The anion–π interaction: naissance and establishment of a peculiar supramolecular bond
Patrick
Gamez
ab
aDepartament de Química Inorgànica, QBI, Universitat de Barcelona, Martí i Franquès 1-11, 08028, Barcelona, Spain. E-mail: patrick.gamez@qi.ub.es; Web: http://www.bio-inorganic-chemistry-icrea-ub.com Fax: +34 934907725
bInstitució Catalana de Recerca i Estudis Avançats (ICREA), Passeig Lluís Companys 23, 08010 Barcelona, Spain
First published on 20th December 2013
Abstract
A decade of thorough theoretical and experimental investigations has established the anion–π interaction as a new and important supramolecular bond. The present review describes the physical nature of this noncovalent interaction, the different (structural) factors that can influence its strength, and three state-of-the-art literature reports illustrating its rational utilization for the development of anion receptors and biological applications.
 Patrick Gamez | Patrick Gamez studied Chemistry at the University of Lyon in France where he obtained his first degree. In 1995, he received his D. Phil. in the field of enantioselective catalysis and was awarded the French Chemical Society Prize for his PhD research. After a period of postdoctoral research at the Max-Planck-Institut für Kohlenforschung (as a Marie-Curie fellow) and at the University of Strasbourg, he joined Leiden University in 1999. Since 2010, he has been ICREA Research Professor at the Universitat de Barcelona. His research interests include the design and preparation of ligands for the development of metal complexes with applications in anticancer and anti-Alzheimer drug research, supramolecular chemistry (anion–π and lone pair–π interactions), gas storage and catalysis (Metal–Organic Frameworks), and molecular switches (spin-transition compounds). |
1. Introduction
Non-covalent interactions are the heart of supramolecular chemistry, the chemistry beyond the molecule, as defined by Lehn.1 Supramolecules are thus generated through the assembly of a number of molecular components that are held together by means of intermolecular contacts, which are not covalent in nature.2 There are four main types of established non-covalent bonds,3 namely the hydrogen bond,4 ionic interactions,5 van der Waals interactions,6 and hydrophobic bonds.7The cation–π interaction, a peculiar type of van der Waals interaction dominated by electrostatic and polarization effects, is a supramolecular bond that is well recognized by the chemistry community.8 Its iniquitousness and importance in biology, chemistry or materials science have been clearly demonstrated.9
Over the last decade, the related “inverse” anion–π interaction (i.e. the interaction of an anion with the positive electrostatic potential on the ring edge of an electron-deficient aromatic group) has emerged as a new supramolecular bond.10 The anion–π bond was first described theoretically,11,12 and the initial experimental evidence was reported in 2004.13–15 Since then, numerous applications of this non-covalent bonding interaction have been reported in diverse areas of chemistry.16,17 For instance, a number of examples have been described in the fields of anion recognition,18 supramolecular chemistry,16 or materials science,19 and its relevance in biological systems is increasingly considered.20
In the present review, the specific characteristics of the anion–π interaction are explained and the different factors influencing their strength are enumerated. Subsequently, three recent examples of the application of the anion–π interaction in anion recognition and in biological systems are presented.
2. Physical nature of the anion–π interaction
Electrostatic forces and ion-induced polarization effects are the main contributors to the bonding energy of the anion–π contact. The electrostatic term is associated with the quadrupole moment, i.e. Qzz, of the aromatic host (Fig. 1a).21 The Qzz value for benzene is −8.45 B (B = Buckingham); therefore, benzene is able to interact with a positively charged entity, such as a cation (cation–π interaction). When the hydrogen atoms of benzene are replaced by electron-withdrawing fluorine atoms, the Qzz value of the resulting hexafluorobenzene molecule amounts to +9.50 B; hence, hexafluorobenzene can favourably interact with electron-rich guests, viz. anions (or lone pairs).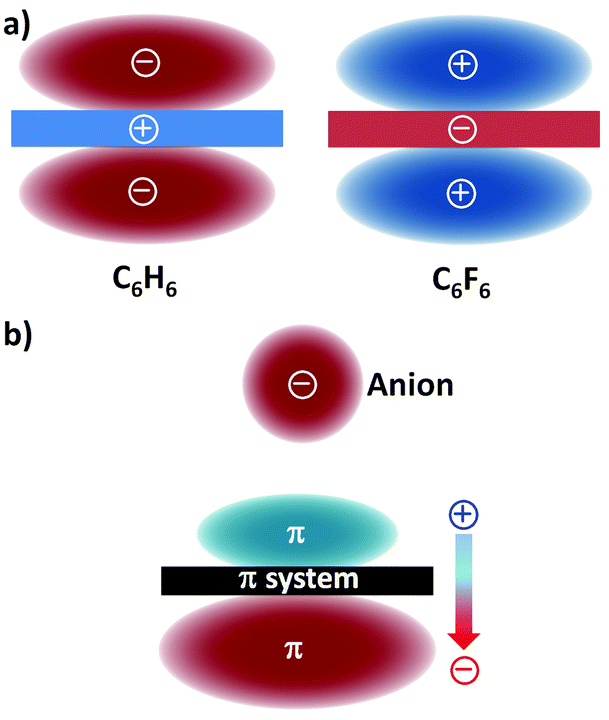 | ||
| Fig. 1 (a) Schematic representation of the quadrupole moments of benzene (C6H6; Qzz = −8.45 B) and hexafluorobenzene (C6F6; Qzz = +9.50 B)21 and (b) the ion-induced dipole22 (the molecular polarizabilities parallel to the main symmetry axis are α∥ = 41.5 and 37.7 a.u. (a.u. stands for atomic units), for benzene and hexafluorobenzene, respectively). | ||
The presence of the interacting anion in the vicinity of the arene gives rise to the polarization of the π system (Fig. 1b); this interaction of the incoming anion with the ion-induced dipole has a significant contribution to the total binding energy.22 It should be mentioned here that dispersion forces, which usually occur in weak interactions involving aromatic rings, only play a minor role in anion–π bonding.23
Because both cation–π and anion–π interactions are driven by electrostatic and polarization effects, aromatic hosts with negligible quadrupole moments (namely with Qzz values close to zero) should be able to interact favourably with both positive and negative guests. Actually, the interaction energies of the s-triazine ring (Qzz = +0.90 B) with the chloride anion and the lithium cation are −5.2 and −6.2 kcal mol−1.22
The interaction between benzene (an electron-rich aromatic ring) and chloride is intuitively expected to be repulsive. However, its unfavourable quadrupole moment (Qzz = −8.45 B) and favourable polarizability (α∥ = 41.5 a.u.) cancel each other; consequently, the interaction energy of the benzene–chloride pair is negligible. The same phenomenon is observed for the interaction of hexafluorobenzene (Qzz = +9.50 B and α∥ = 37.7 a.u.) with sodium.24
The directionality of non-covalent interactions is an important feature since it provides a pre-determined pathway that can be exploited in supramolecular chemistry to generate functional nanostructures, both in the solid-state and in solution. In that context, the hydrogen and halogen bonds are highly directional intermolecular contacts that allow the rational design of supramolecules.25,26 The directionality of the anion–π and lone pair–π interactions has thus been assessed. Comparative theoretical studies have been carried out for the sodium–benzene (cation–π) and chloride–hexafluorobenzene (anion–π) complexes.27
The interaction energies of an ion moving in a plane perpendicular to an aromatic ring (x-axis in Fig. 2) and along the axis perpendicular to the arene plane and passing through the ring centroid (y-axis in Fig. 2) have been calculated for the sodium–benzene and chloride–hexafluorobenzene systems. The interacting ion has been initially placed at the point above the ring centroid corresponding to the minimum potential energy, viz. 2.42 Å for benzene and 3.05 Å for hexafluorobenzene (Fig. 2). In the plane of the ring, i.e. along the x-axis, the energy decrease when the ion moves away from the ring centroid is more pronounced for the cation–π complex. Indeed, about 20% energy loss is observed for Na+–benzene (from −22.2 to −17.1 kcal mol−1; Fig. 2a) while it is only 5% for Cl−–hexafluorobenzene (from −14.1 to −13.1 kcal mol−1; Fig. 2b). These data suggest that cation–π interactions are stronger above the centre of the ring whereas anion–π contacts are equally realized all over the π system. In the y direction (see the y-axis in Fig. 2), the diminution of the interaction energy is more gradual for the Cl−–hexafluorobenzene pair; thus, at the first increment of 0.35 Å, the energy decrease is half (ca. 5%, from −14.1 to −13.4 kcal mol−1; Fig. 2b) that of the Na+–benzene complex (ca. 10%, from −22.2 to −19.9 kcal mol−1; Fig. 2a). This is most likely due to the fact that the chloride anion is significantly bigger than the cation (ionic radius of 1.67 Å for Cl− against 1.16 Å for Na+). Subsequently (i.e. at longer distances from the centroid), the same trend (namely similar energy-loss percentages) is observed for both systems. These calculations clearly indicate that an effective anion–π interaction can be realized above a wider area above the π ring (for the cation–π pair, the strongest interaction is achieved exactly in the middle of the ring, at the minimum potential energy point).
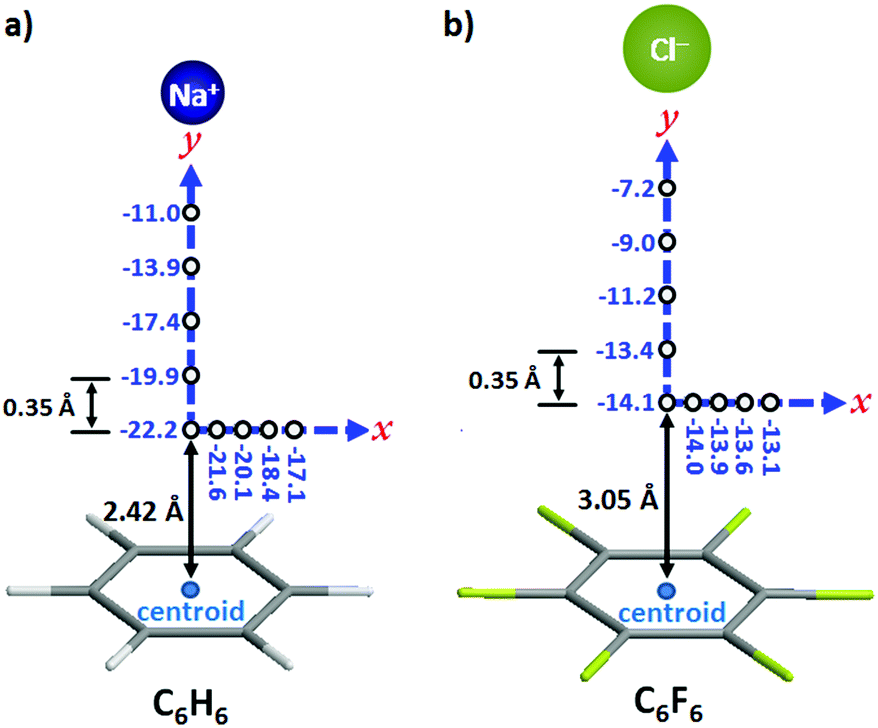 | ||
| Fig. 2 Computed interaction energies (in kcal mol−1) in the plane parallel to the arene ring (x-axis) and along the axis perpendicular to arene plane and passing through the ring centroid (y-axis) for (a) the sodium–benzene complex and (b) the chloride–hexafluorobenzene complex. The interacting ions were initially placed at potential energy minimum point at the centroid, namely 2.42 Å for benzene and 3.05 Å for hexafluorobenzene.27 | ||
The directionality of the anion–π (and the lone pair–π) supramolecular bond has been investigated further using the Cambridge Structural Database (CSD), which is a great source of valuable information since it contains thousands of solid-state structures where such supramolecular contacts may occur.28,29 For the statistical analyses of the CSD data, ConQuest software (version 1.14) and the IsoStar application of the CSD to generate 3D scatterplots have been used.10,30 These studies, applying an original methodical procedure, have clearly revealed that both the anion–π and lone pair–π interactions are directional.31,32 For example, the 3D scatterplots depicted in Fig. 3 have been obtained for the tetrafluoroborate–pentafluorophenyl (anion–π) and acetonitrile–pentafluorophenyl (lone pair–π) pairs.
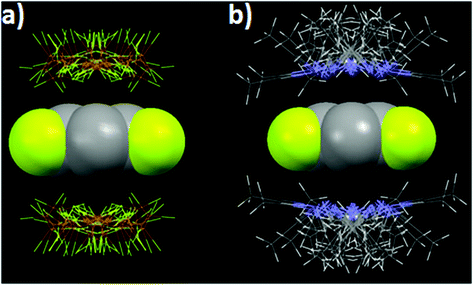 | ||
| Fig. 3 Views of the distribution of the CSD hits characterizing the noncovalent contacts between (a) BF4− anions and the pentafluorophenyl ring and (b) acetonitrile and pentafluorophenyl ring.10,32 C gray, F yellow, N blue, H white, B orange. | ||
For the BF4−–C6F5 pair (anion–π interaction), all anions are located above the ring (Fig. 3a), but not necessarily exactly over the ring centroid. This situation actually is in total agreement with the theoretical calculations, which have shown that anion–π interactions can take place above a wide area of the arene plane (see above and Fig. 2b). Similar circumstances are observed for the CH3CN–C6F5 pair (lone pair–π interaction; Fig. 3b).
In summary, anion–π and lone pair–π interactions are directional non-covalent bonds that can therefore be used to generate supramolecular architectures through rational design.31
3. Factors influencing the strength of the anion–π interaction
Taking into account the considerations described in section 2, it is evident that to favour the creation of strong anion–π contacts, the aromatic host should have a large and positive quadrupole moment Qzz and a large molecular polarizability α∥. The magnitude of these two parameters obviously will depend on various factors that can affect the electron density of the π ring, such as the substituents, the charge or its involvement in other types of supramolecular interactions (Fig. 4).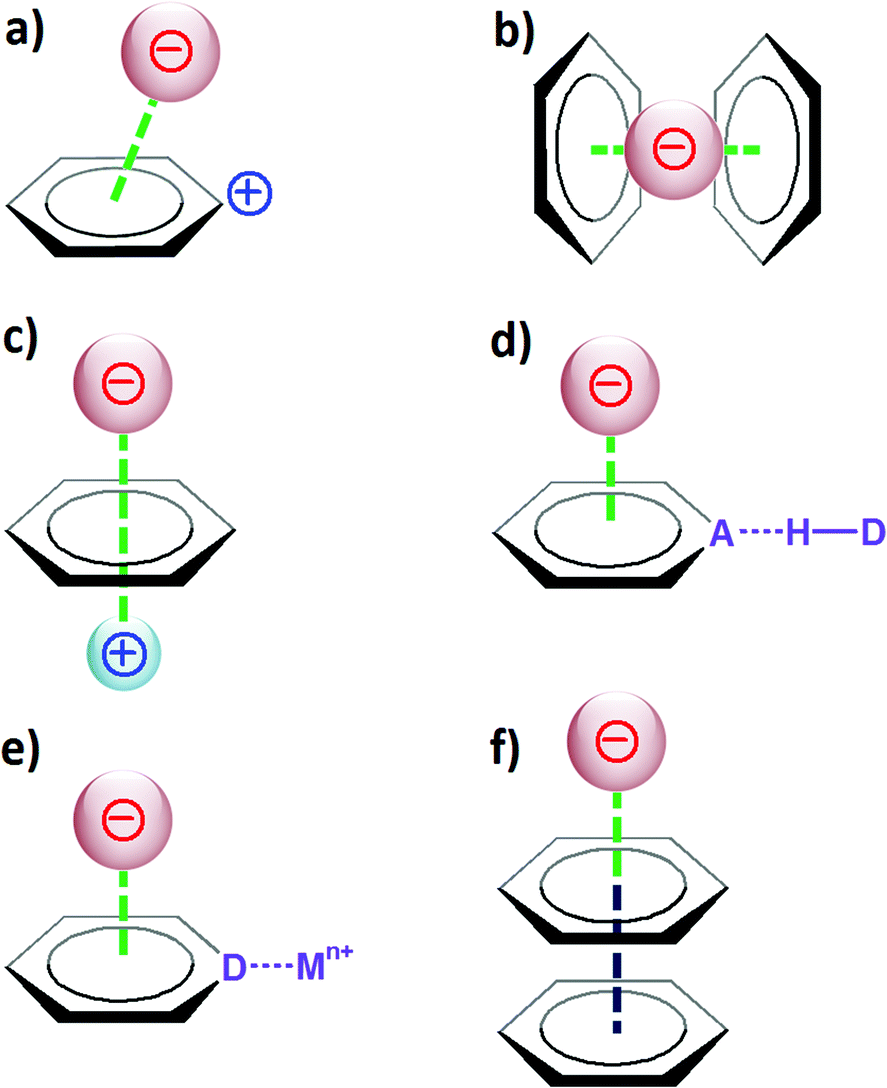 | ||
| Fig. 4 Representation of the different factors that can affect (enhance) the strength of the anion–π interaction; (a) positive charge on the aromatic ring, (b) additivity of the anion–π interaction, (c) cation–π interaction, (d) hydrogen bond (A⋯H–D), (e) coordination bond (D⋯Mn+), (f) π–π interaction. | ||
If the aromatic host is positively charged, the resulting electronic deficiency of the π ring obviously will increment the anion-binding ability of the arene (Fig. 4a). For instance, the calculated binding energy (with BSSE correction; BSSE stands for basis set superposition error) for the interaction between the tropylium cation and fluoride is −94.4 kcal mol−1,33 which is significantly stronger than a common cation–π interaction (for example, the binding energy for K+–benzene is −19 kcal mol−1).34
The additivity of the anion–π interaction is another feature that has an effect on the strength of the host–guest system (Fig. 4b). Thus, the binding energy for the Br−-1,3,5-triazine complex amounts to −5 kcal mol−1, while the corresponding values for the Br−-(1,3,5-triazine)2 and Br−-(1,3,5-triazine)3 systems are −10.2 and −21.7 kcal mol−1, respectively.35
If an arene interacts with both a cation and an anion at the opposite sides of the π system (Fig. 4c), the aromatic ring then mediates the transfer of information between the charged guests, and a stable anion–π–cation complex is obtained. For example, the binding of sodium to benzene is favourable, as reflected by its binding energy of −21.0 kcal mol−1 (this is a typical cation–π interaction). In contrast, the binding of fluoride to benzene is unfavourable, and the corresponding energy is +2.8 kcal mol−1. However, if the benzene host first interacts with Na+ and subsequently binds F−, then the resulting interaction energy, namely for the Na+–C6H6–F− supramolecule, is equal to −93.1 kcal mol−1; therefore, a very stable ternary complex is formed.24
When an aromatic ring acts simultaneously as a hydrogen-bond acceptor and an anion host, it has been shown that such a combination leads to reinforcement of both interactions (Fig. 4d). For instance, the binding energy for fluoride interacting with the electron-deficient arene pyridazino[4,5-d]pyridazine is −20.8 kcal mol−1 (with BSSE correction).36 The hydrogen-bonding interaction of this heteroaromatic compound (containing four nitrogen atoms that can act as hydrogen-bond acceptors) with one molecule of water is characterized as a binding energy of −4.7 kcal mol−1; with two water molecules, the energy increases to −9.1 kcal mol−1. Remarkably, the interaction energy of pyridazino[4,5-d]pyridazine bound to an F− anion and two H2O is −40.5 kcal mol−1.36 This value is greater than the sum of the energies for the independent noncovalent interactions (namely for two H bonds and one anion–π contacts, E = −9.1 to −20.8 = −29.9 kcal mol−1), thus revealing a synergistic effect between the different interactions.
Nitrogen-containing arenes are electron deficient; consequently, they exhibit the ability to bind anions (through anion–π contacts). In addition, since they hold N-donor atoms, they can coordinate to metal ions. Theoretical studies have revealed that upon coordination to a metal ion, the propensity of such N ligands to interact with anions is significantly enhanced (Fig. 4e). Accordingly, the calculated energy for the interaction of pyrazine with chloride is −10.9 kcal mol−1 (with BSSE and ZPE corrections; ZPE stands for zero point energy).37 When the pyrazine ring is coordinated to two silver(I) ions (through its two N donors), the complexation energy rises to −149.6 kcal mol−1.
Finally, the electron-deficient aromatic ring can itself be involved in π–π interactions (Fig. 4f). It has been computationally established that the interplay between π–π and anion–π interactions may give rise to strong cooperative effects.38 For example, the interaction of fluoride with benzene is unfavourable; actually, the corresponding calculated energy is +2.8 kcal mol−1. However, if the benzene ring engaged in this unfavourable anion–π contact on one side further interacts with a hexafluorobenzene molecule on the other side (through π–π stacking), then the binding energy of the ternary complex F−–C6H6–C6F6 becomes negative, i.e. E = −6.5 kcal mol−1 (with BSSE corrections).38
It has to be mentioned here that besides the various factors that affect the binding ability of the aromatic host, the nature of the anionic guest is also of great importance. Small anions are more polarizing; consequently, they generate stronger anion–π contacts. Planar or linear polyatomic anions such as nitrate or azide can interact by means of “π–π stacking-type” interactions with aryl hosts.39 Energetically, the anion–π interaction is less favourable than the cation–π one; in fact, the equilibrium anion⋯arene contact distances are larger than cation⋯arene ones, as the result of the greater van der Waals radii of anions (compared to the cation ones).
It is clear that to engineer a strong anion–π interaction (for instance for the design of anion receptors), all considerations described above should be borne in mind. In particular, the combination of the anion–π interaction with other type(s) of noncovalent bonds is desirable to achieve stable systems.
4. Selected elegant applications of the anion–π interaction reported during the last year
Since the pioneering theoretical and experimental reports about ten years ago,11–14 the anion–π interaction has received increasing attention from the scientific community.17,18 In the present section, a selection of remarkable examples of the rational use of the anion–π interaction, published in the last twelve months, is presented.4.1. Selective ion transport agents
Calix[4]pyrroles of type 1 (Fig. 5) have been applied successfully for the generation of effective anion receptors, which bind negatively charged guests through four hydrogen bonds.40,41 Lately, Matile, Ballester and co-workers have designed calix[4]pyrrole molecules bearing two aromatic groups (Fig. 5).42 Hence, these “two-wall” aryl-extended macrocyles have been used as valuable tools to investigate the potential role played by different π-substituents (molecules 1a–1e; Fig. 5) on the binding of the nitrate ion, which is an essential nutriment in plant biology.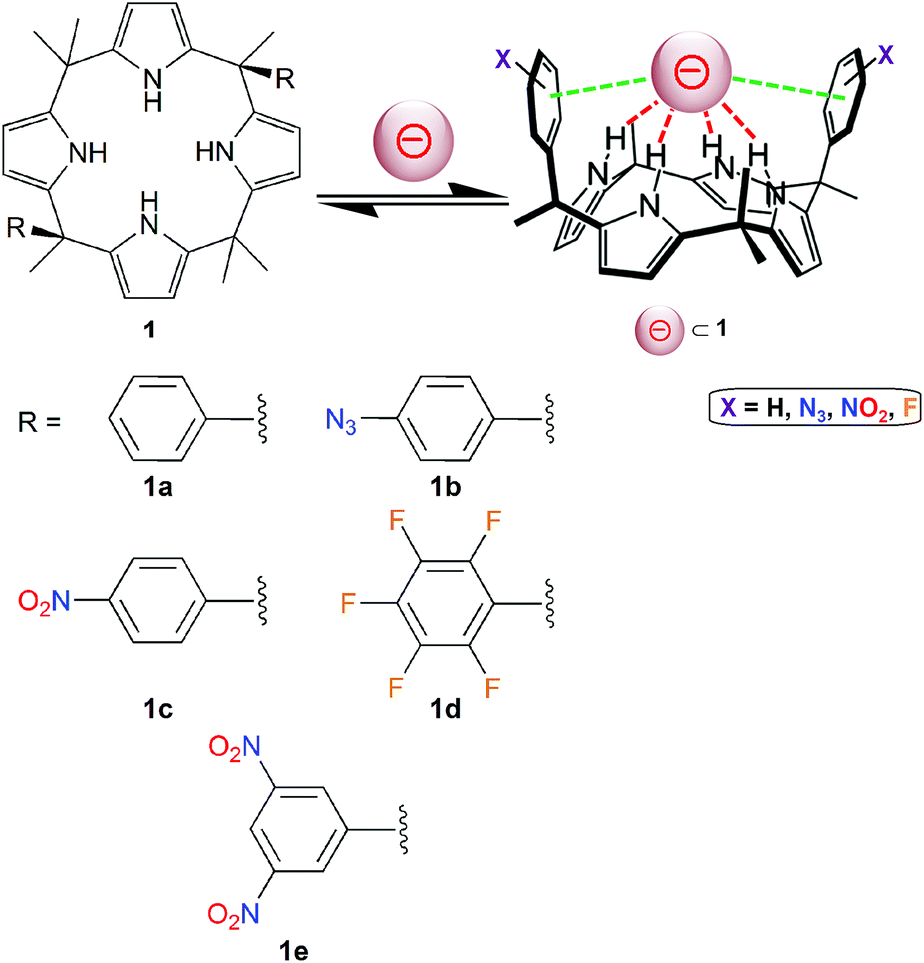 | ||
| Fig. 5 Molecular structures of “two-wall” aryl-extended calix[4]pyrroles 1 (holding different R groups) used for anion-binding studies.42 The pink sphere symbolizes an anion. | ||
The binding affinities of 1a–1e have been assessed quantitatively by 1H NMR, in deuterated acetonitrile and using tetrabutylammonium nitrate as the source of NO3−. Upon addition of nitrate, a comparable shifting of the signal corresponding to the pyrrolic NH proton is observed for the five receptors 1a–1e, which indicates that they can all bind the ion by means of hydrogen-bonding interactions. In addition, for compound 1e, when increasing amounts of [Bu4N][NO3] are added (actually, four equivalents of the nitrate salt are required to induce saturation in the chemical shift changes), the two signals of the 3,5-dinitrophenyl groups shift upfield. This important feature suggests that the NO3− ion does not interact via Carom–H⋯NO3− ion (hydrogen bonding, which would result in a downfield shifting of these signals). Therefore, the nitrate ion appears to interact by means of anion–π interactions with the two dinitro-substituted aryl substituents.
For all complexes, the free energy of binding has been calculated from the NMR data. The ΔG values, ranging from −2.0 kcal mol−1 (receptor 1a) to −4.3 kcal mol−1 (receptor 1e), illustrate the occurrence of attractive interactions. Comparison of these values with that of the calix[4]pyrrole without aryl substituents (namely with R = CH3 in Fig. 5) reveals that 1a and 1b experience repulsive nitrate–π interactions, while 1c shows slightly attractive nitrate–π interactions, and 1d and 1e (with the more π-acidic aromatic groups) clearly undergo the strongest anion–π contacts.
Single crystals of the supramolecular complex [Me4N][NO3]⊂1e have been obtained in acetonitrile; hence, its molecular structure has been determined by X-ray diffraction. These crystallographic studies show that [Me4N][NO3]⊂1e adopts two distinct binding geometries in the solid-state, which both support the proposed structure of the complex in solution.42 In the first geometry, the nitrate anion is situated pseudoparallel to the aromatic walls of the calix[4]pyrrole, while for the second binding motif, the NO3− ion is located almost perpendicular to the aryl groups. Although the π,π-sandwich geometry (pseudoparallel nitrate–π complex) cannot be fully excluded, it is believed that the perpendicular complex is expected to be predominant in solution. The structure of this perpendicular host–guest compound is shown in Fig. 6. This binding geometry is characterized by Npyrrole–H⋯nitrate hydrogen bonds in the range 2.927(2)–2.989(2) Å and NO3−⋯centroid (anion–π) contact distances of 3.220(3) and 3.330(2) Å (Fig. 6).
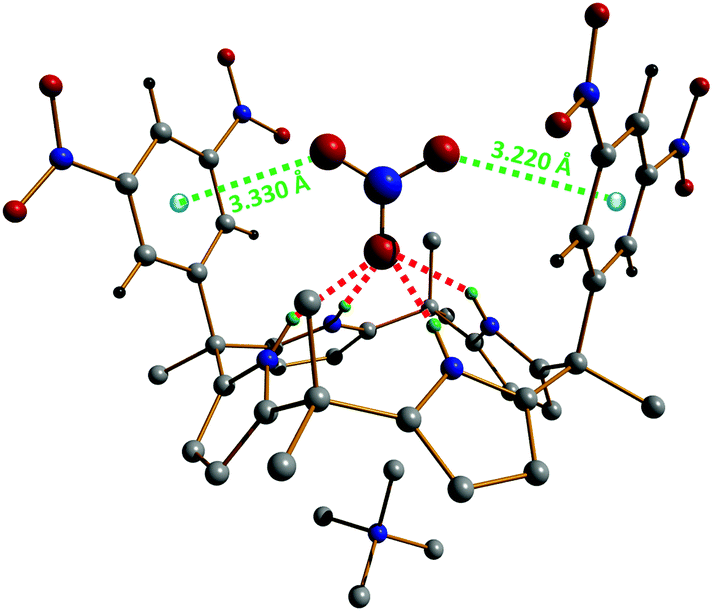 | ||
| Fig. 6 Perpendicular geometry observed in the single-crystal X-ray structure of [Me4N][NO3]⊂1e.42 Hydrogen bonds are shown as red dotted lines and the anion–π interactions as green dotted lines. The nitrate⋯centroid contact distances (anion–π) amount to 3.220(3) and 3.330(2) Å. | ||
The potential ion-transport activity of the calix[4]pyrroles 1 (Fig. 5) has been examined using large unilamellar vesicles (LUVs) containing a fluorophore (viz. pH-sensitive 8-hydroxy-1,3,6-pyrenetrisulfonate(HPTS) or carboxyfluorescein (CF)).42 EC50 values (EC50 corresponds to the effective calix[4]pyrrole concentration required to reach 50% activity in the assay) were determined from the HPTS and CF assays. For all pyrrole-based anionic receptors, the assay results are far better with HPTS than with CF, therefore indicating that the ion movements between the extra- and intravesicular environments take place through ion transport (via an anion/cation symport mechanism) and not vesicle rupture.42 Moreover, these studies have shown that compounds 1c and 1e are the more efficient ion transporters, and that 1e exhibits the greatest proficiency for the transport of nitrate (as expected from the initial NMR and crystallographic experiments). This higher selectivity for NO3− displayed by 1e is obviously due to the fact that the oxoanion is capable of satisfying its three centres of charges; indeed, one of the nitrate oxygen atoms is tightly hydrogen bonded to tetrapyrrole core while the two others favourably interact with the two 3,5-dinitrophenyl groups through anion–π contacts (Fig. 6). Accordingly, the combination of the electron-deficient aromatic walls of 1e with its four pyrrolic H-donor groups create a pocket that is perfectly suitable for the binding and protection of NO3− as it passes through the membrane.
4.2. Anion recognition
During the last ten years, the group of Wang is developing a new family of macrocyclic anion-host molecules based on the electron-deficient 1,3,5-triazine ring.43 Such oxacalix[2]arene[2]triazine derivatives are obtained applying a straightforward three-step procedure. The first two steps involve the reaction between 1,3,5-trichloro-2,4,6-triazine and resorcinol, which produce tetraoxacalix[2]arene[2]triazine 2, depicted in Fig. 7. The final step allows the functionalization of 2 through nucleophilic substitutions of its chlorine atoms.44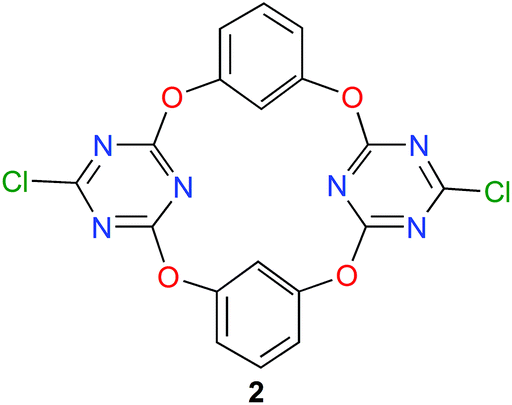 | ||
| Fig. 7 Structure of tetraoxacalix[2]arene[2]triazine 2.45 | ||
This year, Wang and co-workers have reported a beautiful study with the simplest member of this tetraoxacalix[2]arene[2]triazine family, namely the bis-chloro compound 2 (Fig. 7).45
Wang have shown that these heterocalix[4]aromatic molecules usually adopt 1,3-alternate conformation (especially when the triazine-ring substituents are not large).43 This spatial arrangement generates an electron-deficient aromatic cavity that can thus host electron-rich guests (anion–π interactions). For instance, the V-shaped receptor 2 can bind halides in acetonitrile by means of anion–π interactions.46 These encouraging results have prompted Wang and co-workers to use tetraoxacalix[2]arene[2]triazine 2 with different anions displaying distinct geometries, namely the tetrahedral tetrafluoroborate (BF4−), triangular nitrate (NO3−), octahedral hexafluorophosphate (PF6−) and linear thiocyanate (NCS−) anions. The anion-binding properties have been investigated in solution (spectrometric titrations), in the gas phase (mass spectrometry) and in the solid-state (X-ray crystallography).
The fluorescence titrations have been carried out in acetonitrile using tetrabutylammonium salts of the anions investigated.
The addition of increasing amounts of the different anions to an acetonitrile solution of 2 gives rise to the appearance of a new emission band, whose intensity is related to the guest concentration. All titration data point to the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 guest⊂host complexes for the four different anions. The stability of the anion⊂2 complexes follows the order NO3−⊂2 ≫ BF4−⊂2 > PF6−⊂2 > NCS−⊂2, with association constants ranging from 239 (NCS−) to 16950 (NO3−) M−1.45
:1 guest⊂host complexes for the four different anions. The stability of the anion⊂2 complexes follows the order NO3−⊂2 ≫ BF4−⊂2 > PF6−⊂2 > NCS−⊂2, with association constants ranging from 239 (NCS−) to 16950 (NO3−) M−1.45
The formation of the anion⊂2 complexes has also been evidenced in the gas phase through electrospray-ionization mass spectrometry (ESI-MS) measurements. These ESI-MS experiments have revealed the sole generation of 1:1 guest⊂host complexes.45
Next, single crystals of the supramolecular complexes obtained between tetraethylammonium salts of the four anions studied, i.e. [Et4N][X] (with X = BF4−, NO3− and NCS−), and 2 have been obtained and their solid-state structures have been determined by X-ray diffraction. The anionic parts (namely without the tetraethylammonium cation) of the corresponding X⊂2 complexes are shown in Fig. 8. All supramolecular complexes show anion–π interactions between the anion and one or two triazine rings. The tetrafluoroborate anion in BF4−⊂2 establishes anion–π contacts with both electron-deficient heteroarenes (Fig. 8a), one of them being well below the minimum F–triazine centroid distance, i.e. 2.96 Å, characterizing a strong interaction.28 Besides the anion–Cg (Cg = ring centroid) distances of 2.856(6) and 3.106(11) Å (green dotted lines in Fig. 8a), the BF4− anion is hydrogen bonded to one of the resorcinol aryl groups (BF4−⋯H–C distance of 2.650(2) Å; red dotted line in Fig. 8a). The nitrate ion strongly interacts through one of its oxygen atoms with one triazine ring, as evidenced by the O–Cg distance of 3.084(7) Å (green dotted line; Fig. 8b).28 In addition, the NO3−⊂2 complex is stabilized by two short NO3−⋯H–C bonding contacts of 2.440(2) Å (red dotted lines; Fig. 8b); hence, the oxoanion satisfies its three centres of charges, therefore forming a very stable host–guest complex. The hexafluorophosphate anion makes two weak anion–π contacts with both triazine rings; the observed F–Cg distances of 3.703(7) Å (green dotted lines; Fig. 8c) are significantly above the minimum value (namely 2.96 Å)28 for a strong anion–π interaction. Furthermore, the hexafluorophosphate ion is involved in hydrogen-bonding interactions with the two resorcinolyl moieties (PF6−⋯H–C distance of 2.340(7) Å; red dotted lines in Fig. 8c). The thiocyanate anion undergoes an anion–π interaction through its nitrogen atom and a lone pair–π interaction via its sulfur atom (Fig. 8d). The N–Cg distance of 3.050(5) Å matches to the minimum value that illustrates the occurrence of an anion–π contact.28 The lone pair–π distance of 3.643(2) Å (S–Cg; Fig. 8d) is indicative of a weak supramolecular bond; indeed, a strong bonding contact would be described by an S–Cg distance below 3.47 Å.28,29 The NCS guest is the only anion of the series investigated that does not undergo additional hydrogen-bonding interactions.
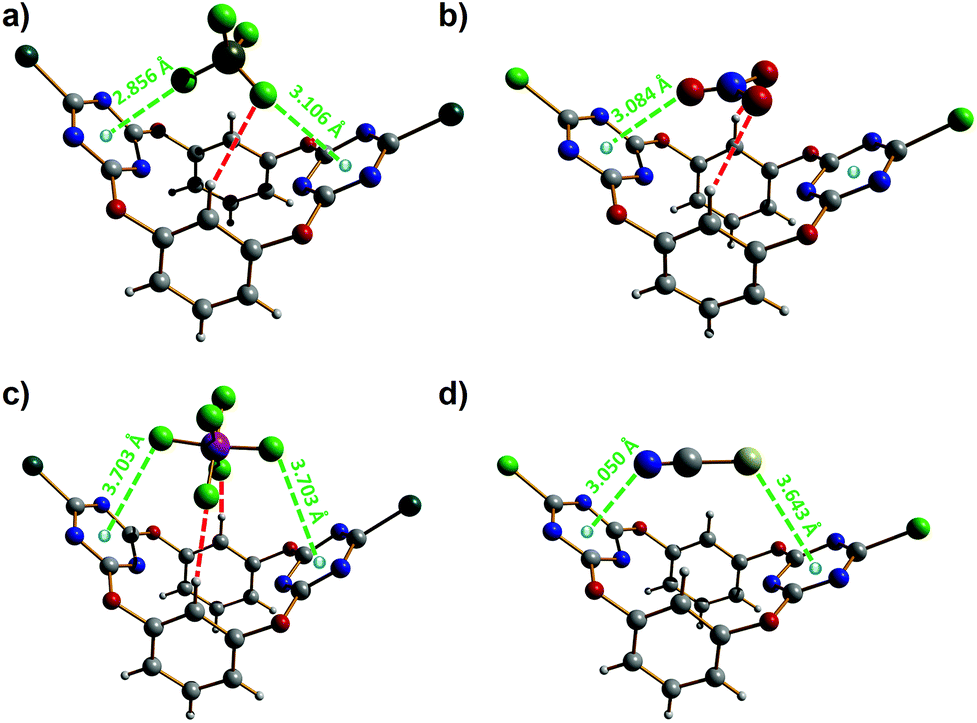 | ||
| Fig. 8 Molecular structures of the different anion⊂2 complexes.45 (a) BF4−⊂2; (b) NO3−⊂2; (c) PF6−⊂2; (d) NCS−⊂2. Hydrogen bonds are shown as red dotted lines and the anion–π interactions as green dotted lines. The anion–π and lone pair–π contact distances are given. | ||
4.3. Biological relevance of the anion–π interaction
As mentioned earlier in the introduction, the importance of the cation–π interaction in biological systems has been clearly recognized.9 In contrast, the demonstration of the significance of the anion–π (and lone pair–π) interaction in biology is still in its infancy.47,48Recently, Sacchettini and co-workers reported an outstanding study on the development of effective antituberculosis drugs, where anion–π interactions play an important role.49
The glyoxylate shunt (or glyoxylate cycle) is a variation of the tricarboxylic acid cycle (TCA cycle, also known as Krebs cycle) that allows bacteria (as well as plants, prokaryotes and fungi) to avoid the carbon dioxide generating steps of the TCA cycle through the use of fatty acids to synthesize carbohydrates. Since this pathway is absent in mammals, the alteration of the glyoxylate shunt is considered as a possible strategy to eradicate Mycobacterium tuberculosis (Mtb), the pathogenic bacteria responsible of most cases of tuberculosis (Tb). Hence, the inhibition of enzymes operating in the glyoxylate cycle represents a potential approach in antituberculosis drug design.49
The glyoxylate shunt involves two enzymes, namely isocitrate lyase (ICL), which mediates the hydrolysis of isocitrate into glyoxylate and succinate; and malate synthase (GlcB) that generates malate from glyoxylate, using one molecule of acetyl-CoA (acetyl coenzyme A).
GlcB is a better target than ICL for drug design because this enzyme displays a larger active site, consisting of a 20 Å × 7 Å cavity capable of hosting the pantothenate tail of the acetyl-CoA molecule, which contains a magnesium(II) ion (for the catalytic reaction). The development of small inhibitors of GlcB thus may allow the production of efficient antitubercular therapeutics.
It has been shown that phenyldiketo acids (PDKAs; Fig. 9) act as efficient inhibitors of GlcB.49
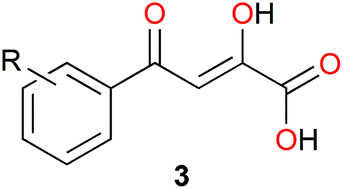 | ||
| Fig. 9 Chemical structure of phenyldiketo acid derivatives 3, PDKAs (R symbolizes various phenyl substituents), which are efficient inhibitors of GlcB.49 | ||
For instance, (Z)-2-hydroxy-4-oxo-4-phenylbut-2-enoic acid (3a, R = H in Fig. 9) exhibits an IC50 value (IC50 is the half maximal inhibitory concentration) of 2.0 μM against GlcB (moreover, it is inactive against ICL).49 Actually, IC50 values ranging from 0.17 to 6.0 μM have been obtained for a series of different aryl-substituted PDKAs, the best inhibitor being (Z)-4-(3-chlorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (3f, R = 3-Cl in Fig. 9).49
The crystal structures of various phenyldiketo acid inhibitors bound to the active site of GlcB could be obtained (Fig. 10). The molecular structure of GlcB complexed with (Z)-2-hydroxy-4-oxo-4-phenylbut-2-enoic acid, the parent member of the PDKA family (3a, R = H in Fig. 9), reveals that the inhibitor coordinates to the magnesium(II) ion in an edge-on fashion (Fig. 10a), which is comparable to that observed for the natural binders glyoxylate or malate.49 The GlcB-inhibitor complex is stabilized by a number of supramolecular interactions, including hydrogen bonds and anion–π contacts (see red and green dotted lines in Fig. 10a). Thus, the β-diketone unit of the PDKA molecule is doubly hydrogen bonded to the guanidinium group of an arginine residue (red dotted lines in Fig. 10a), with Ndonor–Oacceptor contact distances of 2.913 and 2.937 Å. In addition, the inhibitor is involved in anion–π interactions (green dotted lines in Fig. 10a) through (i) its carboxylate moiety that is in close contact with the carbonyl function of a glycine residue (OPDKA–CGly distance of 3.141 Å)50 and (ii) its phenyl ring that interacts with the carboxylate group of aspartic acid residue (characterized by short Cphenyl–OAsp contact distances of 3.265 and 3.387 Å).
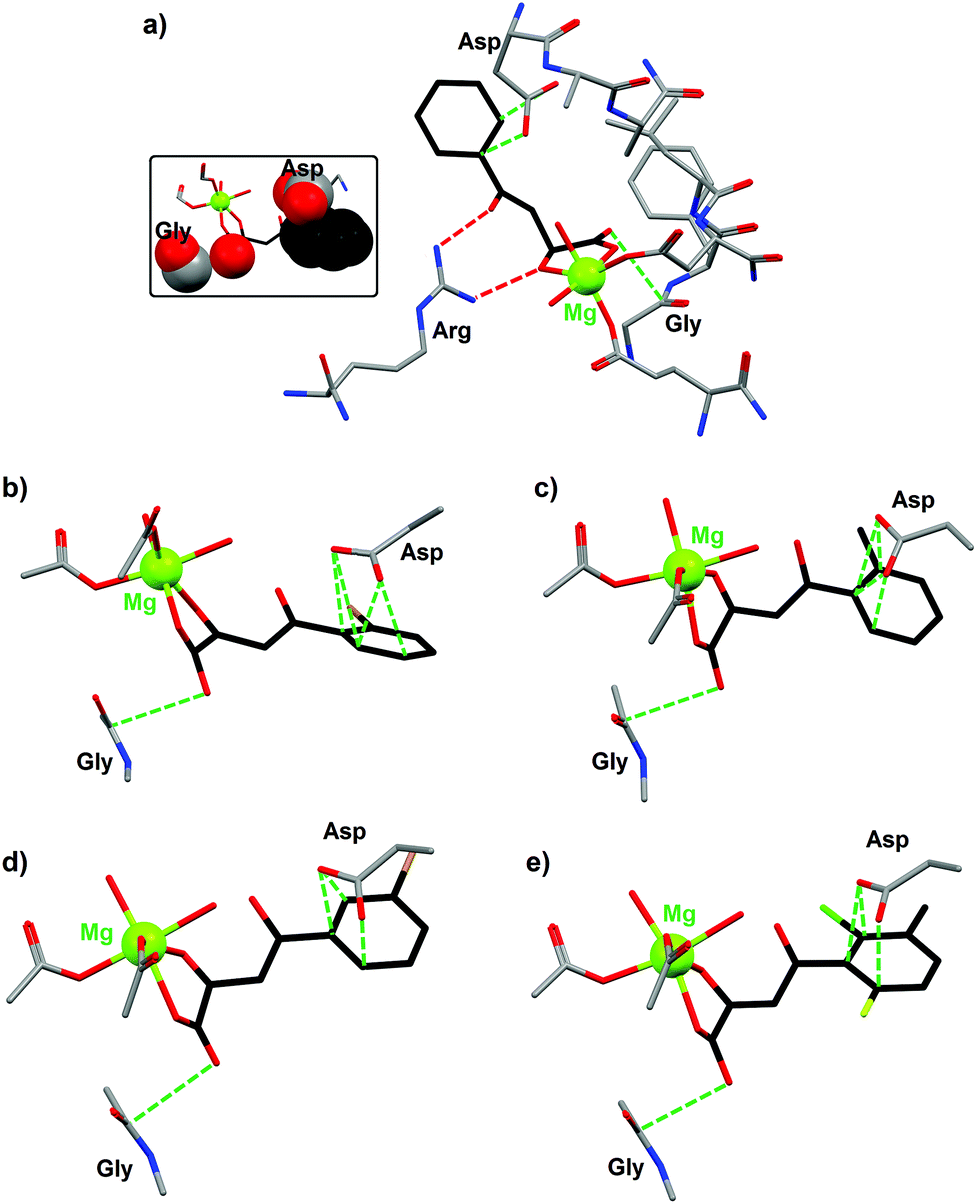 | ||
| Fig. 10 Crystal structures of the active site of malate synthase (GlcB) complexed with the inhibitor (a) (Z)-2-hydroxy-4-oxo-4-phenylbut-2-enoic acid (3a, R = H in Fig. 9; PDB code 3S9I); (b) (Z)-4-(2-bromophenyl)-2-hydroxy-4-oxobut-2-enoic acid (3b, R = 2-Br in Fig. 9; PDB code 3S9Z); (c) (Z)-2-hydroxy-4-oxo-4-o-tolylbut-2-enoic acid (3c, R = CH3 in Fig. 9; PDB code 3SAD); (d) (Z)-4-(3-bromophenyl)-2-hydroxy-4-oxobut-2-enoic acid (3d, R = 3-Br in Fig. 9; PDB code 3SAZ); (e) (Z)-4-(2-chloro-6-fluoro-3-methylphenyl)-2-hydroxy-4-oxobut-2-enoic acid (3e, R = 2-Cl, 3-methyl, 6-F in Fig. 9; PDB code 3SB0).49 Supramolecular interactions between the inhibitor and different amino acid residues of GlcB are shown as dotted lines (red dotted lines for H bonds and green dotted lines for anion–π contacts). The inset figure illustrates the occurrence of strong anion–π interactions (shown in space-filling mode) between (Z)-2-hydroxy-4-oxo-4-phenylbut-2-enoic acid and two amino acid residues of GlcB. | ||
Similar structural features are observed with four other PDKA derivatives; hence, the anion–π interactions for all GlcB–PDKA complexes have been compared. For (Z)-4-(2-bromophenyl)-2-hydroxy-4-oxobut-2-enoic acid (3b, R = 2-Br in Fig. 9), the OPDKA–CGly distance amounts to 3.214 Å and the phenyl–Asp supramolecular pair is stabilized by four short Cphenyl–OAsp bonds, ranging from 2.992 to 3.155 Å (Fig. 10b). For (Z)-2-hydroxy-4-oxo-4-o-tolylbut-2-enoic acid (3c, R = CH3 in Fig. 9), the OPDKA–CGly distance is 3.170 Å and the four Cphenyl–OAsp bonds vary in the range 3.122–3.448 Å (Fig. 10c). For (Z)-4-(3-bromophenyl)-2-hydroxy-4-oxobut-2-enoic acid (3d, R = 3-Br in Fig. 9), the OPDKA–CGly distance is 3.157 Å. The phenyl–Asp interaction is stabilized by three Cphenyl–OAsp bonds, which range from 2.978 to 3.348 Å (Fig. 10d). Finally, for the trisubstituted-aryl molecule (Z)-4-(2-chloro-6-fluoro-3-methylphenyl)-2-hydroxy-4-oxobut-2-enoic acid (3e, R = 2-Cl, 3-methyl, 6-F in Fig. 9), the OPDKA–CGly distance is 3.128 Å and the three Cphenyl–OAsp bonds are in the range 2.885–3.328 Å (Fig. 10e).50
For the monosubstituted PDKA inhibitors 3a–3d, the strength of the phenyl–Asp anion–π interaction follows the order 3d ≈ 3b > 3c > 3a. The PDKA–Gly interaction is comparable for all complexes, within the range 3.141–3.214 Å. Interestingly, the IC50 values respect the same trend, namely 3b (0.6 μM) ≈ 3d (0.8 μM) > 3c (1.1 μM) > 3a (2.0 μM). Therefore, a correlation between the anion–π interactions (phenyl–Asp interactions) and the GlcB-inhibition activities is clearly observed. The trisubstituted inhibitor 3e, although it gives rise to the shortest Cphenyl–OAsp contact (namely 2.885 Å), displays the poorest IC50 value (i.e. 2.5 μM) of the 3a–3e series. This lower efficiency exhibited by 3e is most likely due to steric reasons; indeed, less room is available in the pocket of the 6-position of the aryl group compared to the 2- or 3-positions.49 The significance of the anion–π interactions is further confirmed by the use of the inhibitors (Z)-2-hydroxy-4-(2,6-dimethylphenyl)-4-oxobut-2-enoic acid (3g, R = 2,6-dimethyl in Fig. 9) and (Z)-4-(2-chloro-6-fluorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (3h, 2-Cl, 6-F in Fig. 9), which are of similar size and substitution positioning. Indeed, 3g has an IC50 > 100 μM (i.e. this compound is inactive) whereas 3h gives an IC50 of 2.7 μM.49 Obviously, the influence of the ring substituents of the PDKA derivatives is critical for the inhibition activity; the electron-withdrawing halogen substituents have an effect on ring π–interactions with the carboxylate of the Asp residue (Fig. 10).
5. Conclusions
During its first years of existence, there has been some controversy regarding the actual occurrence of the anion–π interaction.10 Thanks to several worldwide theoretical and experimental studies, this new noncovalent bond has been increasingly acknowledged by the scientific community; hence, the examples of its utilization reported in the present review clearly demonstrate that the anion–π interaction is now considered as a supramolecular bond, just as the related cation–π interaction. It should be mentioned however that while the cation–π interaction has been widely investigated and evidenced in biological structures, there has been comparatively little discussion of the potential structural and functional relevance of the anion–π interaction in biomolecules.47 The last example,49 described in section 4, and more recent ones51,52 unambiguously provide direct evidence for anion–π interactions occurring in biological systems. Therefore, in the coming years, due consideration should be given to the potential participation of anion–π supramolecular bonds in the function of particular biochemical structures or the formation of biological assemblies involving aryl groups (like for instance from amino acids or nucleobases).Acknowledgements
ICREA (Institució Catalana de Recerca i Estudis Avançats) and the Ministerio de Economía y Competitividad of Spain (project CTQ2011-27929-C02-01) are acknowledged.Notes and references
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- C. A. Schalley, Analytical Methods in Supramolecular Chemistry, Wiley-VCH, Weinheim, 2007 Search PubMed.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, in Molecular Cell Biology, ed. W. H. Freeman, New York, 4th edn, 2000 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CrossRef CAS PubMed.
- Ionic interactions in natural and synthetic macromolecules, ed. Alberto Ciferri and Angelo Perico, John Wiley & Sons, Inc., Hoboken, New Jersey, 2012 Search PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Waltham, San Diego, Oxford, Amsterdam, 3rd edn, 2011 Search PubMed.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed.
- D. Quinonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deya, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef CAS.
- M. Mascal, A. Armstrong and M. D. Bartberger, J. Am. Chem. Soc., 2002, 124, 6274–6276 CrossRef CAS PubMed.
- P. de Hoog, P. Gamez, H. Mutikainen, U. Turpeinen and J. Reedijk, Angew. Chem., Int. Ed., 2004, 43, 5815–5817 CrossRef CAS PubMed.
- S. Demeshko, S. Dechert and F. Meyer, J. Am. Chem. Soc., 2004, 126, 4508–4509 CrossRef CAS PubMed.
- The occurrence of short anion⋯arene close contacts has been observed earlier but has not been recognised as anion–π interactions; see for instance: J. W. Steed, R. K. Juneja and J. L. Atwood, Angew. Chem., Int. Ed., 1994, 33, 2456–2457 CrossRef CAS; B. Hasenknopf, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Angew. Chem., Int. Ed. Engl., 1996, 35, 1838–1840 CrossRef.
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed.
- A. Robertazzi, F. Krull, E. W. Knapp and P. Gamez, CrystEngComm, 2011, 13, 3293–3300 RSC.
- A. Frontera, Coord. Chem. Rev., 2013, 257, 1716–1727 CrossRef CAS PubMed.
- G. Aragay, A. Frontera, V. Lloveras, J. Vidal-Gancedo and P. Ballester, J. Am. Chem. Soc., 2013, 135, 2620–2627 CrossRef CAS PubMed.
- S. Marsili, R. Chelli, V. Schettino and P. Procacci, Phys. Chem. Chem. Phys., 2008, 10, 2673–2685 RSC.
- C. Garau, A. Frontera, D. Quinonero, P. Ballester, A. Costa and P. M. Deya, ChemPhysChem, 2003, 4, 1344–1348 CrossRef CAS PubMed.
- C. Garau, D. Quinonero, A. Frontera, P. Ballester, A. Costa and P. M. Deya, Org. Lett., 2003, 5, 2227–2229 CrossRef CAS PubMed.
- D. Y. Kim, N. J. Singh and K. S. Kim, J. Chem. Theory Comput., 2008, 4, 1401–1407 CrossRef CAS.
- C. Garau, D. Quinonero, A. Frontera, P. Ballester, A. Costa and P. M. Deya, New J. Chem., 2003, 27, 211–214 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2013, 15, 4565–4570 RSC.
- C. Estarellas, A. Bauza, A. Frontera, D. Quinonero and P. M. Deya, Phys. Chem. Chem. Phys., 2011, 13, 5696–5702 RSC.
- T. J. Mooibroek, C. A. Black, P. Gamez and J. Reedijk, Cryst. Growth Des., 2008, 8, 1082–1093 CAS.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- IsoStar Knowledge Base from the Cambridge Crystallographic Data Centre (CCDC), http://www.ccdc.cam.ac.uk/products/csd_system/isostar/.
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 3902–3906 RSC.
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 1027–1030 RSC.
- D. Quinonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deya, ChemPhysChem, 2007, 8, 1182–1187 CrossRef CAS PubMed.
- J. Sunner, K. Nishizawa and P. Kebarle, J. Phys. Chem., 1981, 85, 1814–1820 CrossRef CAS.
- C. Garau, D. Quinonero, A. Frontera, P. Ballester, A. Costa and P. M. Deya, J. Phys. Chem. A, 2005, 109, 9341–9345 CrossRef CAS PubMed.
- D. Escudero, A. Frontera, D. Quinonero and P. M. Deya, J. Comput. Chem., 2009, 30, 75–82 CrossRef CAS PubMed.
- D. Quinonero, A. Frontera and P. M. Deya, ChemPhysChem, 2008, 9, 397–399 CrossRef CAS PubMed.
- A. Frontera, D. Quinonero, A. Costa, P. Ballester and P. M. Deya, New J. Chem., 2007, 31, 556–560 RSC.
- H. Casellas, C. Massera, F. Buda, P. Gamez and J. Reedijk, New J. Chem., 2006, 30, 1561–1566 RSC.
- I. W. Park, J. Yoo, S. Adhikari, J. S. Park, J. L. Sessler and C. H. Lee, Chem.–Eur. J., 2012, 18, 15073–15078 CrossRef CAS PubMed.
- R. Custelcean, L. H. Delmau, B. A. Moyer, J. L. Sessler, W. S. Cho, D. Gross, G. W. Bates, S. J. Brooks, M. E. Light and P. A. Gale, Angew. Chem.,Int. Ed., 2005, 44, 2537–2542 CrossRef CAS PubMed.
- L. Adriaenssens, C. Estarellas, A. V. Jentzsch, M. M. Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324–8330 CrossRef CAS PubMed.
- M. X. Wang, Chem. Commun., 2008, 4541–4551 RSC.
- Q. Q. Wang, D. X. Wang, Q. Y. Zheng and M. X. Wang, Org. Lett., 2007, 9, 2847–2850 CrossRef CAS PubMed.
- D. X. Wang and M. X. Wang, J. Am. Chem. Soc., 2013, 135, 892–897 CrossRef CAS PubMed.
- D. X. Wang, Q. Y. Zheng, Q. Q. Wang and M. X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7485–7488 CrossRef CAS PubMed.
- G. J. Jones, A. Robertazzi and J. A. Platts, J. Phys. Chem. B, 2013, 117, 3315–3322 CrossRef CAS PubMed.
- M. Egli and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197–205 CrossRef CAS PubMed.
- I. V. Krieger, J. S. Freundlich, V. B. Gawandi, J. P. Roberts, Q. Sun, J. L. Owen, M. T. Fraile, S. I. Huss, J. L. Lavandera, T. R. Ioerger and J. C. Sacchettini, Chem. Biol., 2012, 19, 1556–1567 CrossRef CAS PubMed.
- A. Das, S. R. Choudhury, C. Estarellas, B. Dey, A. Frontera, J. Hemming, M. Helliwell, P. Gamez and S. Mukhopadhyay, CrystEngComm, 2011, 13, 4519–4527 RSC.
- J. P. Schwans, F. Sunden, J. K. Lassila, A. Gonzalez, Y. Tsai and D. Herschlag, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11308–11313 CrossRef CAS PubMed.
- A. Bauza, D. Quinonero, P. M. Deya and A. Frontera, Chem.–Asian J., 2013, 8, 2708–2713 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |