Asymmetric hydrogenation of α-arylacrylic and β-arylbut-3-enoic acids catalyzed by a Rh(I) complex of a monodentate secondary phosphine oxide ligand†
Kaiwu
Dong
,
Yang
Li
,
Zheng
Wang
and
Kuiling
Ding
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: kding@mail.sioc.ac.cn; Fax: +86-21-6416-6128; Tel: +86-21-5492-5146
First published on 24th January 2014
Abstract
The RhI complexes of chiral secondary phosphine oxide ligands have been disclosed to be efficient for the catalytic asymmetric hydrogenation of α-arylacrylic and β-arylbut-3-enoic acids, providing the corresponding chiral α-arylpropanoic and β-arylbutanoic acids in excellent yields with up to 97% ee, including several anti-inflammatory drugs.
Optically active α-arylpropanoic or 3-arylbutanoic acids and their derivatives represent important classes of chiral chemicals that have found widespread pharmaceutical and synthetic applications. For example, α-arylpropanoic acids such as Naproxen and Ibuprofen are well-known non-steroid anti-inflammatory and analgesic drugs,1
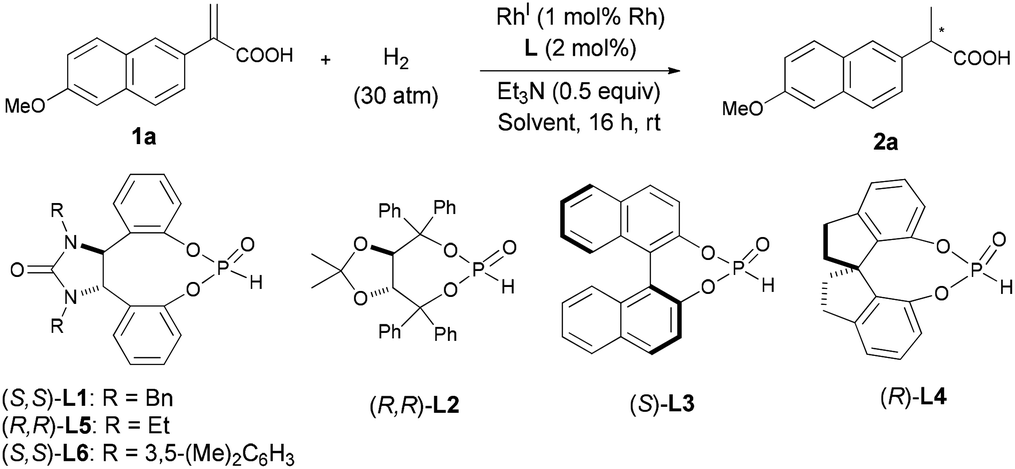
while the 3-arylbutanoic acid derivatives such as chiral 3-arylbutanols can be used as important intermediates for the synthesis of aromatic sesquiterpenes of the bisabolane family.2 Although a variety of methods for the asymmetric syntheses of the α-arylpropanoic acids3 or 3-arylbutanoic acids4 have been developed, catalytic asymmetric hydrogenation (AH) of the corresponding α-arylacrylic or β-arylbut-3-enoic acids represents the most straightforward and environmentally benign approach to these compounds. In the AH of α-arylacrylic acids, high enantioselectivities have been achieved with RuII/diphosphine,5 RhI/diphosphine6 or IrI/phosphine oxazoline catalysts.5m,7 On the other hand, the AH of β-arylbut-3-enoic acids has been relatively less explored, with moderate to high enantioselectivities being achieved using RuII/diphosphine5c,8 or RhI/diphosphine catalysts.9 As far as we know, there is only a single example using a RhI complex of a monodentate chiral phosphine for AH of α-phenylacrylic acid with poor enantioselectivity (15% ee),10 despite their ready preparation and good stability, among other salient features.11 Herein, we report our results on the use of a chiral monodentate secondary phosphine oxide (SPO) ligand for the efficient RhI-catalyzed AH of a wide variety of α-arylacrylic or β-arylbut-3-enoic acids, to give the corresponding α-arylpropanoic and β-arylbutanoic acids in excellent yields with good to excellent enantioselectivities (up to 97% ee) (Scheme 1).

 | ||
| Scheme 1 Catalysts for AH of α-arylacrylic and β-arylbut-3-enoic acids. | ||
Chiral SPOs are a type of readily accessible P ligands that are stable to air oxidation and inert to water, but their application in transition-metal-catalyzed asymmetric reactions has been relatively less explored.12 Very recently, we have demonstrated that some chiral SPO compounds, either used alone or in combination with achiral triarylphosphines, are highly efficient chiral ligands for RhI-catalyzed AH of α-substituted ethenylphosphonic acids, β,β-diarylacrylic acids, and α- or β-trifluoromethyl acrylic acids.13 Encouraged by these results, we sought to extend the RhI/SPO catalyzed AH methodology to the α-arylacrylic and β-arylbut-3-enoic acids. The study was initiated by surveying the effects of several reaction parameters, including chiral SPO ligands (L1–L6), RhI source, solvent, temperatures, as well as base additives, on the RhI-catalyzed AH of 2-(6-methoxynaphthalen-2-yl)acrylic acid (1a), a precursor for Naproxen. The reactions were generally conducted at rt for 16 h under 30 atm of hydrogen, and the results are shown in Table 1. Though full conversions of 1a were achieved in all cases, the enantioselectivity of the product 2a varied to a considerable extent depending on the detailed conditions. For the screening of the SPO ligands L1–L4, the reactions were run in CH2Cl2 with 0.5 equiv. of Et3N as the additive, and the catalyst was generated in situ by mixing 1 mol% [Rh(cod)2]OTf (cod = cycloocta-1,5-diene, OTf− = CF3SO3−) with the SPO ligand in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. Clearly, 1,2-diphenylethylenediamine-derived14 SPO L1 appears to be the most promising (69% ee) among the tested ligands (entries 1–4), which was thus used in further optimization studies. With (S,S)-L1 as the chiral ligand, the cationic [Rh(cod)2]OTf, Rh(cod)2BF4 or Rh(nbd)2BF4 afforded similar enantioselectivities of 2a (69–72% ee, entries 1, 5, and 6), whereas the [Rh(μ-Cl)(cod)]2 led to a substantial enhancement of ee value to 81% (entry 7). The solvents also demonstrated a profound effect on the enantioselectivity of the reaction (entries 7–20). While the reactions performed well in aprotic solvents [CH2Cl2, CHCl3, DCE (dichloroethane), THF, Et2O, dioxane, GDE (glycol dimethyl ether), MTBE (methyl tert-butyl ether), toluene, and EtOAc] to give 2a in moderate to high enantioselectivities (entries 8–17), the protic solvents [CH3OH, iPrOH, and TFE (CF3CH2OH)] only resulted in substantially decreased ee values (entries 18–20) under otherwise identical reaction conditions. Toluene and dioxane turned out to be optimal, affording 2a in excellent enantioselectivities (92% ee, entries 12 and 15). A number of organic or inorganic bases have also been examined as additives for [Rh(μ-Cl)(cod)]2/(S,S)-L1 catalyzed AH of 1a, among which Et3N was found to be optimal in terms of enantioselectivity (for details, see Tables S1 and S2 in ESI†). SPO ligands L5 and L6 with a backbone similar to that of L1 have also been examined in the AH of 1a, to give the product 2a with 93% and 89% ee, respectively (entries 21 and 22). Finally, lowering the reaction temperature to 0 °C led to a further improvement of ee value to 95% for the [Rh(μ-Cl)(cod)]2/L5 catalyzed reaction, albeit in this case the reaction was run under slightly modified conditions (50 atm of H2, 24 h, entry 23). Thus, two sets of optimized conditions for AH of 1a were found using either [Rh(μ-Cl)(cod)]2/(R,R)-L5 (defined as method A shown in entry 23) or [Rh(μ-Cl)(cod)]2/(S,S)-L1 (defined as method B shown in entry 12) as the catalyst, both affording 2a with high ee values albeit slightly varied conditions were used.
:2 molar ratio. Clearly, 1,2-diphenylethylenediamine-derived14 SPO L1 appears to be the most promising (69% ee) among the tested ligands (entries 1–4), which was thus used in further optimization studies. With (S,S)-L1 as the chiral ligand, the cationic [Rh(cod)2]OTf, Rh(cod)2BF4 or Rh(nbd)2BF4 afforded similar enantioselectivities of 2a (69–72% ee, entries 1, 5, and 6), whereas the [Rh(μ-Cl)(cod)]2 led to a substantial enhancement of ee value to 81% (entry 7). The solvents also demonstrated a profound effect on the enantioselectivity of the reaction (entries 7–20). While the reactions performed well in aprotic solvents [CH2Cl2, CHCl3, DCE (dichloroethane), THF, Et2O, dioxane, GDE (glycol dimethyl ether), MTBE (methyl tert-butyl ether), toluene, and EtOAc] to give 2a in moderate to high enantioselectivities (entries 8–17), the protic solvents [CH3OH, iPrOH, and TFE (CF3CH2OH)] only resulted in substantially decreased ee values (entries 18–20) under otherwise identical reaction conditions. Toluene and dioxane turned out to be optimal, affording 2a in excellent enantioselectivities (92% ee, entries 12 and 15). A number of organic or inorganic bases have also been examined as additives for [Rh(μ-Cl)(cod)]2/(S,S)-L1 catalyzed AH of 1a, among which Et3N was found to be optimal in terms of enantioselectivity (for details, see Tables S1 and S2 in ESI†). SPO ligands L5 and L6 with a backbone similar to that of L1 have also been examined in the AH of 1a, to give the product 2a with 93% and 89% ee, respectively (entries 21 and 22). Finally, lowering the reaction temperature to 0 °C led to a further improvement of ee value to 95% for the [Rh(μ-Cl)(cod)]2/L5 catalyzed reaction, albeit in this case the reaction was run under slightly modified conditions (50 atm of H2, 24 h, entry 23). Thus, two sets of optimized conditions for AH of 1a were found using either [Rh(μ-Cl)(cod)]2/(R,R)-L5 (defined as method A shown in entry 23) or [Rh(μ-Cl)(cod)]2/(S,S)-L1 (defined as method B shown in entry 12) as the catalyst, both affording 2a with high ee values albeit slightly varied conditions were used.
|
|
||||
|---|---|---|---|---|
| Entry | RhI | L | Solvent | eeb (%) |
| a Unless otherwise noted, all reactions were conducted under 30 atm of H2 at rt for 16 h. [1a] = 0.1 M, [RhI] = 1.0 mM, [L] = 2.0 mM, solvent = 2.5 mL. All conversions were >99% as determined by 1H NMR analysis. b Determined by HPLC on a chiral AD-H column after esterification with CH2N2, and the absolute configuration was assigned by comparison of the [α]20D with that reported in the literature.7b c Method A: the reaction is carried out in toluene under 50 atm of H2 at 0 °C for 24 h. d Method B: the reaction is carried out in dioxane under 30 atm of H2 at rt for 16 h. | ||||
| 1 | [Rh(cod)2]OTf | (S,S)-L1 | CH2Cl2 | 69 (R) |
| 2 | [Rh(cod)2]OTf | (R,R)-L2 | CH2Cl2 | 0 |
| 3 | [Rh(cod)2]OTf | (S)-L3 | CH2Cl2 | 43 (S) |
| 4 | [Rh(cod)2]OTf | (R)-L4 | CH2Cl2 | 23 (S) |
| 5 | [Rh(cod)2]BF4 | (S,S)-L1 | CH2Cl2 | 72 (R) |
| 6 | [Rh(nbd)2]BF4 | (S,S)-L1 | CH2Cl2 | 71 (R) |
| 7 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | CH2Cl2 | 81 (R) |
| 8 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | CHCl3 | 85 (R) |
| 9 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | DCE | 72 (R) |
| 10 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | Et2O | 57 (R) |
| 11 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | THF | 86 (R) |
| 12d | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | Dioxane | 92 (R) |
| 13 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | GDE | 82 (R) |
| 14 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | MTBE | 80 (R) |
| 15 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | Toluene | 92 (R) |
| 16 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | Acetone | 60 (R) |
| 17 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | EtOAc | 84 (R) |
| 18 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | CH3OH | 24 (R) |
| 19 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | iPrOH | 5 (R) |
| 20 | [Rh(μ-Cl)(cod)]2 | (S,S)-L1 | TFE | 9 (R) |
| 21 | [Rh(μ-Cl)(cod)]2 | (R,R)-L5 | Toluene | 93 (S) |
| 22 | [Rh(μ-Cl)(cod)]2 | (S,S)-L6 | Toluene | 89 (R) |
| 23c | [Rh(μ-Cl)(cod)]2 | (R,R)-L5 | Toluene | 95 (S) |
With these results in hand, we proceeded to examine the substrate scope of the protocol in the AH of α-substituted acrylic acids 1a–o. The reactions were carried out with [Rh(μ-Cl)(cod)]2/(R,R)-L5 or [Rh(μ-Cl)(cod)]2/(S,S)-L1 as the catalyst under the conditions specified by method A (entry 23, Table 1) or method B (entry 12, Table 1), respectively, and the results are summarized in Table 2. Full conversions of the substrates and good to excellent enantioselectivities (83–96% ee) of α-arylpropanoic acids 2a–n were achieved for the AH of various α-arylacrylic acids 1a–n, indicating the less significant stereoelectronic influence of the substituents on the reactions involving this type of substrate (entries 1–14). On the other hand, AH of α-benzylacrylic acid 1o was less satisfactory, with a moderate ee value of 2o being attained using either method A or B (entry 15). Generally speaking, similar levels of enantioselectivity were achieved for most of the substrates using the reaction conditions of methods A and B, but method B showed superior outcomes to method A for some specific substrates (1i–k, 1m–n), indicating mutual complementation of two methods. Especially noteworthy is that several non-steroid anti-inflammatory drugs were synthesized in high optical purities by using this approach. Apart from Naproxen (95% ee, entry 1), Flurbiprofen (90% ee, entry 11), Ibuprofen (93% ee, entry 12), Ketoprofen (90% ee, entry 13) and Suprofen (83% ee, entry 14) were all readily accessible in a straightforward manner via this AH protocol.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate (1a–o) | A/B | Conv.b (%) | eec (%) |
| a Unless otherwise noted, all reactions were conducted using either method A or B with [1] = 0.1 M, [[Rh(μ-Cl)(cod)]2] = 0.5 mM, and 0.5 molar equiv. of Et3N. Conditions for method A: PH2 = 50 atm, [(R,R)-L5] = 2.0 mM, toluene (2.5 mL), 0 °C, 24 h. Conditions for method B: PH2 = 30 atm, [(S,S)-L1] = 2.0 mM, dioxane (2.5 mL), rt, 16 h. b Determined by 1H NMR. c Determined by a chiral HPLC column after esterification with CH2N2, and the absolute configurations were assigned by comparison of their optical rotations with reported values (see the ESI). d The products are non-steroid anti-inflammatory agents. e 2 mol% RhI was used. | ||||
| 1d |

|
A | > 99 | 95 (S) |
| B | >99 | 92 (R) | ||
| 2 |

|
A | >99 | 96 (S) |
| B | >99 | 93 (R) | ||
| 3 |

|
Ae | >99 | 91 (S) |
| B | >99 | 92 (R) | ||
| 4 |
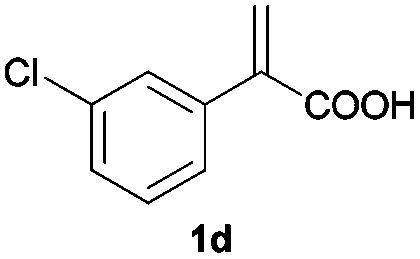
|
A | >99 | 94 (S) |
| 5 |

|
A | >99 | 94 (S) |
| 6 |
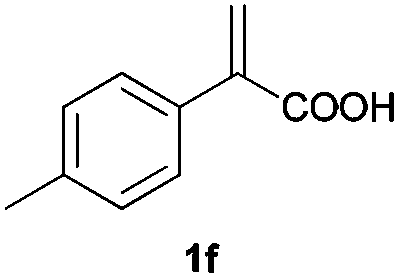
|
A | >99 | 93 (S) |
| 7 |

|
A | >99 | 94 (S) |
| 8 |

|
A | >99 | 94 (S) |
| 9 |
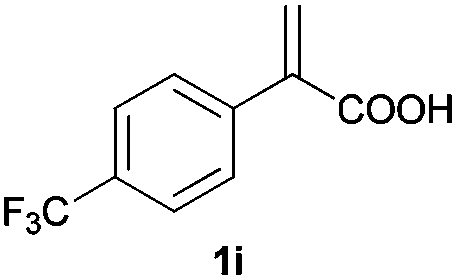
|
A | >99 | 81 (S) |
| B | >99 | 90 (R) | ||
| 10 |

|
A | 50 | 60 (S) |
| B | >99 | 92 (R) | ||
| 11d |

|
A | >99 | 86 (S) |
| B | >99 | 90 (R) | ||
| 12d |
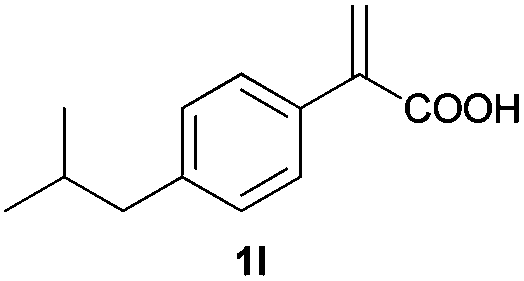
|
A | >99 | 93 (S) |
| 13d |
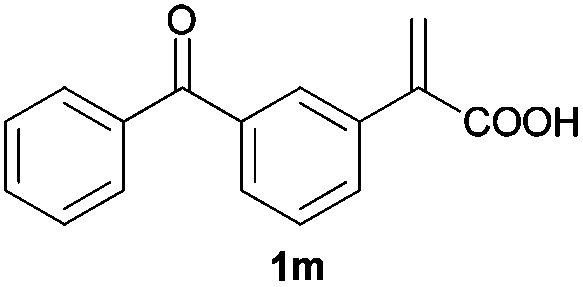
|
A | >99 | 86 (S) |
| B | >99 | 90 (R) | ||
| 14d |

|
A | >99 | 78 (+) |
| B | >99 | 83 (−) | ||
| 15 |

|
A | 50 | 43 (S) |
| B | >99 | 54 (R) | ||

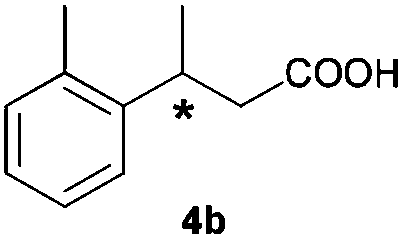
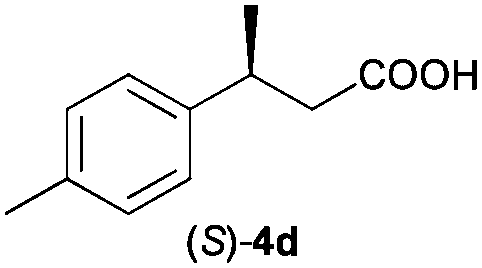
The RhI/SPO catalyzed protocol has also been extended to the AH of β-arylbut-3-enoic acids 3a–g, a class of less explored olefinic substrates. As shown in Table 3, in most cases the [Rh(μ-Cl)(cod)]2/(S,S)-L1 catalyst system was demonstrated to be highly enantioselective for the AH of these substrates, providing the corresponding chiral β-arylbutanoic acids 4a, 4c–e, and 4g in full substrate conversions with excellent enantioselectivities (94–97% ee, entries 1, 3–5, and 7). Exceptions are found for AH of the o-methyl substituted substrate 3b and 3-(1-naphthyl)but-3-enoic acid 3f, wherein substantially declined ee values for 4b (entry 2) and 4f (entry 6) were obtained, probably as a result of unfavourable steric interactions of the o-substituent with the catalyst, although we are unable to give a clear scenario of the actual interaction mode at the moment.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate (3a–g) | A/B | Product (4a–g) | eeb (%) |
| a For reaction conditions, see footnote a in Table 2. All conversions were >99% as determined by 1H NMR. b Determined by HPLC on a chiral column after esterification with CH2N2. The absolute configurations were assigned by comparison of their optical rotations with reported values (see the ESI). c AH of (E)-3-phenylbut-2-enoic acid (the isomer of 3a) using method B afforded only 15% conversion and 51% ee of 4a; thus the possibility of isomerization–hydrogenation can be ruled out. | ||||
| 1c |
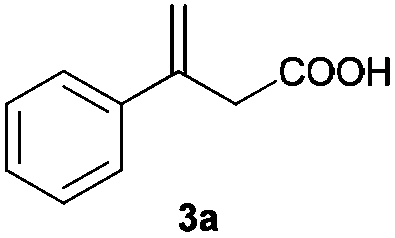
|
A |

|
77 (R) |
| B | 94 (S) | |||
| 2 |
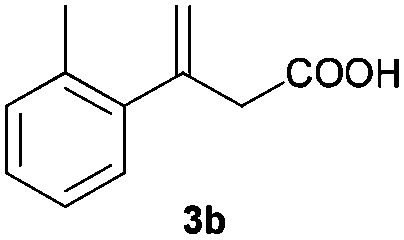
|
B |
|
16 (+) |
| 3 |

|
B |

|
95 (+) |
| 4 |
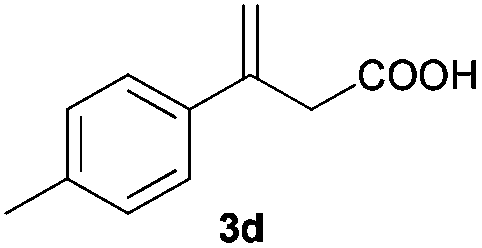
|
B |
|
94 (S) |
| 5 |
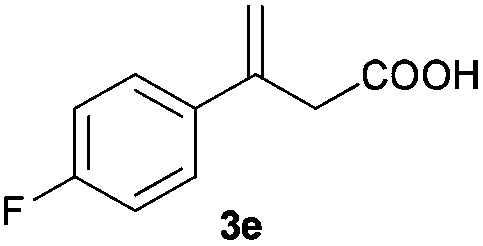
|
B |

|
94 (+) |
| 6 |
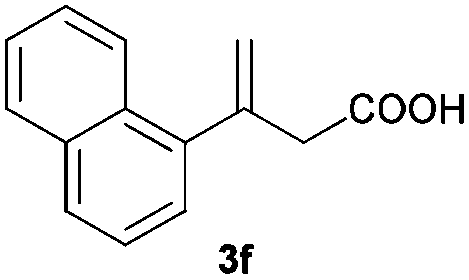
|
B |

|
59 (S) |
| 7 |
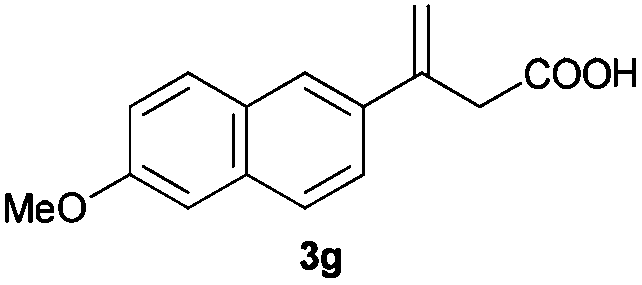
|
B |
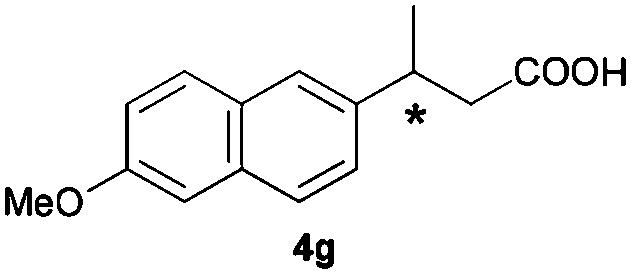
|
97 (+) |

Conclusions
A class of RhI complexes of monodentate chiral SPO ligands were found to be highly enantioselective in the AH of α-arylacrylic acids and β-arylbut-3-enoic acids, affording the corresponding enantioenriched α-arylpropanoic acids and β-arylbutanoic acids, respectively, in full substrate conversions and generally good to excellent enantioselectivities (up to 97% ee). Especially noteworthy is that several non-steroid anti-inflammatory drugs are readily accessible in high enantiopurities by using this AH protocol, which thus may demonstrate further potential in asymmetric synthesis of bioactive compounds.Experimental
General procedures for RhI/SPO catalyzed asymmetric hydrogenation of α-arylacrylic acids or β-arylbut-3-enoic acids
Acknowledgements
We acknowledge financial support of this work from the Major Basic Research Development Program of China (no. 2010CB833300), NSFC (no. 91127041, 21121062, and 21232009), CAS, and the Science and Technology Commission of Shanghai Municipality.Notes and references
- (a) I. T. Harrison, B. Lewis, P. Nelson, W. Rooks, A. Roszkowski, A. Tomolonis and J. H. Fried, J. Med. Chem., 1970, 13, 203 CrossRef CAS; (b) T. Y. Shen, Angew. Chem., Int. Ed. Engl., 1972, 11, 460 CrossRef CAS PubMed; (c) D. Lednicer and L. A. Mitscher, The Organic Chemistry of Drug Synthesis, Wiley, New York, 1977, vol. 1 Search PubMed.
- (a) A. I. Meyers and D. Stoianova, J. Org. Chem., 1997, 62, 5219 CrossRef CAS; (b) C. Fuganti, S. Serra and A. Dulio, J. Chem. Soc., Perkin Trans. 1, 1999, 279 RSC; (c) J.-Q. Li, X. Quan and P. G. Andersson, Chem.–Eur. J., 2012, 18, 10609 CrossRef CAS PubMed.
- (a) X. Dai, N. A. Strotman and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 3302 CrossRef CAS PubMed , and the references cited therein; ; (b) P. M. Lundin and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 11027 CrossRef CAS PubMed; (c) C. R. Smith and T. V. RajanBabu, J. Org. Chem., 2009, 74, 3066 CrossRef CAS PubMed; (d) C. M. Crudden, Y. B. Hleba and A. C. Chen, J. Am. Chem. Soc., 2004, 126, 9200 CrossRef CAS PubMed; (e) K. Ishihara, D. Nakashima, Y. Hiraiwa and H. Yamamoto, J. Am. Chem. Soc., 2003, 125, 24 CrossRef CAS PubMed; (f) J. S. Harvey, S. P. Simonovich, C. R. Jamison and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 13782 CrossRef CAS PubMed.
- (a) B. E. Rossiter and N. M. Swingle, Chem. Rev., 1992, 92, 771 CrossRef CAS; (b) Y. Takaya, T. Senda, H. Kurushima, M. Ogasawara and T. Hayashi, Tetrahedron: Asymmetry, 1999, 10, 4047 CrossRef CAS; (c) S. Sakuma, M. Sakai, R. Itooka and N. Miyaura, J. Org. Chem., 2000, 65, 5951 CrossRef CAS PubMed; (d) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed; (e) M. C. Hillier, J. N. Desrosiers, J. F. Marcoux and E. J. J. Grabowski, Org. Lett., 2004, 6, 573 CrossRef CAS PubMed.
- (a) T. Ohta, H. Takaya, M. Kitamura, K. Nagai and R. Noyori, J. Org. Chem., 1987, 52, 3174 CrossRef CAS; (b) X. Zhang, T. Uemura, K. Matsumura, N. Sayo, H. Kumobayashi and H. Takaya, Synlett, 1994, 501 CrossRef CAS; (c) T. Uemura, X. Zhang, K. Matsumura, N. Sayo, H. Kumobayashi, T. Ohta, K. Nozaki and H. Takaya, J. Org. Chem., 1996, 61, 5510 CrossRef CAS; (d) T. Benincori, E. Brenna, F. Sannicol, L. Trimarco, P. Antognazza, E. Cesarotti, F. Demartin and T. Pilati, J. Org. Chem., 1996, 61, 6244 CrossRef CAS PubMed; (e) R. A. Brown, P. Pollet, E. McKoon, C. A. Eckert, C. L. Liotta and P. G. Jessop, J. Am. Chem. Soc., 2001, 123, 1254 CrossRef CAS; (f) Q.-H. Fan, C.-Y. Ren, C.-H. Yeung, W.-H. Hu and A. S. C. Chan, J. Am. Chem. Soc., 1999, 121, 7407 CrossRef CAS; (g) C.-C. Pai, C.-W. Lin, C.-C. Lin, C.-C. Chen and A. S. C. Chan, J. Am. Chem. Soc., 2000, 122, 11513 CrossRef CAS; (h) L. Qiu, J. Qi, C.-C. Pai, S. Chan, Z. Zhou, M. C. K. Choi and A. S. C. Chan, Org. Lett., 2002, 4, 4599 CrossRef CAS PubMed; (i) L. Qiu, J. Wu, S. Chan, T. T.-L. Au-Yeung, J.-X. Ji, R. Guo, C.-C. Pai, Z. Zhou, X. Li, Q.-H. Fan and A. S. C. Chan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5815 CrossRef CAS PubMed; (j) L. Qiu, F. Y. Kwong, J. Wu, W. H. Lam, S. Chan, W.-Y. Yu, Y.-M. Li, R. Guo, Z. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2006, 128, 5955 CrossRef CAS PubMed; (k) Q.-H. Fan, Y.-M. Chen, X.-M. Chen, D.-Z. Jiang, F. Xi and A. S. C. Chan, Chem. Commun., 2000, 789 RSC; (l) L. Qiu, Y.-M. Li, F. Y. Kwong, W.-Y. Yu, Q.-H. Fan and A. S. C. Chan, Adv. Synth. Catal., 2007, 349, 517 CrossRef CAS; (m) J. Liu, Y. Feng, B. Ma, Y.-M. He and Q.-H. Fan, Eur. J. Org. Chem., 2012, 6737 CrossRef CAS; (n) A. Scrivanti, S. Bovo, A. Ciappa and U. Matteoli, Tetrahedron Lett., 2006, 47, 9261 CrossRef CAS PubMed.
- (a) A. S. C. Chan, Chem. Abstr., 1991, 115, 28896 Search PubMed; (b) F. Robin, F. Mercier, L. Ricard, F. Mathey and M. Spagnol, Chem.–Eur. J., 1997, 3, 1365 CrossRef CAS; (c) W.-H. Hu, C. C. Pai, C. C. Chen, G.-P. Xue and A. S. C. Chan, Tetrahedron: Asymmetry, 1998, 9, 3241 CrossRef CAS; (d) B. Zupancic, B. Mohar and M. Stephan, Adv. Synth. Catal., 2008, 350, 2024 CrossRef CAS; (e) M. Stephan, D. Sterk and B. Mohar, Adv. Synth. Catal., 2009, 351, 2779 CrossRef CAS; (f) B. Zupancic, B. Mohar and M. Stephan, Org. Lett., 2010, 12, 1296 CrossRef CAS PubMed; (g) B. Zupancic, B. Mohar and M. Stephan, Org. Lett., 2010, 12, 3022 CrossRef CAS PubMed; (h) M. Stephan, D. Sterk, B. Zupancic and B. Mohar, Org. Biomol. Chem., 2011, 9, 5266 RSC.
- (a) Y. Zhang, Z. Han, F. Li, K. Ding and A. Zhang, Chem. Commun., 2010, 156 RSC; (b) S.-F. Zhu, Y.-B. Yu, S. Li, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8872 CrossRef CAS PubMed; (c) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin., 2012, 70, 1427 CAS . Ir(I) complexes with other P–N ligands have also provided high selectivities in the AH of α-methyl cinnamic esters, see, for example: ; (d) D. H. Woodmansee, M.-A. Mueller, M. Neuburger and A. Pfaltz, Chem. Sci., 2010, 1, 72 RSC.
- (a) M. Saburi, H. Takeuchi, M. Ogasawara, T. Tsukahara, Y. Ishii, T. Ikariya, T. Takahashi and Y. Uchida, J. Organomet. Chem., 1992, 428, 155 CrossRef CAS; (b) K. Yamamoto, K. Ikeda and L. L. Yin, J. Organomet. Chem., 1989, 370, 319 CrossRef CAS.
- X. Sun, L. Zhou, C.-J. Wang and X. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2623 CrossRef CAS PubMed.
- W. S. Knowles and M. J. Sabacky, Chem. Commun., 1968, 1445 RSC.
- (a) M. van den Berg, A. J. Minnaard, E. P. Schudde, J. van Esch, A. H. M. de Vries, J. G. de Vries and B. L. Feringa, J. Am. Chem. Soc., 2000, 122, 11539 CrossRef CAS; (b) C. Claver, E. Fernandez, A. Gillon, K. Heslop, D. J. Hyett, A. Martorell, A. G. Orpen and P. G. Pringle, Chem. Commun., 2000, 961 Search PubMed; (c) M. Reetz and T. Sell, Tetrahedron Lett., 2000, 41, 6333 CrossRef CAS; (d) M. T. Reetz and G. Mehler, Angew. Chem., Int. Ed., 2000, 39, 3889 CrossRef CAS.
- (a) M. T. Reetz, T. Sell and R. Goddard, Chimia, 2003, 57, 290 CrossRef CAS; (b) X. Jiang, A. J. Minnaard, B. Hessen, B. L. Feringa, A. L. L. Duchateau, J. G. O. Andrien, J. A. F. Boogers and J. G. de Vries, Org. Lett., 2003, 5, 1503 CrossRef CAS PubMed; (c) X. Jiang, M. van den Berg, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Tetrahedron: Asymmetry, 2004, 15, 2223 CrossRef CAS PubMed; (d) H. Landert, F. Spindler, A. Wyss, H.-U. Blaser, B. Pugin, Y. Ribourduoille, B. Gschwend, B. Ramalingam and A. Pfaltz, Angew. Chem., Int. Ed., 2010, 49, 6873 CrossRef CAS PubMed . For a highlight on asymmetric catalysis with chiral SPO preligands, see: ; (e) N. V. Dubrovina and A. Börner, Angew. Chem., Int. Ed., 2004, 43, 5883 CrossRef CAS PubMed.
- (a) K. Dong, Z. Wang and K. Ding, J. Am. Chem. Soc., 2012, 134, 12474 CrossRef CAS PubMed ; and related work, see: ; (b) Y. Li, K. Dong, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 6748 CrossRef CAS PubMed; (c) K. Dong, Y. Li, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 14191 CrossRef CAS PubMed.
- (a) Y. Liu, Z. Wang and K. Ding, Acta Chim. Sin., 2012, 70, 1464 CAS; (b) Y. Liu and K. Ding, J. Am. Chem. Soc., 2005, 127, 10488 CrossRef CAS PubMed; (c) Y. Liu, C. A. Sandoval, Y. Yamaguchi, X. Zhang, Z. Wang, K. Kato and K. Ding, J. Am. Chem. Soc., 2006, 128, 14212 CrossRef CAS PubMed; (d) J. Zhang, Y. Li, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2011, 50, 11743 CrossRef CAS PubMed; (e) J. Zhang, K. Dong, Z. Wang and K. Ding, Org. Biomol. Chem., 2012, 10, 1598 RSC; (f) Y. Liu, Z. Wang and K. Ding, Tetrahedron, 2012, 68, 7581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00042g |
| This journal is © the Partner Organisations 2014 |