 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of polymer chemistry on globular protein–polymer block copolymer self-assembly†
Dongsook
Chang
,
Christopher N.
Lam
,
Shengchang
Tang
and
Bradley D.
Olsen
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: bdolsen@mit.edu
First published on 30th June 2014
Abstract
Bioconjugates of the model red fluorescent protein mCherry and synthetic polymer blocks with different hydrogen bonding functionalities show that the chemistry of the polymer block has a large effect on both ordering transitions and the type of nanostructures formed during bioconjugate self-assembly. The phase behaviours of mCherry-b-poly(hydroxypropyl acrylate) (PHPA) and mCherry-b-poly(oligoethylene glycol acrylate) (POEGA) in concentrated aqueous solution show that changes in polymer chemistry result in increase in the order–disorder transition concentrations (CODTs) by approximately 10–15 wt% compared to a previously studied globular protein–polymer block copolymer, mCherry-b-poly(N-isopropylacrylamide) (PNIPAM). The CODTs are always minimized for symmetric bioconjugates, consistent with the importance of protein–polymer interactions in self-assembly. Both mCherry-b-PHPA and mCherry-b-POEGA also form phases that have not previously been observed in other globular protein–polymer conjugates: mCherry-b-PHPA forms a cubic phase that can be indexed to Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d and mCherry-b-POEGA displays coexistence of lamellae and a cubic Iad structure over a narrow range of concentration and temperature. Several common behaviours are also revealed by comparison of different polymer blocks. With increasing concentration and temperature, ordered phases always appear in the order lamellar, cubic/PL, and hexagonal, although not all phases are observed in all materials. High concentration solutions (near 80 wt%) also undergo a re-entrant order–disorder transition to form nematic liquid crystalline phases, regardless of the polymer block chemistry.
d and mCherry-b-POEGA displays coexistence of lamellae and a cubic Iad structure over a narrow range of concentration and temperature. Several common behaviours are also revealed by comparison of different polymer blocks. With increasing concentration and temperature, ordered phases always appear in the order lamellar, cubic/PL, and hexagonal, although not all phases are observed in all materials. High concentration solutions (near 80 wt%) also undergo a re-entrant order–disorder transition to form nematic liquid crystalline phases, regardless of the polymer block chemistry.
Introduction
There is great interest in incorporating a variety of proteins into bio-based materials to capture their high catalytic activity or specific molecular recognition ability. Potential applications include biocatalysts,2,3 biosensors,4–6 and bioelectronic devices using electrocatalytic or photoelectric materials.7,8 These protein-based biomaterials can benefit from control over the nanoscale arrangement of the proteins because it promotes proper orientation of proteins, facile transport of materials (substrates, products, or charges), and improved stability and longevity of folded protein structures.10,11 Both top-down approaches that combine lithographic techniques with protein immobilization chemistry and bottom-up approaches including block copolymer templating methods12–16 have been demonstrated to pattern proteins on the nanometer length scale in 2-D and 3-D. While lithographic techniques are capable of achieving very precise spatial control, patterning is limited to two dimensions, and the resultant protein density per unit area is relatively low. Methods using diblock copolymers or lipids to template proteins can achieve relatively high protein density, but the majority of the material is typically template polymer, reducing the total functional density of protein.The direct self-assembly of bioconjugates where a protein and a water-soluble polymer block are conjugated to form a block copolymer is a promising method that can potentially meet the design criteria for materials incorporating globular proteins and enzymes.18–20 It can achieve high three-dimensional density, connectivity of domains, and control over nanoscale morphology and protein orientation.18 In addition, the need for the use of harsh organic solvents is eliminated. Despite many advances in protein–polymer bioconjugate chemistry,21–23 the majority of studies of globular protein–polymer conjugates have focused on dilute solution self-assembly behaviour. Proteins conjugated to thermoresponsive polymers form spherical aggregates24–26 including micelles and vesicles for applications in drug delivery. More complex architectures including toroids and figure eight structures have been reported from triblock conjugates composed of a globular protein and a diblock copolymer.27
Introduction of a globular protein as one block in a diblock copolymer results in significant changes in the phase behaviour of the materials due to the effects of the globular protein shape and surface interactions (hydrophobic, ionic, and hydrogen bonding interactions).28 Using a model protein–polymer conjugate block copolymer, mCherry-b-poly(N-isopropylacrylamide) (PNIPAM), it was observed that lamellar phases are favoured for coil fractions greater than 0.5 while hexagonal phases are favoured at lower coil fractions. A small region of hexagonally perforated lamellae was also observed near a coil fraction of 0.5. The order–disorder transition concentration (CODT) for mCherry-b-PNIPAM reaches a minimum for near-symmetric polymer volume fractions, suggesting that repulsive protein–polymer interactions contribute to the self-assembly of globular protein–polymers. Unexpectedly, re-entrant order–disorder transition (ODT) behaviour is observed such that the bioconjugate becomes disordered at very high solution concentrations. This phase behaviour is significantly different than traditional coil–coil block copolymers.29–31
Herein, the role of polymer chemistry and protein–polymer interactions on the self-assembly of mCherry–polymer bioconjugates is explored. Because hydrogen bonding is one of the key interactions between proteins and polymers, it is hypothesized that changing hydrogen bonding may enable differences in self-assembly to be observed due to changes in protein–polymer interactions. Therefore, conjugate block copolymers are prepared from poly(hydroxypropyl acrylate) (PHPA) and poly(oligoethylene glycol acrylate) (POEGA), polymers specifically chosen to have lower critical solution temperature (LCST) behaviour similar to PNIPAM but different hydrogen bonding patterns. PHPA is a hydrogen bond donor and acceptor based on hydroxyl and ester groups instead of amide groups, and POEGA is a hydrogen bond acceptor only. PHPA and POEGA have lower critical solution temperatures (LCSTs) which enable control of solvent-mediated polymer interactions using temperature. It is shown that these changes in polymer chemistry have a large effect on the protein–polymer conjugate phase behaviour, yielding an important path for structure control in bioconjugate materials and insights into the fundamental physics governing their self-assembly.
Experimental details
Materials
2-Hydroxypropyl acrylate (95%, mixture of isomers) and 2-methoxyethyl acrylate (98%) (MEA) were purchased from Sigma-Aldrich. Inhibitors were removed by passing the liquid monomers through a basic alumina column. Azobisisobutyronitrile (AIBN) was recrystallized from methanol prior to use. 2-(2-Methoxyethoxy)ethyl acrylate (MEEA) monomer was synthesized according to a previously reported method.32 Details of this synthesis are provided in the ESI.† All other reagents and solvents were purchased from Sigma-Aldrich and used without further purification.Synthesis and characterization of mCherry–b–PHPA and mCherry–b–POEGA bioconjugates
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.5 molar ratio of monomer–CTA–AIBN for polymers with Mn of 17.7 and 27.2 kg mol−1, and at a ratio of 1600:1:1.45 for Mn of 57.0 kg mol−1. All reactions were started with 40 g monomer at 2 M monomer concentration. Reaction mixtures were degassed by three freeze–pump–thaw cycles, initiated by heating to 55 °C, and stopped before reaching a conversion of 30% to minimize incorporation of the protected maleimide endgroup into the growing polymer chains. RAFT polymerization of POEGA was carried out in dioxane with all other conditions the same as PHPA polymerization. Feed ratios of MEA–MEEA–CTA–initiator were 270:270:1:0.5 for polymers with Mn of 18.4 and 26.1 kg mol−1 and 627:660:1:0.5 for Mn of 57.6 kg mol−1. PHPA and POEGA were each precipitated three times after polymerization, in a 1:1 mixture of hexanes–ethyl acetate and pure hexanes, respectively. The maleimide-protecting group was removed by a retro Diels–Alder reaction by heating the polymers to 120 °C for 2 hours under vacuum. To eliminate all detectable coupling between maleimide groups during deprotection, approximately 10 wt% of butylated hydroxytoluene and hydroxyquinone were each added to the polymers prior to deprotection. Added inhibitors were removed by repeating the same precipitation steps three times followed by a thermal precipitation in water.
:1:0.5 molar ratio of monomer–CTA–AIBN for polymers with Mn of 17.7 and 27.2 kg mol−1, and at a ratio of 1600:1:1.45 for Mn of 57.0 kg mol−1. All reactions were started with 40 g monomer at 2 M monomer concentration. Reaction mixtures were degassed by three freeze–pump–thaw cycles, initiated by heating to 55 °C, and stopped before reaching a conversion of 30% to minimize incorporation of the protected maleimide endgroup into the growing polymer chains. RAFT polymerization of POEGA was carried out in dioxane with all other conditions the same as PHPA polymerization. Feed ratios of MEA–MEEA–CTA–initiator were 270:270:1:0.5 for polymers with Mn of 18.4 and 26.1 kg mol−1 and 627:660:1:0.5 for Mn of 57.6 kg mol−1. PHPA and POEGA were each precipitated three times after polymerization, in a 1:1 mixture of hexanes–ethyl acetate and pure hexanes, respectively. The maleimide-protecting group was removed by a retro Diels–Alder reaction by heating the polymers to 120 °C for 2 hours under vacuum. To eliminate all detectable coupling between maleimide groups during deprotection, approximately 10 wt% of butylated hydroxytoluene and hydroxyquinone were each added to the polymers prior to deprotection. Added inhibitors were removed by repeating the same precipitation steps three times followed by a thermal precipitation in water.
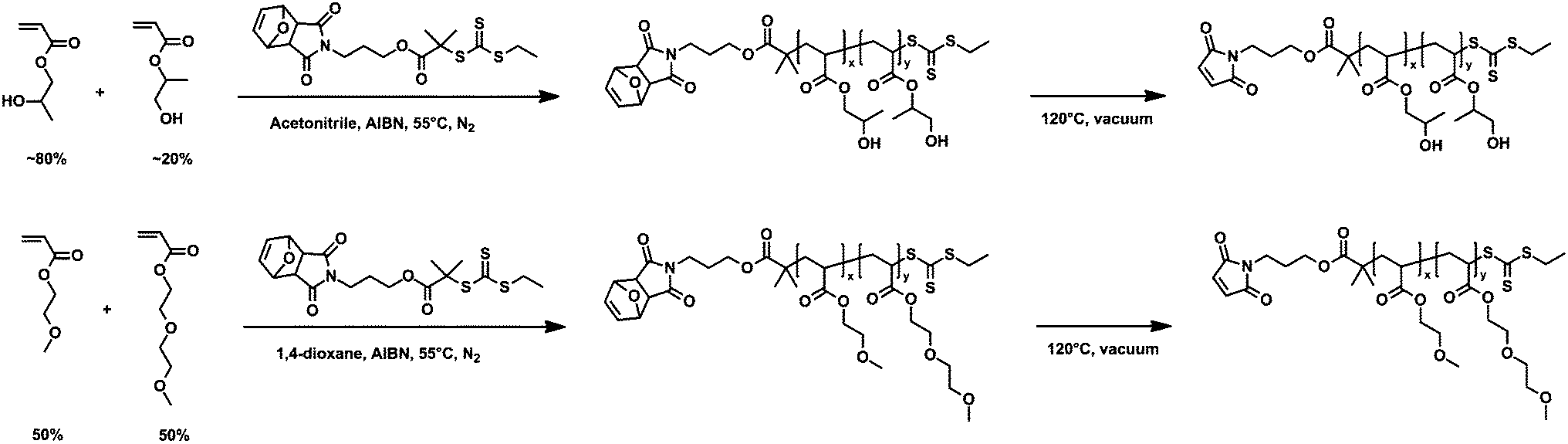 | ||
| Scheme 1 Synthesis of poly(hydroxypropyl acrylate) (PHPA; top) and poly(oligoethyleneglycol acrylate) (POEGA; bottom) by RAFT polymerization. | ||
The molar masses and polydispersity indexes (PDI) of the synthesized polymers were analysed by gel permeation chromatography before and after deprotection, showing no observable change. An Agilent Technologies 1260 Infinity system using two ResiPore, 7.5 × 300 mm (Agilent Technologies) columns in N,N-dimethylformamide (DMF) with 0.02 M lithium bromide (LiBr) as the mobile phase was used for chromatography, and signals were collected from a Wyatt DAWN HELEOS II multi-angle light scattering detector and a Wyatt Optilab T-rEX refractometer. The dn/dc values of PHPA and POEGA were measured using a built-in method; experimentally determined values of 0.053 and 0.042 mL g−1 (Fig. S2†) were used to obtain absolute molar masses. The reported molar masses and PDI values in Table 1 were measured after the deprotection step.
| Abbreviation | Copolymer | Polymer Mn (kg mol−1) | PDI | Bioconjugate Mn (kg mol−1) | ϕ polymer | Compositionb | Cloud pointc (°C) |
|---|---|---|---|---|---|---|---|
| a Density of PHPA 1.16,1 POEGA 1.05,9 and mCherryS131C 1.35 g cm−3 (ref. 17) were used to calculate volume fraction of polymers in bioconjugates. b The composition shown is the molar ratio of MEA to MEEA monomer in POEGA. c The cloud point was measured for homopolymers in water. | |||||||
| mChPH18 | mCherry–b–PHPA 18 kDa | 17.7 | 1.20 | 45.8 | 0.42 | — | 11.6 |
| mChPH27 | mCherry–b–PHPA 27 kDa | 27.2 | 1.22 | 55.4 | 0.53 | — | 11.4 |
| mChPH57 | mCherry–b–PHPA 57 kDa | 57.0 | 1.23 | 85.1 | 0.70 | — | 11.4 |
| mChPOE18 | mCherry–b–POEGA 18 kDa | 18.4 | 1.18 | 46.5 | 0.46 | 54/46 | 18.7 |
| mChPOE26 | mCherry–b–POEGA 26 kDa | 26.1 | 1.19 | 54.2 | 0.54 | 53/47 | 21.7 |
| mChPOE57 | mCherry–b–POEGA 57 kDa | 57.6 | 1.25 | 85.7 | 0.72 | 49/51 | 23.0 |
H1-NMR was recorded on a Bruker 400 MHz spectrometer in CDCl3 to determine the monomer composition of POEGA. Composition was calculated based on the ratio of acrylate methylene (δ4.23, –COO–CH2–) and oxymethylene groups (δ3.55–3.69, –COO–CH2CH2O– and –OCH2CH2O–) (Fig. S5†). The calculated monomer composition (Table 1) matched well with the approximately equimolar feed ratio, with a slightly higher incorporation of MEA monomer observed. The cloud points of the polymers were determined by measuring the transmittance of 1 wt% polymer solutions in water using UV-vis spectrometry with a temperature ramp from 5 °C to 20 °C and from 10 °C to 30 °C for PHPA and POEGA, respectively, at 0.2 °C per minute. The temperature at which a 50% drop in transmittance was observed was recorded as the cloud point according to reported methods in the literature.34 The cloud point of PHPA measured is lower than reported values,34,35 which can be attributed to the presence of the hydrophobic maleimide end group.
Results and discussion
In comparison to previously reported mCherry-b-PNIPAM bioconjugates, mCherry-b-PHPA and mCherry-b-POEGA diblock copolymers show phase transitions at significantly different concentrations and temperatures as well as the appearance of previously unobserved nanostructures. Phase diagrams are constructed for both polymers at three different polymer volume fractions (Fig. 1) by using a complementary set of characterization techniques including SAXS (Fig. 4 and S7†), DPLS, and turbidimetry (Table 2).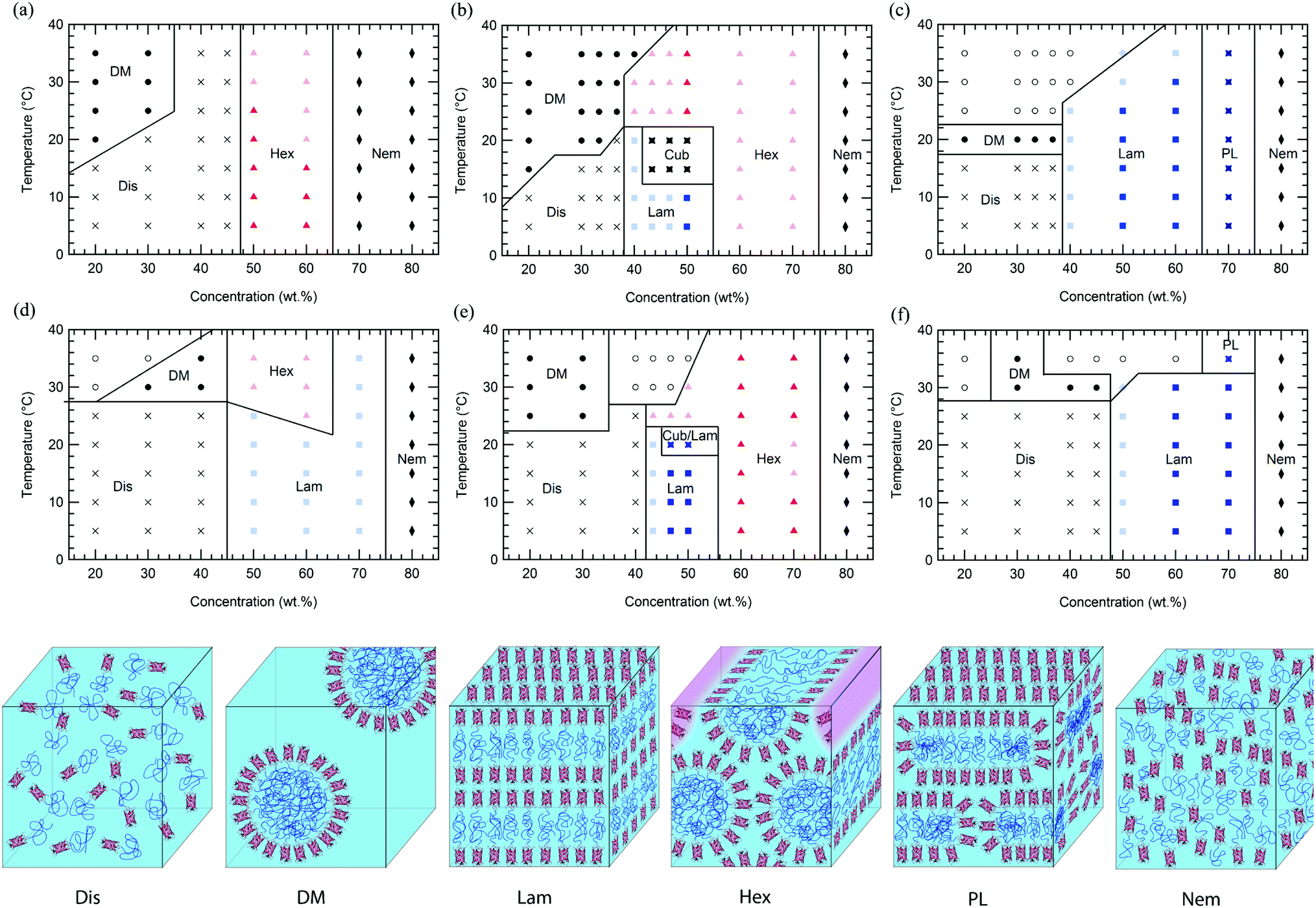 | ||
| Fig. 1 Phase diagrams of mCherry-b-PHPA in aqueous solution are shown at the top row at polymer fraction (a) 0.42, (b) 0.53, and (c) 0.70, and mCherry-b-POEGA at the bottom row at polymer fraction (d) 0.46, (e) 0.54, and (f) 0.72 as a function of concentration and temperature. Dis: disordered phase, DM: disordered micellar phase, Hex: hexagonally packed cylindrical phase, Lam: lamellar phase, Cub: bicontinuous cubic phase, PL: perforated lamellar phase, Nem: nematic phase, and Cub/Lam: cubic and lamellar phase coexistence. Light coloured symbols represent non-birefringent lamellae or hexagonally packed cylinders, while dark symbols represent birefringent phases. Closed symbols denote a homogeneous phase, and open symbols denotes macrophase separated phases. | ||
| T DPLS (°C) | T t (°C) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mCherry-b-PHPA | mCherry-b-POEGA | mCherry-b-PHPA | mCherry-b-POEGA | |||||||||
| ϕ polymer | 0.4 | 0.5 | 0.7 | 0.4 | 0.5 | 0.7 | 0.4 | 0.5 | 0.7 | 0.4 | 0.5 | 0.7 |
| a Temperature at which bioconjugates lost birefringence. b ○: sample displayed no birefringence, ●: sample remained birefringent at all temperatures, —: sample did not macrophase separate. | ||||||||||||
| 20 wt% | ○ | ○ | ○ | ○ | ○ | ○ | — | — | 20.5 | 28.1 | — | 27.0 |
| 30 wt% | ○ | ○ | ○ | ○ | ○ | ○ | — | — | 21.5 | 31.7 | — | — |
| 40 wt% | ○ | ○ | ○ | ○ | ○ | ○ | — | — | 25.8 | — | 25.2 | 32.0 |
| 50 wt% | 29.3 | ● | 29.1 | ○ | 19.8 | ○ | — | — | — | — | 31.1 | 29.4 |
| 60 wt% | 20.1 | ○ | 34.4 | ○ | ● | 31.8 | — | — | — | — | — | 34.8 |
| 70 wt% | ● | ○ | ● | ○ | ● | ● | — | — | — | — | — | — |
| 80 wt% | ● | ● | ● | ● | ● | ● | — | — | — | — | — | — |
Regardless of the polymer coil fraction or polymer chemistry, phase diagrams have several common features. At low temperatures below the thermoresponsive transition temperature of the bioconjugates, water is a nonselective solvent for both the protein and polymer. In this regime, low concentration solutions of mCherry-b-PHPA and mCherry-b-POEGA form disordered phases. Both bioconjugates microphase separate with increasing concentration, exhibiting ordered regions in the general range of 40 to 70 wt%. They form hexagonally packed cylinders or lamellae depending upon coil fraction and type of polymer, and lyotropic order–order transitions between the two phases can be observed within this concentration window. At very high concentrations, both bioconjugates display a re-entrant order–disorder transition (ODT) behaviour, transitioning gradually from an ordered phase to a disordered nematic phase at 80 wt%.
Above the thermoresponsive temperature where water is a selective solvent for mCherry, low concentration solutions form a disordered micellar phase (DM) due to the thermoresponsive behaviour of the polymers. A region of macrophase separation often accompanies this thermoresponsive transition, indicating coexistence between a high conjugate concentration and low conjugate concentration phase. It is likely that the phases formed show kinetic effects on structure formation due to the high thermodynamic barrier for polymer diffusion across water-rich nanodomains above the thermoresponsive transition temperature, as previously observed.19 At concentrations between 40 and 70 wt%, most bioconjugates maintain their nanostructures with increasing temperature but display a decrease in degree of ordering, evidenced by broader SAXS peaks and decreased birefringence. Symmetric bioconjugates show multiple thermotropic order–order transitions within this microphase separated region and display the most complex phase behaviours. High concentration phase behaviour is strongly affected by solvent selectivity towards the polymer at high temperature. Heating reduces the degree of hydration of the polymer coil, leading to an effective reduction in polymer coil fraction.
Lyotropic order–disorder transitions in a nonselective solvent
Although the overall shapes of the phase diagrams are similar for all polymers, the lyotropic order–disorder transition of the bioconjugates is strongly affected by the type of polymer and the polymer coil fraction. The CODT is defined as the lowest concentration at which ordering is observed at temperatures below the block copolymer thermoresponsive temperature, where both blocks are well hydrated. Representative SAXS curves between 30 and 60 wt% at 5 °C show lyotropic disorder to order transitions with increasing concentration (Fig. 4). mChPH18 starts to form hexagonally packed cylinders at 50 wt% (Fig. 4a), while mChPH27 and mChPH57 form lamellar phases at 40 wt% (Fig. 4b and c). mChPOE18 forms a poorly ordered lamellar phase at 50 wt% (Fig. 4d), while mChPOE26 forms a lamellar phase at 43 wt% (Fig. 4e, curve not shown); mChPOE57 also forms lamellae at 50 wt% (Fig. 4f).A plot of CODT for the three different polymer block types (mCherry conjugated to PNIPAM, PHPA, and POEGA) (Fig. 2) shows a trend in CODT as a function of polymer block chemistry. The CODT of mCherry-b-PNIPAM is 5–10 wt% lower than that of mCherry-b-PHPA, and the CODT of mCherry-b-PHPA is 5–10 wt% lower than that of mCherry-b-POEGA at all three coil fractions studied. This suggests that changing polymer block chemistry, which changes protein–polymer interactions, has a large effect on this transition. The results support the conclusion that PNIPAM is the most repulsive to the protein while POEGA is the least repulsive. The trend in CODT does not follow the trend in thermoresponsive transition temperature (Tt) for the three polymers. Despite PNIPAM having the highest Tt of the three polymers, mCherry-b-PNIPAM is observed to have the lowest CODT, while mCherry-b-POEGA, which has the intermediate Tt value, has the highest CODT. It is observed that mCherry conjugates with polymers capable of both accepting and donating hydrogen bonds, PNIPAM and PHPA, have the lowest CODTs, although a more quantitative understanding of the interactions in this system is required to draw firm conclusions regarding the origin of this effect.
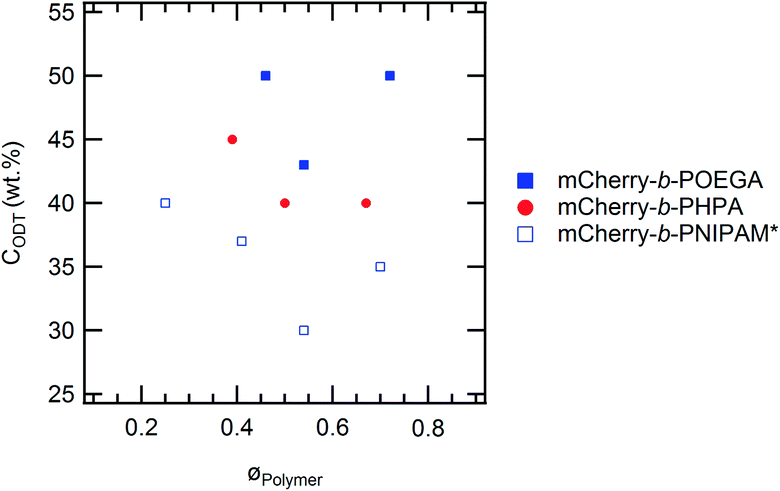 | ||
| Fig. 2 Order–disorder transition concentration (CODT) values for the three types of bioconjugates versus polymer volume fraction. *mCherry-b-PNIPAM data are adapted from Fig. 5 of ref. 20. | ||
The CODT is minimized at near-symmetric coil fraction for which protein–polymer interactions are maximized (Fig. 2). While mCherry-b-POEGA and mCherry-b-PNIPAM show clear minima as a function of coil fraction, the minimum for mCherry-b-PHPA likely lies around 0.6, as the CODTs of the conjugates with coil fractions of 0.50 and 0.67 are the same to within the resolution of our measurement. In all cases, the small number of coil fractions sampled does not allow precise resolution of the coil fraction at which the CODT is minimized. This non-monotonic trend in CODT with coil fraction further supports the hypothesis that solvent-mediated repulsive interactions between protein and polymer blocks are responsible for microphase separation near order–disorder transition concentrations.20
Re-entrant order–disorder transition
Surprisingly, both mCherry-b-PHPA and mCherry-b-POEGA become disordered upon increasing concentration for all coil fractions studied, but in all cases remain in an anisotropic phase, as evidenced by strong optical birefringence. At concentrations between 60 and 80 wt%, the primary SAXS peaks of both mCherry-b-PHPA and mCherry-b-POEGA bioconjugates significantly broaden as concentration increases (Fig. 3a and d). This loss of ordering is consistent with our recent observation of re-entrant transitions in mCherry-b-PNIPAM.28 However, birefringence of mCherry-b-PHPA and mCherry-b-POEGA is maintained or becomes stronger as concentration nears 80 wt% (Fig. 3b and e), which, in combination with the disordered SAXS pattern, is characteristic of a nematic liquid crystalline phase. Polarized optical microscopy images shown in Fig. 3c and f show birefringence of 80 wt% solution samples of mChPH18 and mChPOE18. Because these materials are prepared by adding solvent to a solid-state material that is kinetically trapped in a more ordered state (Fig. S9†), the loss of microphase separation and formation of a nematic phase appears to be an equilibrium effect.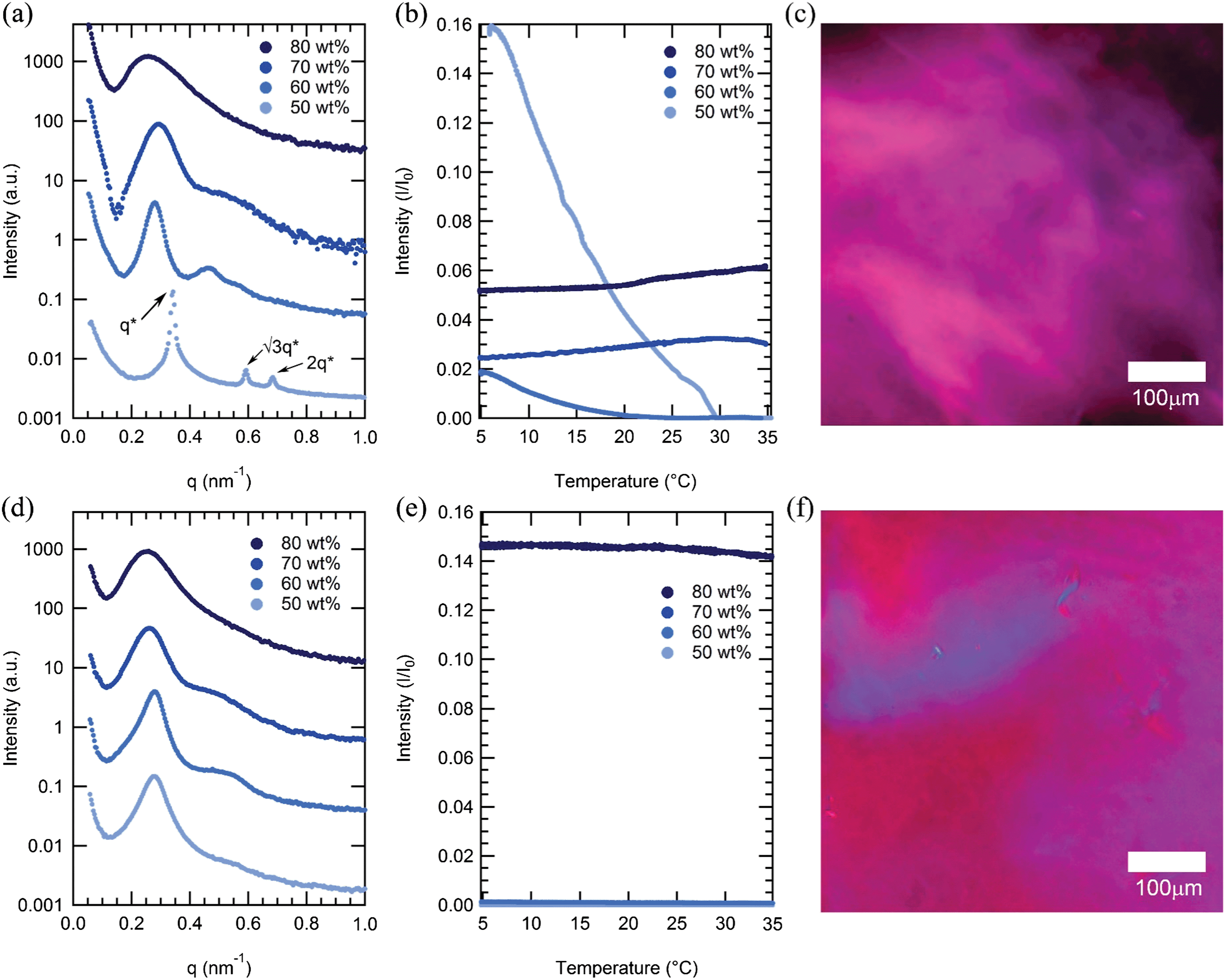 | ||
| Fig. 3 Top row: SAXS curves (a), DPLS curves (b), and a polarized optical microscopy image (c) of mCherry-b-PHPA 18 kDa (mChPH18) sample. Bottom row: SAXS curves (d), DPLS curves (e), and a polarized optical microscopy image (f) of mCherry-b-POEGA 18 kDa (mChPOE18). For mChPH18, a birefringent hexagonally packed cylindrical phase (Hex) at 50 wt% gradually loses ordering as concentration increases to 80 wt% by SAXS, while birefringence begins increasing at 70 wt%. The nonbirefringent lamellar phase of mChPOE18 between 50 and 70 wt% becomes gradually disordered as concentration increases to 80 wt% while becoming birefringent. Polarized light microscopy images are shown for 80 wt% samples. | ||
The observed disordering at high concentration is unexpected in block copolymers, where the dilution approximation36 predicts that repulsion between two blocks increases as concentration increases in a nonselective solvent. Re-entrant ODT behaviour has been previously observed in polystyrene-b-polyisoprene (PS-PI) diblock copolymers in decane which is a selective solvent for polyisoprene.37 A loss of micellar nanostructure and transition to a disordered phase as solutions become highly concentrated is hypothesized to result from a loss of repulsive pair interaction potential between micelles. A triblock copolymer of poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) (PPO-PEO-PPO) in water, selective for PEO, does not order in the bulk melt state while it microphase separates in the presence of a selective solvent.38,39 The observation of re-entrant order–disorder transitions in mCherry–polymer systems in water which is only slightly selective for polymer as shown in a later section, suggests that interactions between protein and polymer become net attractive with increasing concentration. The fact that microphase separated order is lost with increasing concentration regardless of the polymer chemistry suggests that this may be a universal property of mCherry–polymer conjugate materials.
The presence of optical anisotropy in the absence of microphase separation suggests that at high concentration the mCherry-b-PHPA and mCherry-b-POEGA conjugates form nematic phases. Such phases are commonly observed in rod-coil, main-chain, and side-chain liquid crystalline block copolymers,40 including such polymers where the liquid crystalline motif is composed of a simple polypeptide secondary structure (e.g., α-helix).41 Formation of a nematic phase by biological molecules was recognized as early as 1936 for rod-shaped tobacco mosaic viruses.42 Many other rod-shaped biological molecules, including Pf1 bacteriophage43 and amyloid protein fibers,44,45 were also shown to form nematic phases. Phase transitions to a nematic phase above a critical concentration are entropically driven due to excluded volume effects.46 However, globular proteins themselves rarely exhibit liquid crystalline phases.47,48 It is surprising that mCherry conjugates form liquid crystalline phases given the protein's low aspect ratio. From its crystal structure (PDB 2H5Q), mCherry has a β-barrel structure with dimensions of approximately 4.2 nm in height and 2.5 nm in diameter, giving an aspect ratio of 1.7, well below the de Gennes criteria of 4.49 Although mCherry–polymer conjugates have previously been shown to be aspherical in dilute solution,19 the highly overlapped nature of polymer chains in concentrated solution is known to result in significantly different chain configurations that prevent comparison between chain configurations in dilute solution and in the nematic phase. It is possible that anisotropic electrostatic interactions between proteins50 in addition to excluded volume effects contribute to the formation of anisotropic phases. In addition, conjugation to the polymer can result in a more anisotropic macromolecular structure resembling classical small molecule mesogens such as 5CB that are capable of forming nematic phases.
Birefringence is strongest at ϕ = 0.4 for both mCherry-b-PHPA and mCherry-b-POEGA, where the volume fraction of protein is largest. This supports the notion that formation of the nematic phase is governed by interactions between mCherry blocks. mChPOE18 has especially high birefringence compared to mChPOE26 and mChPOE57 as well as other bioconjugates. The fact that mChPOE18 has the strongest optical anisotropy correlates with its relatively small region of microphase separated structures. When protein and polymer blocks are most miscible, the mCherry liquid crystalline interactions can have the largest effect on phase behaviours.
Low concentration in selective solvents
At high temperature where water is a selective solvent for mCherry, the primary SAXS peak of low concentration solutions of all bioconjugates shifts to a lower q value compared to that at low temperature. The shifting primary peak at high temperature is correlated with the formation of larger structures, presumably micelles formed due to the thermoresponsive desolvation of the polymer blocks. The micellar transition occurs at consistently higher temperatures in mCherry-b-POEGA than in mCherry-b-PHPA, which is consistent with the observed higher cloud points of POEGA homopolymers. However, for both types of conjugate, the transition from disordered to micellar phases occurs at a significantly higher temperature than the homopolymer cloud point temperature, indicating the enhanced solubility imparted by the large protein block and the effect of increased concentration.The stability of a homogeneous micellar phase leading to macrophase separation depends on the polymer block type and coil fraction (transition temperatures are listed in Table 2). mCherry-b-PHPA and mCherry-b-POEGA both show a narrower concentration and temperature range of macrophase separation than mCherry-b-PNIPAM conjugates, despite PHPA and POEGA homopolymers having lower cloud point temperatures than PNIPAM. mChPH18 and mChPH27 form macroscopically homogeneous phases throughout all concentrations and temperatures measured, and a macrophase separated region is only observed in mChPH57. While equilibrium thermodynamics suggests that the two phases should have compositions equal to those on the boundaries of the two phase region, the structure observed by SAXS does not show signs of lamellar ordering, suggesting that the material may be kinetically trapped in a disordered micellar phase as previously observed for other conjugates.19 The mCherry-b-POEGA bioconjugate solutions show two types of macrophase separation behaviour. At low concentration, two phases of water-rich and micelle-rich solutions are formed, as shown in mChPOE18 and mChPOE57 (Fig. 1d and f). At intermediate concentration, macrophase separated regions flanked by a homogeneous micellar phase in mChPOE26 or a hexagonally packed cylindrical phase in mChPOE57 suggest that a true equilibrium state is a coexistence of two phases between the neighbouring homogeneous phases. mCherry-b-PNIPAM solutions show much larger regions of macrophase separation, which is correlated with the low stability of mCherry-b-PNIPAM micelles in dilute solution compared to mCherry-b-PHPA and mCherry-b-POEGA micelles.20
Macrophase separation temperatures (Tt) summarized in Table 2 are lowest in mCherry-b-PHPA and highest in mCherry-b-PNIPAM,20 which is also consistent with the trend in cloud points of homopolymers. The macrophase separation temperature increases with increasing concentration, similar to the trend observed in mCherry-b-PNIPAM. However, unlike mCherry-b-PNIPAM, thermal transitions are undetectable by differential scanning calorimetry.
Structure of the microphase separated region
For all coil fractions, ordered phases are observed from approximately 40 to 70 wt%, although the type of nanostructure and quality of order are affected by coil fraction, temperature, and concentration. Representative SAXS curves at low and high temperature are shown in Fig. 4 and S7,† respectively. Complementary DPLS results are summarized in Table 2.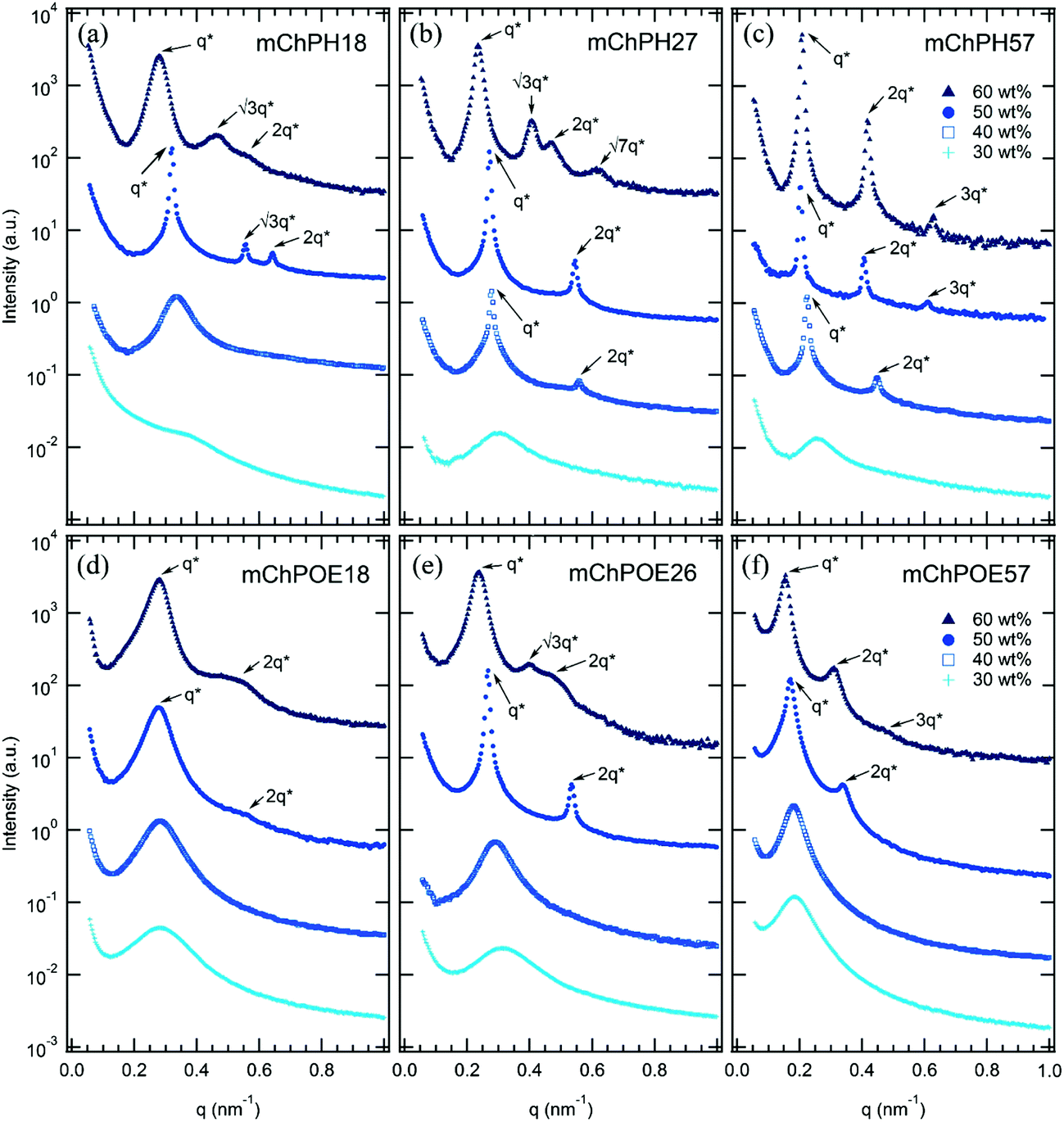 | ||
| Fig. 4 SAXS curves of mCherry-b-PHPA (a–c) and mCherry-b-POEGA (d–f) solutions from 30 to 60 wt% for three polymer coil fractions at 5 °C where both blocks are well solvated. Lyotropic disorder to order transitions occur between 30 and 50 wt% for all bioconjugates ((a) 50 wt% (b) 40 wt% (c) 40 wt% (d) 50 wt% (e) 43 wt% (not shown) (f) 50 wt%). Bioconjugates with ϕ = 0.5 (mChPH27 and mChPOE26) further undergo lyotropic order–order transition from lamellae to hexagonally packed cylinders as concentration increases from 50 to 60 wt% (b and e). Curves are vertically offset for clarity. | ||
At a coil fraction of 0.4, mCherry-b-PHPA forms well-ordered hexagonally packed cylinders, whereas mCherry-b-POEGA shows only weakly ordered lamellar regions (Fig. 1a and d). At low temperature where both blocks are well solvated, mChPH18 starts to order at 50 wt% into a hexagonal phase, which is indicated by a sharp primary peak with clear higher order reflections at √3q* and √4q* (Fig. 4a) and strong birefringence. At high temperature, broadening of the SAXS peaks is observed and birefringence is lost, suggesting a partial loss of order in the hexagonal phase. In contrast, mChPOE18 has very weak ordering throughout all the concentrations and temperatures explored, demonstrating only a weak secondary reflection by SAXS at 2q* that can be assigned to a lamellar phase. No birefringence is observed for mChPOE18 samples except for a nematic phase at 80 wt%, consistent with poor ordering observed by SAXS. Above the thermoresponsive transition temperature between 20 and 25 °C in the concentration range of 50–60 wt%, mChPOE18 solutions form weakly ordered hexagonal phases at 50 and 60 wt% (Fig. S7d†).
The solution phase behavior of bioconjugates with symmetric coil fractions shows the most complex lyotropic and thermotropic order–order transitions, as well as phases that are not observed in previously studied materials (Fig. 4b and e). At low temperatures, mChPH27 and mChPOE26 first form nonbirefringent lamellar phases upon ordering, with birefringence appearing upon increasing order with increasing concentration.51,52 Birefringent lamellae exist in a narrow concentration range up to 50 wt%, above which the solution forms a hexagonally packed cylindrical phase with three or four reflections at q*, √3q*, √4q*, and √7q* observed in SAXS. While for mChPH27 this phase is nonbirefringent at low temperatures, in mChPOE26 there is sufficient order to produce a birefringent cylinder phase. Above the thermoresponsive transition temperature, both conjugates form cylinder phases across the entire ordered region, with no birefringence observed at lower concentrations. This thermotropic order–order transition results from an effectively reduced polymer volume fraction in the bioconjugate above the thermoresponsive temperature. In the case of mChPH27, this phase becomes birefringent over a narrow concentration range then loses birefringence as concentration is further increased, suggesting maximal order at an intermediate concentration. However, in mChPOE26, birefringence appears at 60 wt% and continues to the end of the ordered region.
In a narrow region between 15 and 20 °C and 43 and 50 wt%, mChPH27 forms an intermediate phase with cubic symmetry (Cub) between a lamellar phase at low temperature and a hexagonally packed cylindrical phase at high temperature. Bragg reflections corresponding to √6, √8, √14, √16, √20, √21, √24, and √26q* are indexed (Fig. 5a, top curve), corresponding to the first 8 allowed reflections for an Iad phase.53,54 A plot of reciprocal d spacings versus m = (h2 + k2 + l2)1/2 (where h, k, and l are the Miller indices) (Fig. 5b) shows good agreement between the observed reflections and a crystallographic structure with Iad symmetry. SAXS analysis alone is generally not sufficient for phase assignment; however, considering the fact that Iad corresponds to the bicontinuous cubic phase in many different types of block copolymers53,55,56 and that this phase is located between the hexagonally packed cylindrical phase and lamellar phase, it is likely that this is the first observed gyroid phase made of globular protein–polymer block copolymer.
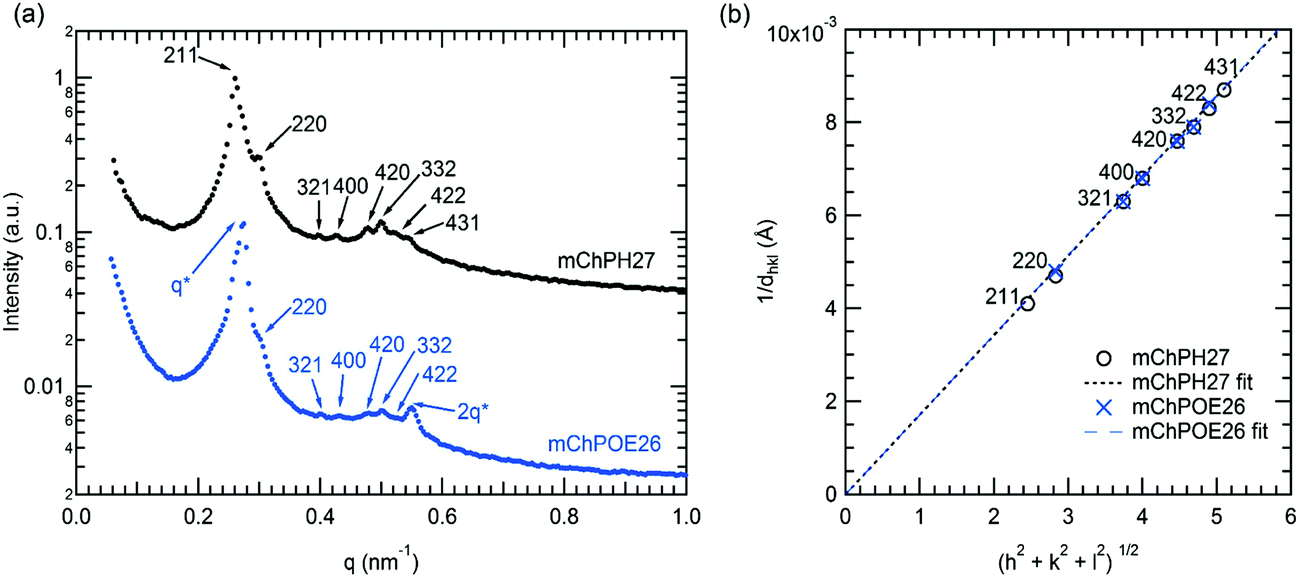 | ||
| Fig. 5 (a) SAXS curve of mChPH27 50 wt% solution at 15 °C (top) and of mChPOE26 50 wt% at 20 °C (bottom). (b) A plot of m vs. 1/dhkl based on the reflections observed in the samples showing the validity of the space group assignment. The fitted lines are forced to intercept the origin. The lattice parameter of 58.4 nm is obtained for mChPH27 from the slope value of 0.00171 with R2 = 0.9996, and lattice parameter is 58.3 nm for mChPOE26 from the slope 0.00172 with R2 = 0.9994. | ||
mChPOE26 displays similar structure at 20 °C over the range 47–50 wt% (Fig. 5a). The two most prominent reflections at 0.275 and 0.5497 nm−1 have a ratio 1:2 and are compatible with lamellar structure, while the rest of the peaks have a peak position ratio of √8:√14:√16:√20:√21:√24. A plot of 1/dhklvs. m shows the second set of peaks matches well with Iad symmetry as for mChPH27 (Fig. 5b). The observed reflections suggest the material exhibits coexistence of lamellar and cubic structures. Coexisting lamellar and cubic phases have been observed in other literature,57,58 and such coexistence is allowed by the phase rule since the copolymer–water mixture is a two component system. Reflections corresponding to √6 and √26 (211 and 431), which are observed in mChPH27 but not in mChPOE26, are expected to be present at 0.2615 and 0.5443 nm−1. However, these peaks are not observed, likely due to an overlap in these regions with the more pronounced peaks from the lamellar phase. The lattice parameter obtained from the plot is 58.3 nm which is very close to the value obtained for mChPH27 (58.4 nm).
At the largest coil fraction of ϕpolymer = 0.7, a lamellar morphology is observed for both conjugates across most of the ordered region (Fig. 1c and f). Phase diagrams of mChPH57 and mChPOE57 are qualitatively similar with slight differences in the CODT and the location of the perforated lamellar (PL) phase. mChPH57 and mChPOE57 initially order into nonbirefringent lamellae upon increasing concentration, and the CODT of mChPH57 is observed to be 10 wt% lower than that of mChPOE57. The nonbirefringent lamellar phase becomes birefringent as concentration increases, and the lamellar ordering dominates the phase behaviour up to 60 wt%. Although lamellae are formed independent of temperature in this region, ordering decreases slightly with increasing temperature, as evidenced by smaller peak intensities in SAXS, the disappearance of third order reflections, and the gradual loss of birefringence on heating. Both display a perforated lamellar phase at 70 wt% suggested by an asymmetric primary peak and additional reflections between q* and 2q* (Fig. S10†) as previously observed in mCherry-b-PNIPAM.18,20 mChPH57 forms a perforated lamellar phase (PL) at all temperatures at 70 wt%, while mChPOE57 maintains the lamellar ordering for most temperatures at 70 wt% with appearance of the perforated lamellar phase at 35 °C.
Common phase behaviours between different protein–polymer conjugates are revealed by the comparison of mCherry-b-PHPA, mCherry-b-POEGA, and previously reported mCherry-b-PNIPAM. Conjugates with the smallest coil fractions exhibit large regions of weakly ordered lamellar or hexagonally packed structures. Conjugates with near symmetric coil fractions display the richest phase behaviours, undergoing a lyotropic ODT from a disordered phase to a lamellar morphology and lyotropic and thermotropic OOTs to form hexagonally packed cylinders. At large polymer volume fractions, the phase behaviours of the conjugates are represented by large regions of lamellar ordering. A re-entrant order–disorder transition to form a nematic phase in highly concentrated solutions is also observed.
Comparison of ordered nanostructures between mCherry-b-PHPA and mCherry-b-POEGA conjugates and previously studied PNIPAM conjugates shows that changing the chemistry of the polymer block results in both changes in the location of the phase transition lines and the appearance of a cubic phase not observed in previous studies. Concentrated solutions of symmetric mCherry-b-PNIPAM bioconjugates tend to form perforated lamellar phases (PL) in a very narrow range between lamellae and hexagonally packed cylinders instead of the cubic phases observed in mCherry-b-PHPA and mCherry-b-POEGA. In traditional coil–coil block copolymer systems, nonequilibrium perforated lamellar phases or equilibrium gyroid phases occur within a narrow range of volume fractions between lamellae and cylinders.59,60 It is possible that perforated lamellae are at least kinetically preferred in mCherry-b-PNIPAM20,28 because of its higher segregation strength and greater packing frustration, consistent with hypothesized stronger repulsive interactions between mCherry and PNIPAM than between mCherry and the other polymers considered.
Irrespective of the specific polymer block chemistry, the order of appearance of the nanostructured morphologies is similar for all block copolymers: lamellar, perforated lamellar or cubic, and hexagonal phases appear in that order with increasing temperature and concentration, although not all phases are observed in each material. These thermotropic and lyotropic order–order transitions can be understood based upon water partitioning. Hydration of the protein and polymer domains is observed to be similar for the different polymer blocks PNIPAM, PHPA, and POEGA, measured by SANS contrast variation (Fig. S11†). SANS contrast variation was performed using a number of D2O%/H2O% solvent compositions from 10/90 to 70/30, but due to macrophase separation observed in solvent compositions comprising a majority percentage of D2O, data analysis was limited to the results obtained in solvent compositions in which the H2O/D2O volume percent ratio was at least 1. Using calculated scattering length densities for each molecular component (Table S1†), the scattering contrast between protein and polymer domains was calculated as a function of water partitioning in the block copolymer using a two-parameter model as reported previously for mCherry-b-PNIPAM.20 From SANS contrast variation, fitting results (Fig. 6) show that approximately 59% of the water is located within both the PHPA and POEGA domains for mChPH27 and mChPOE26, respectively, similar to previous calculations of approximately 64% of the water hydrating the PNIPAM domains for mCherry-b-PNIPAM27k. Calculations show that the ratio of the water volume fraction in the polymer domain to that in the protein domain (ϕwater,polymer/ϕwater,protein) is 0.75/0.67 for mCherry-b-PNIPAM, 0.75/0.70 for mCherry-b-PHPA, and 0.72/0.68 for mCherry-b-POEGA.
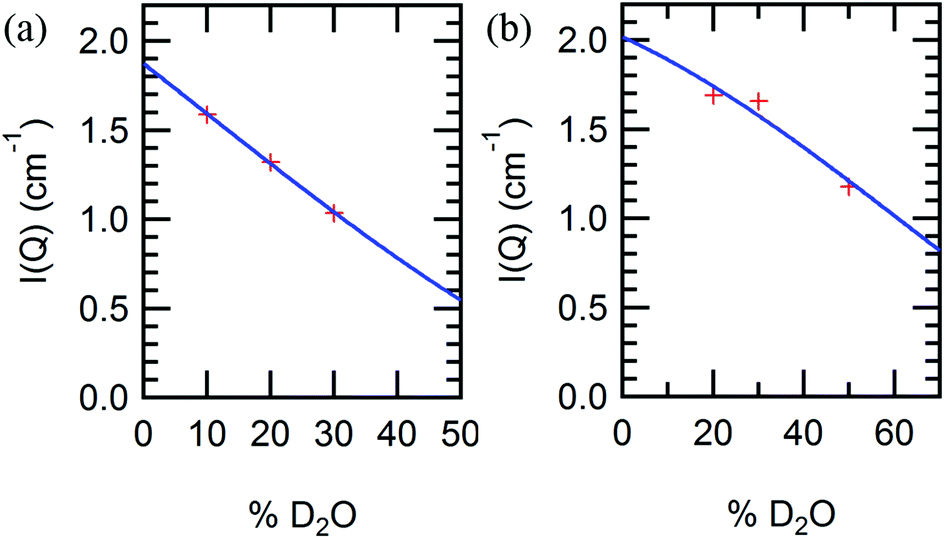 | ||
| Fig. 6 Small-angle neutron scattering peak intensities (data points) and curve fits (solid line) for (a) mChPH27 and (b) mChPOE26 in different D2O/H2O blend compositions at 10 °C. | ||
Due to the slightly preferential hydration of the polymer block, as concentration increases, the volume fraction of the polymer domain decreases faster than that of the protein domains. This leads to an order–order transition to a morphology in which the polymer volume fraction is effectively smaller. Because increasing temperature also results in desolvation of the polymer block, increasing temperature and concentration trigger similar phase transitions. Minimal differences in hydration between mCherry and all three different polymer domains at low temperature suggest that the differences in protein–polymer interactions have a greater effect on the observed trend in CODT than differences in selective hydration.
While not quantitative, the application of polymer solution theory concepts to this system further suggests that a major impact of changing polymer is to tune the protein–polymer interaction. Changing monomers used in the synthetic polymer block is not expected to create a large change in entropy of mixing because coarse-grained theories of polymer solutions (i.e. Flory Huggins theory61 and Leibler's RPA theory62 of block copolymers) show that the dominant entropy of mixing contribution is a function of only the relative volume fraction (degree of polymerization) of the two blocks. Because polymer volume fractions in bioconjugates are carefully controlled and the protein block has the same colloidal size and shape in each conjugate, little change in mixing entropy is expected. Each of the polymers also has a similar degree of polymerization and is studied in the vicinity of its thermoresponsive transition temperature, where the Flory–Huggins interaction parameter between polymer and solvent is expected to cross from less than 0.5 to greater than 0.5 upon increasing temperature. Therefore, although POEGA and PHPA are expected to have a somewhat higher χ parameter as a function of temperature than PNIPAM due to their lower transition temperatures, the investigation of all polymers at temperatures in the vicinity of the responsive transition temperature suggests that solvent-mediated polymer–polymer interactions are not a large difference between systems. This is consistent with the similar hydration observed by SANS.
The asymmetric nature of phase space as a function of polymer volume fraction is observed for all three polymers. Based on 50–60 wt% solution nanostructures where the best ordering is observed, conjugates with a polymer fraction less than approximately 0.5 tend to form hexagonally packed cylinders, while conjugates with polymer volume fractions greater than 0.5 tend to form lamellae. Cylinders are not observed on the high coil fraction side of the phase diagram, and the body-centered cubic spherical phase (BCC) is not observed in all three polymers studied. This is further supported by a detailed study on phase behaviour as a function of concentration and coil fraction for mCherry-b-PNIPAM where no BCC phase or cylinders with protein core were observed.28 The asymmetry as well as no occurrence of a spherical phase can be attributed to the geometric molecular asymmetry in the protein–polymer diblock copolymers arising from the presence of the globular and rigidly folded mCherry. The observation that the lamellar phase is thermodynamically favored when the polymer volume fraction is much higher than 0.5 is consistent with previous observations of asymmetrically shaped block copolymers.63–66
Conclusions
The self-assembly of globular protein–polymer conjugates comprising the red fluorescent protein mCherry and the thermoresponsive polymers PHPA and POEGA shows that changing polymer–protein interactions by changing polymer block chemistry has a large impact on the type of self-assembled phases formed, the location of phase transitions, and the presence of macrophase separation at high temperatures. Changing polymer chemistry and the resultant polymer–protein interactions affect the order–disorder transition concentration: mCherry-b-PNIPAM is observed to have the lowest CODT, while mCherry-b-POEGA conjugates have the highest CODT. The occurrence of the minimum CODT at symmetric compositions suggests that this behavior is driven by changes in the strength of effective repulsive interactions between protein and polymer. A new phase with Iad symmetry is observed in mCherry-b-PHPA and mCherry-b-POEGA, instead of the perforated lamellar (PL) phase formed by near-symmetric mCherry-b-PNIPAM conjugates. Macrophase separation occurs more readily in mCherry-b-PNIPAM than either mCherry-b-PHPA or mCherry-b-POEGA, suggesting that the different polymer chemistries and their respective thermoresponsive behaviours affect the stability of a two-phase region at high temperatures.
General features common to the self-assembly of all three conjugates are also revealed. All three bioconjugates have a microphase separation region between approximately 40 and 70 wt% with lamellar or cylindrical phases, which are commonly observed in traditional coil–coil diblock copolymers, and bioconjugate solutions with higher concentration display a re-entrant ODT behavior. Weakly ordered nematic phases formed by mCherry-b-PHPA and mCherry-b-POEGA suggest that liquid crystalline ordering at high concentration arises from the presence of protein. The asymmetric nature of the phase diagram as a function of coil fraction is also common among the three bioconjugates. Regardless of coil fraction, bioconjugates always form ordered phases in the order lamellar, perforated lamellar or cubic, and hexagonal as concentration or temperature is increased.
Acknowledgements
This work was supported by Air Force Office of Scientific Research (award number FA9550-12-0259). Department of Energy Office of Basic Energy Sciences (award number DE-SC0007106) supported neutron scattering studies of polymer hydration. We thank Dr Lixia Rong and Dr Maya Koga for experimental assistance at NSLS beamline X27C at Brookhaven National Laboratory, and Dr William T. Heller for experimental assistance with SANS at EQ-SANS at Oak Ridge National Laboratory. The authors are grateful to the Keating lab at MIT for use of CD instrument. NMR was measured in MIT DCIF and polarized optical microscopy was conducted in the Institute for Soldier Nanotechnologies.Notes and references
- A. F. M. Barton, Handbook of Poylmer–Liquid Interaction Parameters and Solubility Parameters, Taylor & Francis, 1990 Search PubMed.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef CAS PubMed.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS PubMed.
- A. Heller, J. Phys Chem., 1992, 96, 3579–3587 CrossRef CAS.
- G. F. Drevon and A. J. Russell, Biomacromolecules, 2000, 1, 571–576 CrossRef CAS.
- M. Ferreira, P. A. Fiorito, O. N. Oliveira and S. I. C. de Torresi, Biosens. Bioelectron., 2004, 19, 1611–1615 CrossRef CAS PubMed.
- J. A. He, L. Samuelson, L. Li, J. Kumar and S. K. Tripathy, Langmuir, 1998, 14, 1674–1679 CrossRef CAS.
- R. Das, P. J. Kiley, M. Segal, J. Norville, A. A. Yu, L. Y. Wang, S. A. Trammell, L. E. Reddick, R. Kumar, F. Stellacci, N. Lebedev, J. Schnur, B. D. Bruce, S. G. Zhang and M. Baldo, Nano Lett., 2004, 4, 1079–1083 CrossRef CAS.
- A. Zengin and T. Caykara, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4443–4450 CrossRef CAS.
- B. D. Olsen, Macromol. Chem. Phys., 2013, 214, 1659–1668 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- N. Kumar and J. I. Hahm, Langmuir, 2005, 21, 6652–6655 CrossRef CAS PubMed.
- A. D. Presley, J. J. Chang and T. Xu, Soft Matter, 2011, 7, 172–179 RSC.
- W. J. Shan, P. L. He and N. F. Hu, Electrochim. Acta, 2005, 51, 432–440 CrossRef CAS PubMed.
- S. S. Jia, J. J. Fei, J. J. Deng, Y. L. Cai and J. A. Li, Sens. Actuators, B, 2009, 138, 244–250 CrossRef CAS PubMed.
- M. B. Cardoso, D. Smolensky, W. T. Heller, K. L. Hong and H. O'Neill, Energy Environ. Sci., 2011, 4, 181–188 CAS.
- C. N. Lam, M. Kim, C. S. Thomas, D. Chang, G. Sanoja, C. Okwara and B. D. Olsen, Biomacromolecules, 2014, 15, 1248–1258 CrossRef CAS PubMed.
- C. S. Thomas, M. J. Glassman and B. D. Olsen, ACS Nano, 2011, 5, 5697–5707 CrossRef CAS PubMed.
- C. S. Thomas, L. Z. Xu and B. D. Olsen, Biomacromolecules, 2012, 13, 2781–2792 CrossRef CAS PubMed.
- C. N. Lam and B. D. Olsen, Soft Matter, 2013, 9, 2393–2402 RSC.
- A. Chilkoti, G. H. Chen, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 1994, 5, 504–507 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS PubMed.
- C. Lavigueur, J. G. Garcia, L. Hendriks, R. Hoogenboom, J. J. L. M. Cornelissen and R. J. M. Nolte, Polym. Chem., 2011, 2, 333–340 RSC.
- M. J. Boerakker, N. E. Botterhuis, P. H. H. Bomans, P. M. Frederik, E. M. Meijer, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem.–Eur. J., 2006, 12, 6071–6080 CrossRef CAS PubMed.
- Y. Xia, S. C. Tang and B. D. Olsen, Chem. Commun., 2013, 49, 2566–2568 RSC.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327–2332 CrossRef CAS PubMed.
- C. S. Thomas and B. D. Olsen, Soft Matter, 2014, 10, 3093–3102 RSC.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS PubMed.
- M. W. Matsen and F. S. Bates, J. Chem. Phys., 1997, 106, 2436–2448 CrossRef CAS PubMed.
- F. J. Hua, X. G. Jiang, D. J. Li and B. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2454–2467 CrossRef CAS.
- E. Bays, L. Tao, C. W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1777–1781 CrossRef CAS PubMed.
- T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R. Hoogenboom and U. S. Schubert, Macromolecules, 2008, 41, 5132–5140 CrossRef CAS.
- L. D. Taylor and L. D. Cerankowski, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2551–2570 CrossRef CAS.
- E. Helfand and Y. Tagami, J. Chem. Phys., 1972, 56, 3592–3601 CrossRef CAS PubMed.
- G. A. McConnell and A. P. Gast, Macromolecules, 1997, 30, 435–444 CrossRef CAS.
- K. Zhang and A. Khan, Macromolecules, 1995, 28, 3807–3812 CrossRef CAS.
- J. Noolandi, A.-C. Shi and P. Linse, Macromolecules, 1996, 29, 5907–5919 CrossRef CAS.
- V. P. Shibaev and N. A. Plate, Adv. Polym. Sci., 1984, 60–61, 173–260 CrossRef.
- C. Robinson, Tetrahedron, 1961, 13, 219–234 CrossRef CAS.
- F. C. Bawden, N. W. Pirie, J. D. Bernal and I. Fankuchen, Nature, 1936, 138, 1051–1052 CrossRef.
- M. Zweckstetter and A. Bax, J. Biomol. NMR, 2001, 20, 365–377 CrossRef CAS.
- A. M. Corrigan, C. Müller and M. R. H. Krebs, J. Am. Chem. Soc., 2006, 128, 14740–14741 CrossRef CAS PubMed.
- J.-M. Jung and R. Mezzenga, Langmuir, 2009, 26, 504–514 CrossRef PubMed.
- L. Onsager, Ann. N. Y. Acad. Sci., 1949, 51, 627–659 CrossRef CAS PubMed.
- A. C. Dumetz, A. M. Chockla, E. W. Kaler and A. M. Lenhoff, Biophys J., 2008, 94, 570–583 CrossRef CAS PubMed.
- J. A. Thomson, P. Schurtenberger, G. M. Thurston and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7079–7083 CrossRef CAS.
- P. G. d. Gennes and J. Prost, The Physics of Liquid Crystals, Clarendon Press, 1995 Search PubMed.
- A. Kurut, B. A. Persson, T. Akesson, J. Forsman and M. Lund, J. Phys. Chem. Lett., 2012, 3, 731–734 CrossRef CAS.
- N. P. Balsara, B. A. Garetz and H. J. Dai, Macromolecules, 1992, 25, 6072–6074 CrossRef CAS.
- N. P. Balsara, D. Perahia, C. R. Safinya, M. Tirrell and T. P. Lodge, Macromolecules, 1992, 25, 3896–3901 CrossRef CAS.
- D. A. Hajduk, P. E. Harper, S. M. Gruner, C. C. Honeker, G. Kim, E. L. Thomas and L. J. Fetters, Macromolecules, 1994, 27, 4063–4075 CrossRef CAS.
- M. E. Vigild, K. Almdal, K. Mortensen, I. W. Hamley, J. P. A. Fairclough and A. J. Ryan, Macromolecules, 1998, 31, 5702–5716 CrossRef CAS.
- M. W. Matsen, J. Chem. Phys., 1998, 108, 785–796 CrossRef CAS PubMed.
- P. Alexandridis, U. Olsson and B. Lindman, Langmuir, 1998, 14, 2627–2638 CrossRef CAS.
- C. E. Conn, O. Ces, X. Mulet, S. Finet, R. Winter, J. M. Seddon and R. H. Templer, Phys. Rev. Lett., 2006, 96, 108102 CrossRef.
- A. Crisci, D. N. T. Hay, S. Seifert and M. A. Firestone, Macromol. Symp., 2009, 281, 126–134 CrossRef CAS.
- S. Forster, A. K. Khandpur, J. Zhao, F. S. Bates, I. W. Hamley, A. J. Ryan and W. Bras, Macromolecules, 1994, 27, 6922–6935 CrossRef.
- D. A. Hajduk, H. Takenouchi, M. A. Hillmyer, F. S. Bates, M. E. Vigild and K. Almdal, Macromolecules, 1997, 30, 3788–3795 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953 Search PubMed.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- E. L. Thomas, J. T. Chen, M. J. E. ORourke, C. K. Ober and G. P. Mao, Macromol. Symp., 1997, 117, 241–256 CrossRef CAS.
- B. D. Olsen and R. A. Segalman, Macromolecules, 2005, 38, 10127–10137 CrossRef CAS.
- B. D. Olsen and R. A. Segalman, Macromolecules, 2007, 40, 6922–6929 CrossRef CAS.
- B. D. Olsen, M. Shah, V. Ganesan and R. A. Segalman, Macromolecules, 2008, 41, 6809–6817 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py00448e |
| This journal is © The Royal Society of Chemistry 2014 |