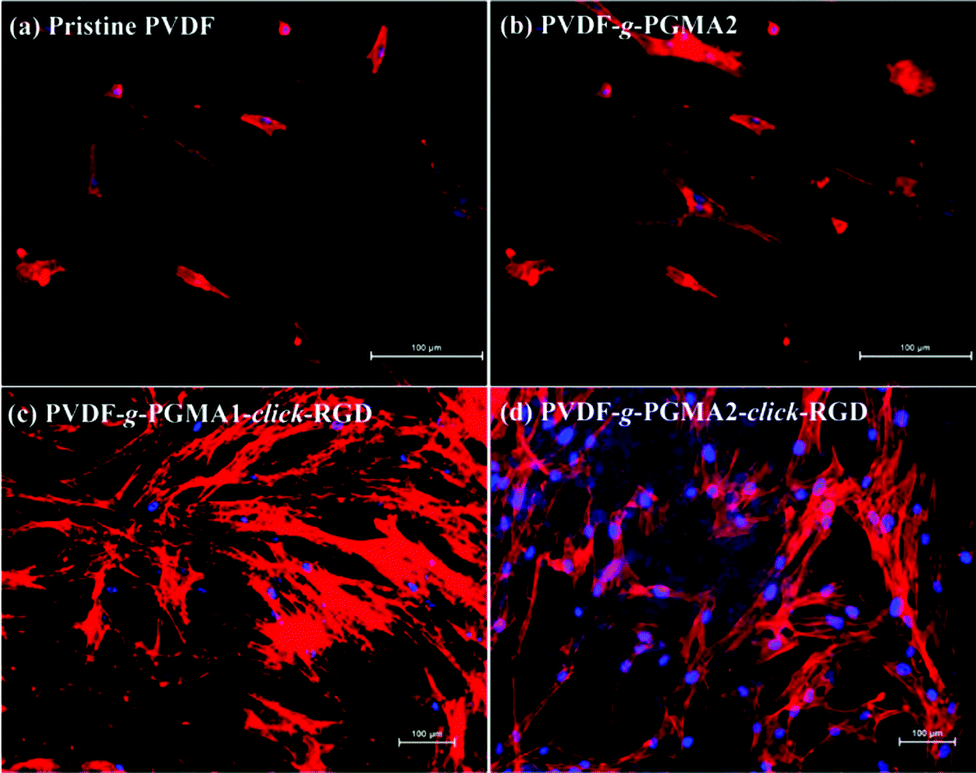 | ||
| Fig. 9 Fluorescence images of DAPI and F-actin stained ASCs adhered on the (a) pristine PVDF, (b) PVDF-g-PGMA2, (c) PVDF-g-PGMA1-click-RGD, and (d) PVDF-g-PGMA2-click-RGD surfaces after 24 h of culture at a seeding density of 104 cells per cm2. The adhered cells were fluorescently stained to label parallin and nuclei for relative adherent cell numbers and spreading areas of ASCs. | ||
3.6 RGD Peptide functionalization improved cell proliferation
The effect of RGD peptide functionalization on cell proliferation was investigated using the AlamarBlue® (AB) assay by seeding ASCs onto different PVDF film surfaces, and the cell proliferation profiles are shown in Fig. 10. The pristine PVDF surface exhibits a poor cell affinity and growth of ASCs, as the only marginal increase in the cell numbers between 1 day and 7 days of cell culture. This result further confirms the poor cytocompatibility of the pristine PVDF surface due to its intrinsic hydrophobicity. It has been widely reported that polymer surfaces with a moderate hydrophilicity of water contact angles in a range of 40–70° and rougher nanostructured topography are favorable for cell adhesion and proliferation.65 Although the surface hydrophilicity and roughness undergo a significant improvement for the PGMA-grafted PVDF surface as compared to the pristine PVDF surface, no positive effect on cell proliferation can be observed on the PVDF-g-PGMA1 and PVDF-g-PGMA2 surfaces.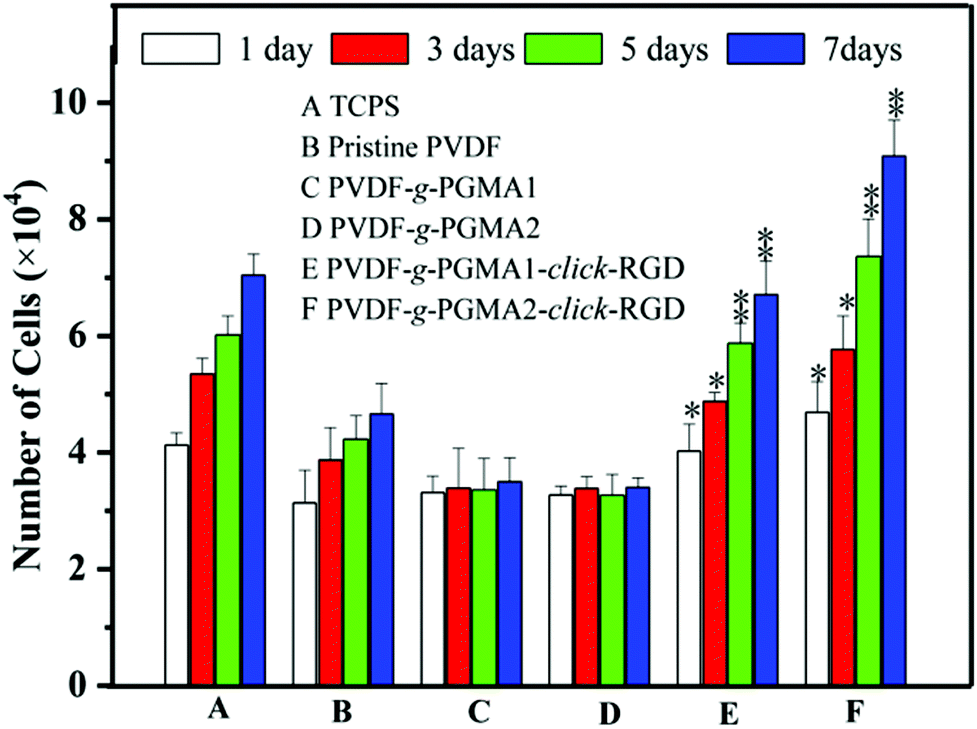 | ||
| Fig. 10 Cell proliferation profiles of the ASCs cultured on different film surfaces after 1, 3, 5 and 7 days of cell culture at 37 °C in a 5% CO2 atmosphere as determined by the AlamarBlue™ assay. TCPS was used as a control. Data is represented as mean ± SD. * p < 0.05 and ** p < 0.01 refer to the statistically significant difference compared with the pristine PVDF. | ||
The grafting of PGMA brushes onto the PVDF film surface appears to have a detrimental impact on cell proliferation, since the number of cells on the PVDF-g-PGMA1 and PVDF-g-PGMA2 surfaces is even less than that on the pristine PVDF surfaces. This result is consistent with the fact that the epoxy groups are usually cytotoxic and mutagenic to cells.66 However, the cell proliferation rates of the RGD peptide functionalized PVDF surfaces, including the PVDF-g-PGMA1-click-RGD and the PVDF-g-PGMA2-click-RGD surfaces, are comparable to that of the TCPS positive control. Furthermore, the number of ASCs on the PVDF-g-PGMA2-click-RGD surface (with a RGD concentration of 2.19 ± 0.87 μg cm−2) is significantly higher than that on the PVDF-g-PGMA1-click-RGD surface (with a RGD concentration of 0.97 ± 0.34 μg cm−2), implying that the cell proliferation rate is positively correlated with the surface concentration of immobilized RGD peptide on the PVDF surfaces. Since the RGD peptides are clicked to the azide-terminated GMA unit by forming 1,4-substituted 1,2,3-triazole, the RGD peptides immobilized on the PGMA brushes remain in a well-defined dispersed manner rather than in a continuous surface layer. Thus, the higher surface density of RGD peptide on the PVDF-g-PGMA2-click-RGD surface results in a surface with a moderate hydrophilicity (47 ± 4°), a rougher nano-structured topography (37.24 nm) and abundant epitopes or ligands for cell adhesion and growth (2.19 ± 0.87 μg cm−2), which are all associated with the dramatic improvement in cell proliferation rate on the PVDF-g-PGMA2-click-RGD surface.
3.7 Cell imaging
After 7 days of incubation, LIVE/DEAD staining was performed to qualitatively evaluate the viability and coverage of ASCs on the different PVDF film surfaces, and the representative fluorescence images are shown in Fig. 11. The pristine PVDF surface shows poor cell affinity and growth behavior, since ASCs are sparsely distributed over the entire film surface (Fig. 11b). Similarly, the PGMA-grafted PVDF film surfaces show no improvement in cell attachment and growth as compared to the pristine PVDF films (Fig. 11 and d). The even lower number of attached ASCs and poor spreading behavior on the PVDF-g-PGMA1 and the PVDF-g-PGMA2 surfaces further confirms the cytotoxicity of epoxy groups to cells. However, ASCs attach and spread readily on the RGD-functionalized PVDF film surfaces (Fig. 11e and f), forming a confluent monolayer, indicating that the cell attachment and proliferation on the RGD peptide-immobilized film surfaces are substantially improved (Fig. 11e and f). Moreover, the coverage extent of ASCs on the PVDF-g-PGMA2-click-RGD surface is evidently higher than that on the PVDF-g-PGMA1-click-RGD surface, suggesting that cell proliferation of ASCs is positively correlated to the surface concentration of the immobilized RGD peptide on the film surface. Hence, an optimally functionalized inert-biomaterial surface, such as the PVDF-g-PGMA2-click-RGD surface, by the combination process of surface-initiated ATRP and click reaction is able to trigger the rapid cell attachment and proliferation and is potentially useful for biomedical applications.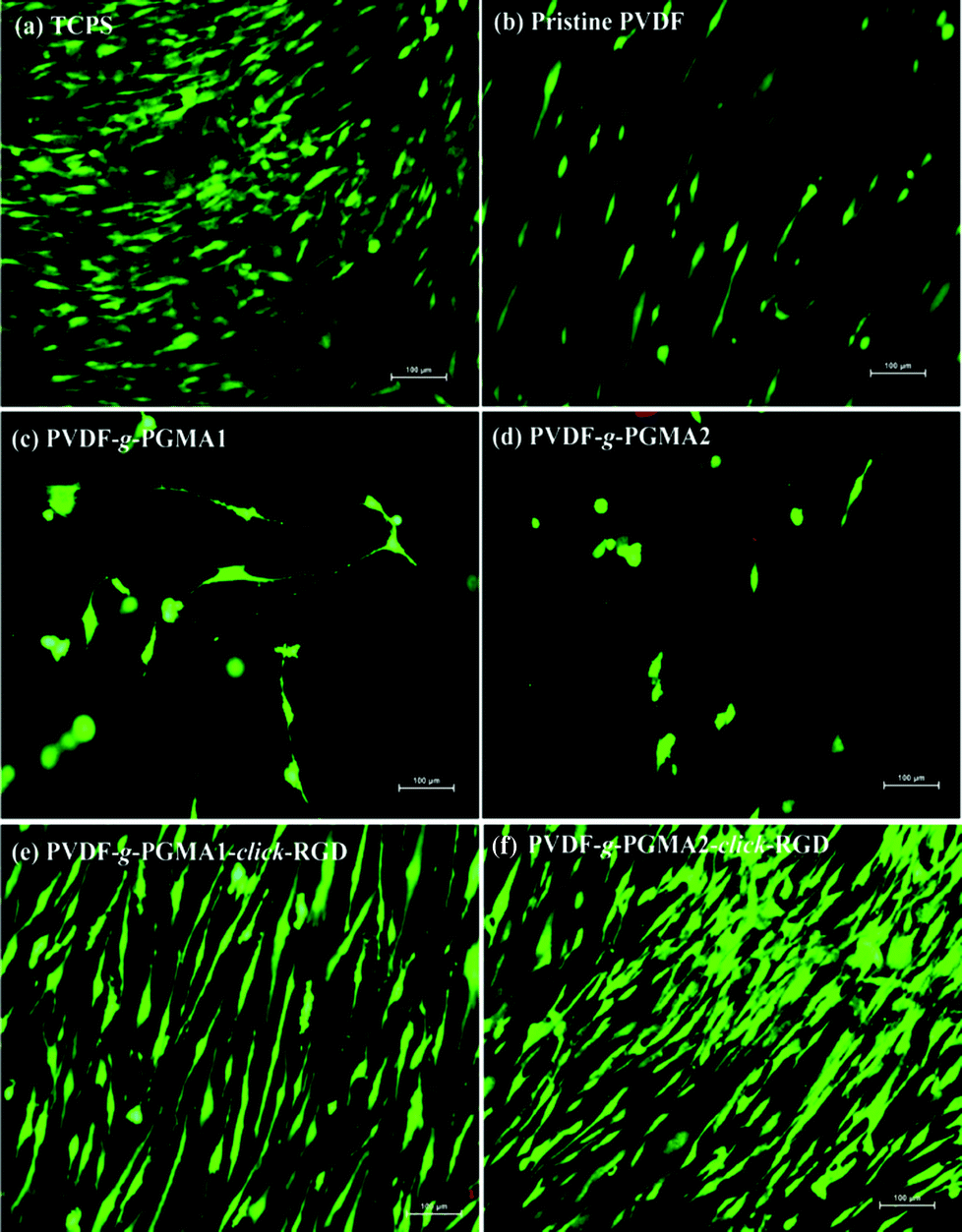 | ||
| Fig. 11 Fluorescence images of LIVE/DEAD-stained cells on the (a) TCPS, (b) pristine PVDF, (c) PVDF-g-PGMA1, (d) PVDF-g-PGMA2, (e) PVDF-g-PGMA1-click-RGD and (f) PVDF-g-PGMA2-click-RGD surfaces after 7 days of cell culture. Viable cells emit a green fluorescence, whilst dead cells emit a red fluorescence. Scale bar: 100 μm. | ||
4. Conclusions
The modification of the PVDF film surface by direct surface-initiated ATRP of GMA from the secondary fluorine atoms on the PVDF backbone was successfully performed to create new surface with tunable functional brushes containing pendant active epoxy groups. The grafting density of PGMA brushes was consistent with a controlled manner to increase linearly with the polymerization time. The ring-open reaction of the oxirane groups with sodium azide was allowed to introduce the azide groups onto the PGMA side chains for the subsequent conjugation of RGD peptide via alkyne-azide click reaction. The as-synthesized PVDF-g-PGMA-click-RGD surfaces were found to promote the cell attachment and proliferation of adipose-derived stem cells (ASCs), as compared to the pristine PVDF and PGMA-grafted film surfaces. In vitro cell studies also demonstrated that the cell proliferation rate and coverage of ASCs on the film surface are positively correlated to the surface concentration of the clicked RGD peptide on the PGMA side chains. The current RGD peptide functionalized PVDF surface provided a rational combination of the required components for biomedical applications, which includes surface-bearing biological active motifs of RGD peptide and superior mechanical strength and durability of PVDF backbone.Acknowledgements
The authors would like to acknowledge the financial assistance of key project of National Natural Science Foundation of China (no. 21236004) and the Singapore National Research Foundation under the CREATE program: the Regenerative Medicine Initiative in Cardiac Restoration Therapy (NRF-Technion).Notes and references
- J. Heuts, J. Salber, A. M. Goldyn, R. Janser, M. Moller and D. Klee, J. Biomed. Mater. Res., Part A, 2010, 92, 1538–1551 Search PubMed.
- H. Scheidbach, C. Tamme, A. Tannapfel, H. Lippert and F. Köckerling, Surg. Endosc., 2004, 18, 211–220 CrossRef CAS PubMed.
- A. Prudente, C. L. Z. Riccetto, M. M. de Souza Godoy Simões, B. M. Pires and M. G. de Oliveira, Colloids Surf., B, 2013, 108, 178–184 CrossRef CAS PubMed.
- C. A. Nava-Ortíz, C. Alvarez-Lorenzo, E. Bucio, A. Concheiro and G. Burillo, Int. J. Pharm., 2009, 382, 183–191 CrossRef PubMed.
- Z. Ma, M. Kotaki, T. Yong, W. He and S. Ramakrishna, Biomaterials, 2005, 26, 2527–2536 CrossRef CAS PubMed.
- Z. Fang, Y. Qiu and Y. Luo, J. Phys. D: Appl. Phys., 2003, 36, 2980 CrossRef CAS.
- A. Nandakumar, Z. T. Birgani, D. Santos, A. Mentink, N. Auffermann, K. van der Werf, M. Bennink, L. Moroni, C. van Blitterswijk and P. Habibovic, Biofabrication, 2013, 5, 015006 CrossRef CAS PubMed.
- H. Hjort, T. Mathisen, A. Alves, G. Clermont and J. Boutrand, Hernia, 2012, 16, 191–197 CrossRef CAS PubMed.
- G. Laroche, Y. Marois, R. Guidoin, M. W. King, L. Martin, T. How and Y. Douville, J. Biomed. Mater. Res., 1995, 29, 1525–1536 CrossRef CAS PubMed.
- U. Klinge, B. Klosterhalfen, A. Öttinger, K. Junge and V. Schumpelick, Biomaterials, 2002, 23, 3487–3493 CrossRef CAS PubMed.
- E. Vinard, R. Eloy, J. Descotes, J. R. Brudon, H. Guidicelli, J. L. Magne, P. Patra, R. Berruet, A. Huc and J. Chauchard, J. Biomed. Mater. Res., 1988, 22, 633–648 CrossRef CAS PubMed.
- Y. Gustafsson, J. Haag, P. Jungebluth, V. Lundin, M. L. Lim, S. Baiguera, F. Ajalloueian, C. Del Gaudio, A. Bianco, G. Moll, S. Sjoqvist, G. Lemon, A. I. Teixeira and P. Macchiarini, Biomaterials, 2012, 33, 8094–8103 CrossRef CAS PubMed.
- A. Asatekin, S. Kang, M. Elimelech and A. M. Mayes, J. Membr. Sci., 2007, 298, 136–146 CrossRef CAS.
- Y. H. Zhao, B. K. Zhu, L. Kong and Y. Y. Xu, Langmuir, 2007, 23, 5779–5786 CrossRef CAS PubMed.
- M. Moghareh Abed, S. Kumbharkar, A. Groth and K. Li, Sep. Purif. Technol., 2012, 106, 47–55 CrossRef.
- D. Klee, Z. Ademovic, A. Bosserhoff, H. Hoecker, G. Maziolis and H. J. Erli, Biomaterials, 2003, 24, 3663–3670 CrossRef CAS PubMed.
- T. Cai, R. Wang, W. J. Yang, S. Lu, K. G. Neoh and E. T. Kang, Soft Matter, 2011, 7, 11133 RSC.
- Y. Chang, Y. J. Shih, C. Y. Ko, J. F. Jhong, Y. L. Liu and T. C. Wei, Langmuir, 2011, 27, 5445–5455 CrossRef CAS PubMed.
- V. M. Kochkodan and V. K. Sharma, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2012, 47, 1713–1727 CrossRef CAS PubMed.
- M. C. Clochard, J. Begue, A. Lafon, D. Caldemaison, C. Bittencourt, J. J. Pireaux and N. Betz, Polymer, 2004, 45, 8683–8694 CrossRef CAS.
- T. R. Dargaville, G. A. George, D. J. Hill and A. K. Whittaker, Prog. Polym. Sci., 2003, 28, 1355–1376 CrossRef CAS.
- L. Matienzo, J. Zimmerman and F. Egitto, J. Vac. Sci. Technol., A, 1994, 12, 2662–2671 CAS.
- A. Rahimpour, S. Madaeni, S. Zereshki and Y. Mansourpanah, Appl. Surf. Sci., 2009, 255, 7455–7461 CrossRef CAS.
- S. J. You, G. U. Semblante, S. C. Lu, R. A. Damodar and T. C. Wei, J. Hazard. Mater., 2012, 238, 10–19 CrossRef PubMed.
- S. Liang, Y. Kang, A. Tiraferri, E. P. Giannelis, X. Huang and M. Elimelech, ACS Appl. Mater. Interfaces, 2013, 5, 6694–6703 CAS.
- N. A. Hashim, F. Liu and K. Li, J. Membr. Sci., 2009, 345, 134–141 CrossRef CAS.
- A. Takahashi and H. Hisatomi, Mol. Med. Rep., 2009, 2, 749–752 CAS.
- Y. Chang, W. J. Chang, Y. J. Shih, T. C. Wei and G. H. Hsiue, ACS Appl. Mater. Interfaces, 2011, 3, 1228–1237 CAS.
- Q. Li, Q. Y. Bi, B. Zhou and X. L. Wang, Appl. Surf. Sci., 2012, 258, 4707–4717 CrossRef CAS.
- A. Olivier, F. Meyer, J.-M. Raquez, P. Damman and P. Dubois, Prog. Polym. Sci., 2012, 37, 157–181 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H. A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- N. Singh, S. M. Husson, B. Zdyrko and I. Luzinov, J. Membr. Sci., 2005, 262, 81–90 CrossRef CAS.
- J. Hester, P. Banerjee, Y. Y. Won, A. Akthakul, M. Acar and A. Mayes, Macromolecules, 2002, 35, 7652–7661 CrossRef CAS.
- M. Zhang and T. P. Russell, Macromolecules, 2006, 39, 3531–3539 CrossRef CAS.
- Y. Chen, D. Liu, Q. Deng, X. He and X. Wang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3434–3443 CrossRef CAS.
- Y. Chen, J. Xiao, W. Zhou, Q. Deng, H. Nie, M. Wan and F. Bai, Surf. Rev. Lett., 2009, 16, 111–121 CrossRef CAS.
- J. Yuan, J. Q. Meng, Y. I. Kang, Q. Y. Du and Y. F. Zhang, Appl. Surf. Sci., 2012, 258, 2856–2863 CrossRef CAS.
- Y. W. Kim, D. K. Lee, K. J. Lee and J. H. Kim, Eur. Polym. J., 2008, 44, 932–939 CrossRef CAS.
- Z. Ke, B. Dai, L. Li, G. Yan and D. Zhou, J. Appl. Polym. Sci., 2010, 115, 976–980 CrossRef CAS.
- C. J. Galvin and J. Genzer, Prog. Polym. Sci., 2012, 37, 871–906 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- H. Bao, L. Li, L. H. Gan, Y. Ping, J. Li and P. Ravi, Macromolecules, 2010, 43, 5679–5687 CrossRef CAS.
- G. K. Such, A. P. Johnston, K. Liang and F. Caruso, Prog. Polym. Sci., 2012, 37, 985–1003 CrossRef CAS.
- A. Bayraktar, B. Saraçoğlu, Ç. Gölgelioğlu and A. Tuncel, J. Colloid Interface Sci., 2012, 365, 63–71 CrossRef CAS PubMed.
- F. Reyes-Ortega, F. J. Parra-Ruiz, S. E. Averick, G. Rodríguez, M. R. Aguilar, K. Matyjaszewski and J. San Román, Polym. Chem., 2013, 4, 2800–2814 RSC.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- C. Huang, K. G. Neoh and E. T. Kang, Langmuir, 2011, 28, 563–571 CrossRef PubMed.
- T. Cai, W. J. Yang, K.-G. Neoh and E.-T. Kang, Ind. Eng. Chem. Res., 2012, 51, 15962–15973 CrossRef CAS.
- A. Lotz, M. Heller, J. Brieger, M. Gabriel and R. Förch, Plasma Processes Polym., 2012, 9, 10–16 CrossRef CAS.
- M. C. Boivin, P. Chevallier, C. A. Hoesli, J. Lagueux, R. Bareille, M. Rémy, L. Bordenave, M. C. Durrieu and G. Laroche, J. Biomed. Mater. Res., Part A, 2013, 101, 694–703 CrossRef PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491–497 CAS.
- L. Y. Santiago, R. W. Nowak, J. Peter Rubin and K. G. Marra, Biomaterials, 2006, 27, 2962–2969 CrossRef CAS PubMed.
- J. Lu, M. Shi and M. S. Shoichet, Bioconjugate Chem., 2008, 20, 87–94 CrossRef PubMed.
- S. J. Yuan, G. Xiong, X. Y. Wang, S. Zhang and C. Choong, J. Mater. Chem., 2012, 22, 13039–13049 RSC.
- T. Cai, K. G. Neoh, E. T. Kang and S. L. Teo, Langmuir, 2011, 27, 2936–2945 CrossRef CAS PubMed.
- J. Q. Meng, C. L. Chen, L. P. Huang, Q. Y. Du and Y. F. Zhang, Appl. Surf. Sci., 2011, 257, 6282–6290 CrossRef CAS.
- C. D. Wagner and G. E. Muilenberg, Handbook of x-ray photoelectron spectroscopy, Physical Electronics Division, Perkin-Elmer Corp., 1979 Search PubMed.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS PubMed.
- S. E. D'Souza, M. H. Ginsberg and E. F. Plow, Trends Biochem. Sci., 1991, 16, 246–250 CrossRef.
- N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS.
- D. Mangindaan, I. Yared, H. Kurniawan, J. R. Sheu and M. J. Wang, J. Biomed. Mater. Res., Part A, 2012, 100, 3177–3188 CrossRef PubMed.
- P. Hamerli, T. Weigel, T. Groth and D. Paul, Biomaterials, 2003, 24, 3989–3999 CrossRef CAS PubMed.
- S. J. Yuan, G. Xiong, A. Roguin and C. Choong, Biointerphases, 2012, 7, 30 CrossRef CAS PubMed.
- H. Marquardt, T. Kuroki, E. Huberman, J. K. Selkirk, C. Heidelberger, P. L. Grover and P. Sims, Cancer Res., 1972, 32, 716–720 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra44789h |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2014 |