 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced copper-mediated polymerization of methyl acrylate in continuous flow reactors†
Benjamin
Wenn‡
a,
Matthias
Conradi‡
a,
Andre Demetrio
Carreiras
ab,
David M.
Haddleton
c and
Tanja
Junkers
*a
aPolymer Reaction Design Group, Institute for Materials Research (IMO-IMOMEC), Universiteit Hasselt, Agoralaan Building D, B-3590 Diepenbeek, Belgium. E-mail: tanja.junkers@uhasslet.be; Fax: +32 (11) 26 8399; Tel: +32 (11) 26 8318
bEscola Politecnica da Universidade de Saõ Paulo, Associação de Engenharia Química, Avenida Professor Luciano Gualberto, Travessa 3, no 380, Butantã – São Paulo, 05508-010, Brazil
cDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
First published on 24th January 2014
Abstract
Photo-induced copper-mediated radical polymerization of methyl acrylate (MA) is carried out in DMSO at 15 °C in a tubular photo-flow reactor as well as in a glass-chip based microreactor. Polymerization reactions proceed rapidly to approximately 90% monomer conversion within 20 minutes of reactor residence time. Control of reactions is high as evidenced by ideal polymerization kinetics, low dispersities of the obtained polymers (in the range of 1.1) and linear evolution of number average molecular weights during polymerization reactions. Poly(MA) with average molecular weights between a few hundred and ∼5000 g mol−1 was obtained under retention of pristine end group fidelity. Besides homopolymers, block copolymers can also be successfully synthesized and poly(methyl acrylate)-b-poly(butyl acrylate) block copolymers with a similar low dispersity are obtained. Reactions proceed under homogeneous reaction conditions. This feature allows the reaction to be carried out in milli- and also in microflow devices. In both cases, equally good control is achieved with only minimal adaptation of the reaction protocol, underpinning the simplicity and fast adaptability of the protocol to different flow reactors.
Introduction
Flow (micro)reactor technology (MRT) has recently gained increasing attention since its offers a wide range of new possibilities for academic research and industrial production in the polymer field.1–4 Flow reactions feature several advantages over batch processes. The high surface to volume ratio of the flow (micro)reactor technology avoids disadvantages of classical tubular or batch reactors, such as difficulties in the control of highly exothermic reactions or the formation of “hot spots” due to poor heat dissipation, allowing uncomplicated heat exchange and constantly stable reaction conditions and also thermal control in highly exothermic reactions.5 The efficient thermal management in flow (micro)reactors can also be employed to avoid side reactions and accelerate slow reactions by increasing the temperature and pressure.6,7 This fact allows the use of a wide range of conventional solvents which cannot always be utilized in classical batch reactor processes under similar reaction conditions.8 With MRT good control and adjustment possibilities for reaction parameters including temperature, residence time, and reactant stoichiometry are obtained. In recent years, the advantages of flow (micro)reactor technology were demonstrated by different research groups, with most examples originating from the realm of organic and pharmaceutical research.9 With respect to polymerization reactions, MRT also features distinct advantages. Application of (micro or milli) flow conditions to polymer reactions intrinsically allows for simple upscaling of processes and likewise acceleration of reactions in general. At the same time – and this is a distinct advantage of chain reactions – also materials of higher quality and overall yields can be obtained from MRT due to the higher stability that is offered by flow systems combined with the more defined reaction conditions that can be achieved. Thus, polymer materials with highly precise structures that are not easily matched by their batch reaction counterparts can be synthesized.1 So far, polymers have been produced in laboratory flow reactors for a variety of reactions, including anionic polymerization, ring opening polymerization and diverse methods in the realm of controlled radical polymerization.5 More recently, also polymer click conjugations and click-like polymer modifications have been added to the available portfolio of flow reactions.10,11Ligation of polyacrylates with other polymer counterparts via copper catalyzed azide–alkyne conjugation (CuAAC) has been demonstrated on a flow chip reactor with high efficiency and comparatively fast reaction times.11 A distinct problem of such reaction, however, is the use of certain copper species, which in this case tend to be only partially soluble. These inhomogeneities inevitably lead to blockages and fouling in the reactor channels, prohibiting stable flow conditions and eventual failure of the reactor setup. While these problems can be overcome by careful choice of reaction conditions and by adjusting the physical parameters of the employed reactors, fewer possibilities exist to translate traditional copper-catalyzed reactions to flow. Within the realm of polymerization reactions, one of the most important copper-mediated processes is copper-mediated radical polymerization, namely atom-transfer radical polymerization (ATRP)12–15 and single electron transfer-living radical polymerization (SET-LRP).16–20 Both techniques require equilibrium between oxidation states of copper species which allows for a reversible activation/deactivation of growing macroradicals and end-capping of the latter with a halide atom, typically bromine. Both ATRP and SET-LRP yield polymers of a similar structure and the difference between both processes mostly stems from the choice of reaction conditions such as solvent polarity and temperature. For the distinct differences and specific reaction mechanisms of the two processes, the reader is referred to the literature.12–23 For both ATRP and SET-LRP, flow reactions have been developed and in principle, production of polymers via these methodologies under continuous conditions is possible.4,24,25 Yet, in all cases, the handling of the copper species and disproportionation of Cu(I) to solid Cu(0) and often insoluble Cu(II) complexes as well as precipitation of copper salts and complexes make establishment of stable flow conditions challenging.26
Until recently only a few transition metal mediated, controlled radical polymerization reactions – unlike free-radical polymerization reactions – have been efficiently initiated by UV-light.27–31 Also reversible addition fragmentation radical transfer polymerization (RAFT)32,33 could be initiated by the use of conventional UV-initiators. However, these reactions are prone to side reactions due to the RAFT typical dithioester moieties acting as chromophores. Newer developments demonstrated that development of UV-labile alkoxyamines could also be used to perform UV-initiated nitroxide-mediated polymerization (NMP) reactions.34–39 Subsequently, Hawker and coworkers demonstrated that also ATRP can be successfully directly initiated by UV light if the copper catalyst is replaced by an iridium species.36 Copper-mediated ATRP could be performed under UV-conditions by applying reverse ATRP conditions, thus making use of conventional initiators.40–44 However, also direct activation of the copper species via UV-light was reported, enabling a true UV-induced ATRP process comparable to the iridium system.45 Also for a system closer to SET-LRP, photo-induced polymerization could be successfully carried out.24 For copper-catalyzed azide–alkyne cycloadditions, photo-generation of the Cu(I) species was likewise reported.46
Herein, we focus on the photo-induced polymerization of methyl acrylate in polar solvents under reaction conditions typical for a SET-LRP reaction (or SARA-ATRP, supplemental activators and reducing agents-ATRP). Haddleton and coworkers have reported on the successful polymerization of a variety of monomers by this process and have demonstrated an efficient and fast light induced polymerization process. Thus, while photo-induced controlled polymerization is usually of highest interest for surface modification and surface patterning,47,48 in this case the photo-process might also be an interesting alternative for the synthesis of polymers by ATRP/SET-LRP on a large scale in a solution process. However, even though this is favorable from an economic point of view as UV-irradiation is cheaper than thermal heating, UV-induced polymerization is not unproblematic. Reaction efficiencies depend largely on the nature of the light source and also on the reactor geometry. Light intensity gradients throughout a batch reactor are unavoidable due to absorption of light. As a consequence, such reactions can be difficult to scale up and reaction kinetics vary widely with the type of reaction vessel chosen. A possibility to overcome this problem is the application of MRT to photo-reactions. UV flow reactors have been shown to be highly efficient and to accelerate several reactions to reach full conversion from reaction times of days to minutes by increasing the light intensity and efficiency.49–52 While gaining more and more popularity in the field of organic chemistry synthesis, no polymerization reactions have, however, to the best of our knowledge, been reported for such lab-scale devices in homogeneous phase, even though reactors are comparatively cheap and easy to handle. In MRT flow reactors, channel widths and thus optical path-lengths are very small, allowing for very reproducible and easily upscalable reaction conditions. Employing UV-flow reactors does not only allow for continuous production of materials, but also for an increased efficiency of the reactions. Since residence times are usually short in micro- and milliflow reactors, interfering reactions stemming from degradation of materials from extended UV-illumination are generally avoided, thus leading to less side products and overall increased product quality.
Based on the novel photo-polymerization protocol introduced by Haddleton and coworkers, we demonstrate for the case of methyl acrylate, how this polymerization can be optimized for micro- and milliflow with high efficiency and short reaction times. To the best of our knowledge, this is the first report on photo-induced controlled radical polymerization employing microreactors. Thus, while the focus is herein put on a specific polymerization protocol, adaption of the described reactors to other (photo)polymerization methodologies is expected.
Experimental
Materials
Ethyl 2-bromoisbutyrate (EBiB, Alfa Aesar, 98+%), copper(II) bromide (CuBr2, Sigma-Aldrich, 99%), and dimethyl sulfoxide (DMSO, Merck, pro analysis) were all used as received. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesized according to a literature procedure.53 Methyl acrylate (MA, Acros, 99%) and butyl acrylate (BA, Acros, 99%) were deinhibited over a column of activated basic alumina, prior to use.Analytical techniques
1H NMR spectra were recorded in deuterated chloroform applying a pulse delay of 12 s with two NMR spectrometers (300 and 400 MHz) from Oxford Instruments Ltd. using a Varian probe (9 mm 4-nucleus AutoSWPFG).Analytical SEC (Size Exclusion Chromatography) was performed on a Tosoh EcoSEC HLC-8320GPC, comprising an autosampler, a PSS guard column SDV (50 × 7.5 mm), followed by three PSS SDV analytical linear XL (5 μm, 300 × 7.5 mm) columns thermostatted at 40 °C (column molecular weight range: 1 × 102 to 1 × 106 g mol−1), and a differential refractive index detector (Tosoh EcoSEC RI) using THF as the eluent with a flow rate of 1 mL min−1. Toluene was used as a flow marker. Calibration was performed using linear narrow polystyrene (PS) standards from PSS Laboratories in the range of 470–7.5 × 106 g mol−1. For the analysis MHKS parameters (α = 0.74, K = 10.2 × 10−5 dL g−1, THF 30 °C)54 were applied.
Electrospray ionization mass spectrometry (ESI-MS) was performed on an LCQ Fleet mass spectrometer (ThermoFischer Scientific) equipped with an atmospheric pressure ionization source operating in the nebulizer-assisted electrospray mode. The instrument was calibrated in the m/z range 220–2000 using a standard solution containing caffeine, MRFA, and Ultramark 1621. A constant spray voltage of 5 kV was used, and nitrogen at a dimensionless auxiliary gas flow rate of 3 and a dimensionless sheath gas flow rate of 3 was purged. The capillary voltage, the tube lens offset voltage, and the capillary temperatures were set to 25 V, 120 V, and 275 °C, respectively. A 250 μL aliquot of polymer solution with a concentration of 10 μg mL−1 was injected. A mixture of THF and methanol (THF–MeOH = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), all HPLC grade, was used as solvent.
:2), all HPLC grade, was used as solvent.
Continuous tubular reactor setup
The tubular flow UV-reactor consists of a brown laboratory bottle with a GL-45 screw cap with a nitrogen inlet, a HPLC pump (Knauer BlueShadow 20P), a quartz cooling mantle (Photochemical Reactors Ltd., UK) which was wrapped tightly by 25 m fluorinated gastight transparent PFA tubing (VICI, 1/16′′ × 0.75 mm, Vtubing = 11 mL), a 400 W medium pressure UV-lamp (λmax = 365 nm) and a glass vial to collect the mixture. The whole tubing was irradiated equally by using the UV-lamp. The reaction temperature of 15 °C was controlled by using a cryostat (Grant LTD6/20) filled with water. Furthermore the irradiation source and reactor were kept in a sealed wooden box for safety reasons. Pictures of the described reactor can be found in the supporting information in the publication of Conradi et al.52 which used a similar reactor setup.Microreactor setup
The microreaction was performed in the Labtrix® Start R2.2 system (Chemtrix BV, NL), fitted with a glass microreactor (3227, reactor volume = 19.5 μL) containing an SOR-2 static micromixer. Reaction solutions were introduced into the reactor through two 1 mL gas-tight syringes (SGE) capable of delivering two solutions at flow rates between 0.1 and 25 μL min−1. The flow rates were controlled via a syringe pump (Chemyx) and the reactor temperature was controlled via a thermoelectric cooler temperature controller MTTC1410 (Melcor Thermal Solutions, temperature range −15 to 195 °C). The same setup was used by Vandenbergh et al. for thermal polymerization reactions.1 An OMNICURE Series 1000 system was used as a UV-light source. The OMNICURE system was equipped with a 100 W high pressure mercury vapor short arc lamp (320–500 nm) at an iris setting of 50%.General procedure for the synthesis of pMA using the continuous tubular flow reactor
Methyl acrylate (23.723 g, 47 eq., 275.29 mmol), EBiB (1.165 g, 1 eq., 5.90 mmol), CuBr2 (0.025 g, 0.02 eq., 0.11 mmol) and Me6TREN (0.157 g, 0.12 eq., 0.69 mmol) were mixed in a 250 mL volumetric flask and filled up to a volume of 250 mL with DMSO. The polymerization targeted a number average molecular weight (Mn) of 4000 g mol−1. The mixture was poured into a 250 mL brown laboratory bottle with a GL-45 screw cap and purged with nitrogen for approximately 45 min before starting the polymerization process. Polymerization reactions with other target molecular weights were carried out with appropriate adjustment of concentrations.Procedure for the synthesis of pMA using the microreactor setup
Methyl acrylate (0.415 g, 44 eq., 4.82 mmol), EBiB (0.021 g, 1 eq., 0.11 mmol), CuBr2 (0.001 g, 0.02 eq., 0.01 mmol) and Me6TREN (0.003 g, 0.12 eq., 0.01 mmol) were mixed in a 5 mL volumetric flask and filled up to a volume of 5 mL with DMSO. The polymerization targeted a number average molecular weight (Mn) of 3800 g mol−1. Prior to use the mixture was purged with nitrogen for 3 min and then transferred into two 1 mL gas tight syringes and inserted into the syringe pump.Procedure for the synthesis of a pMA-b-pBA block-copolymer using the microreactor setup
Poly(methyl acrylate) (0.105 g, 1 eq., 0.03 mmol, Đ = 1.1, Mn = 3100 g mol−1) was used as a macroinitiator and mixed with CuBr2 (0.001 g, 0.02 eq., 0.01 mmol) and Me6TREN (0.001 g, 0.12 eq., 0.01 mmol) in a 5 mL volumetric flask and filled up to a volume of 5 mL with DMSO. The polymerization targeted a number average molecular weight of 7500 g mol−1. Prior to use the mixture was purged with nitrogen for 3 min and then transferred into two 1 mL gas tight syringes and inserted into the syringe pump.Results and discussion
The application of photo-induced copper-mediated polymerization allows for a fundamental struggle with respect to these polymerization types to be directly solved when translating the reaction protocol to a flow system. Due to the rather low concentrations of copper that are required to mediate polymerization (in the present case about 0.02 eq. compared to the initiator), strictly homogeneous reaction conditions are established and reaction blockage or fouling is eliminated. Thus the reactor channel widths play no important role and the polymerization can be applied to micro- and milliflow conditions. For the work as described herein, we have chosen two different flow systems to compare: (i) a true microreactor glass-chip reactor (19.5 μL reactor volume, borosilicate) and (ii) a tubular UV-flow system with an internal volume of 11 mL. Each system features distinct advantages. The microflow setup is best suited for kinetic studies under very economic conditions, and – due to the very small channel width – can be used with UV-light sources of intermediate power. Here we applied a UV-light source with 100 W power, which was, however, not used to full extent and usually attenuated to lower intensities. Conversely, the tubular reactor is less sophisticated (the reactor itself consists of transparent PFA tubing wrapped around an immersion well photo-reactor), but allows synthesis of materials at a significant scale due to the much higher internal volume. For the tubular reactor, a UV-light source with 400 W was chosen to achieve fast polymerization reactions. The peak wavelength in both cases was λ = 365 nm, whereby the irradiation spectrum was relatively broad. It should be noted that in the case of the microreactor, a conventional glass chip was used, thus optical transparency is limited at wavelengths below approximately 350 nm. For the present case study, we used methyl acrylate as a monomer and dimethyl sulfoxide as a solvent as good results were reported for this combination in batch processing. Thereby, the active copper species was generated from a Cu(II)Br2/tris(2-(dimethylamino)ethyl)amine (Me6TREN) system (see Scheme 1). While the exact mechanism has not yet been elucidated, photo-activation of the Me6TREN ligand followed by carbon–halogen bond breakage appears to be the dominant mechanism for chain initiation. In this process, initiator radicals are formed which are deactivated by Cu(II) to yield active Cu(I).31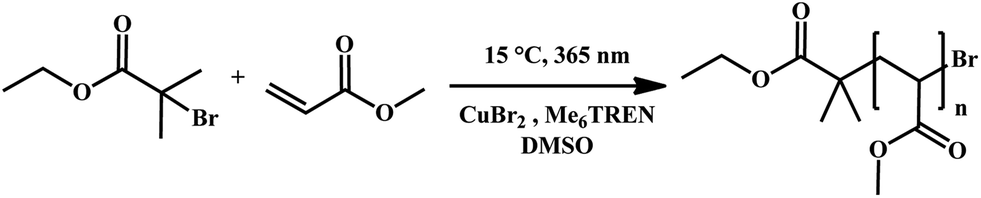 | ||
| Scheme 1 UV-induced copper-mediated polymerization of methyl acrylate (MA) in DMSO using CuBr2 and Me6TREN as a catalyst/ligand pair. | ||
Polymerization in a tubular milli-flow reactor
Fig. 1 depicts the outcome of MA polymerization reactions at 15 °C in the tubular reactor setup. It should be mentioned that the temperature was kept deliberately low. Polymerization rates would be significantly higher at slightly elevated temperatures (and due to heat transfer issues and the high exothermicity of the polymerization reactions also most thermal SET-LRP reactions in non-aqueous solvents are carried out significantly above ambient temperature), nevertheless we opted for low reaction temperatures to underpin the photoinitiated character of the reactions. Additionally, this presents an even higher motivation, low temperatures help to avoid the formation of midchain radicals, as usually observed in any acrylate polymerization reaction.54,55 Midchain radicals inevitably lead to short chain and long chain polymer branching. Choosing low reaction temperatures thus allows for synthesis of primarily linear chains with minimum branch points giving additional value to this photo-polymerization process.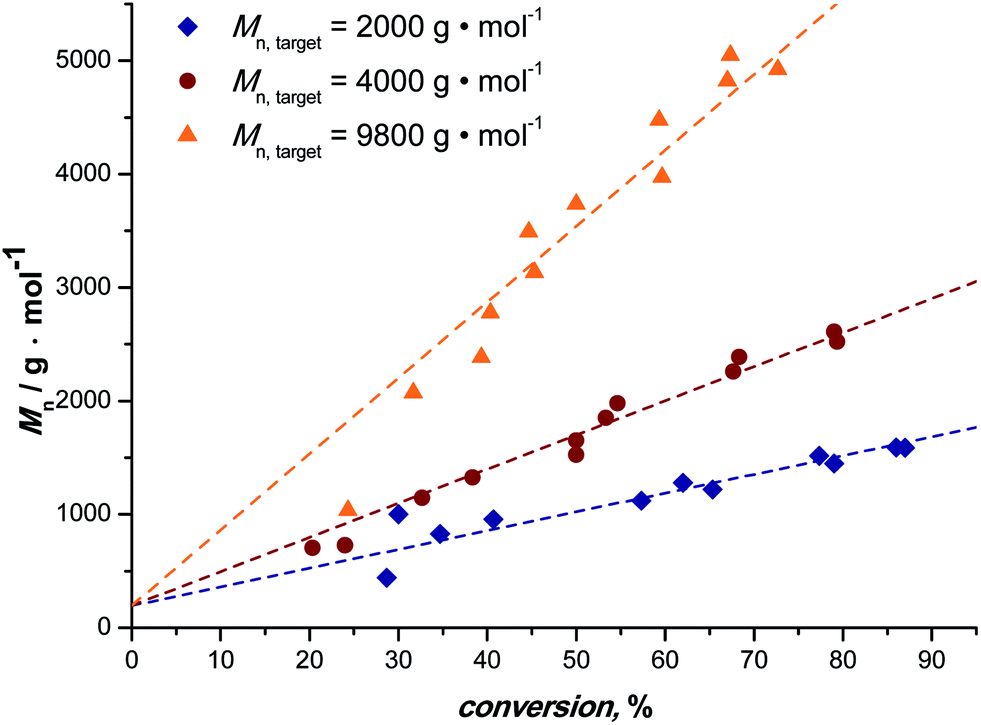 | ||
| Fig. 1 Development of the Mn of UV initiated copper-mediated polymerization of MA, with targeted Mn = 2000 (◆), 4000 (●) and 9800 (▲) g mol−1, in a tubular milli-flow reactor. The dotted lines are best fits of the data whereby the axis intercept was set to the molar mass of the initiator. | ||
Three series of polymerization reactions were performed by variation of the target molecular weight, Fig. 1. Polymerization reactions were set to ideally reach 2000, 4000 and 9800 g mol−1 at full conversion (with cmonomer/cinitiator = 23, 47 and 116, respectively). No higher molecular weights have been targeted since this range covers already a broad range of materials typically synthesized via controlled polymerization reactions. Higher molecular weights will lead to increased viscosities, which was avoided in the current study in order to cancel out complex viscosity effects. All polymerization reactions show a linear increase in Mn with respect to conversion and very high conversions were reached in all cases in a relatively short reaction time (maximum 20 minutes of residence time, see discussion below). The reaction in flow is thus significantly faster compared to all previously reported batch photo-polymerization reactions. It should be noted that the rate of reaction may be influenced by the choice of light source and that the rate increase may not only be due to the enhanced light efficiency of the flow reactor, but also due to usage of a more powerful UV-lamp.
Generally, conversion and thereby degree of polymerization are in a flow reaction influenced by variation of the residence time. With increasing flow rates lower residence times are directly achieved and the data shown in Fig. 1 can simply be constructed from continuous polymerization by variation of the pump flow rate. At the same time, larger reactor volumes can be used at proportionally higher flow rates. Thus, increasing the reactor volume by a factor of two with concomitant increase of the flow rate by the same factor leaves the reaction product unaffected, but increases the overall yield of polymer twofold. For the present reactor setup, a production of pMA of ∼60 g per day can be realized at a residence time of 20 min. Upscaling of the reaction is, as described, simple and can be realized by using longer PFA tubing being wrapped around the light source.
The molecular weight evolution of the three polymerization reactions can be fitted linearly. The slopes of all three reactions are slightly lower than the theoretical value (18, 31 and 67, respectively). The reason for the molecular weights remaining lower than expected remains unclear. Deviation from ideal behavior in controlled polymerization is often observed and the outcome of the reactions can be regarded to be within the usual limits of deviation.
In line with the observation of well-controlled polymerization, a decrease in the dispersity of the polymers is observed (see ESI Fig. S2†). With higher monomer conversion and longer reaction time the dispersity indices are decreasing for all the targeted molecular masses (from roughly 1.2–1.3 to about 1.1). Also in good agreement with expectations, slightly higher initial dispersities are observed for increasing monomer to initiator ratios.
To unambiguously demonstrate that the reaction proceeds under high control the first-order kinetic character of the polymerization was determined, Fig. 2. All reactions show good linearity indicating that the concentrations of the growing radicals remained approximately constant over the course of reactions with only very small deviations from first order kinetics seen for the highest target molecular weight reaction. It may thus be assumed that radical concentrations in all three polymerization reactions were of a similar order. Additionally, in combination with the linear increase in the average molecular weight and low dispersity of the polymers obtained it may be assumed that only very little transfer and termination events disturb the livingness of the reactions. Additionally, it can be observed that no inhibition period occurs for the polymerization and that already at very low residence times significant polymerization occurs. It must be mentioned at this point that the preparation of the reaction solution plays an important role. Of course, natural light is also able to trigger polymerization reactions, thus in principle, a reaction could occur before the solution is injected into the reactor. For the reactions described herein this is prevented by using only dark containers, minimizing light contact of the substances as much as possible. Tests show that directly after deoxygenation of the solution (and thus right before injection into the reactor) no reaction has yet taken place as no polymerization occurs in the dark.
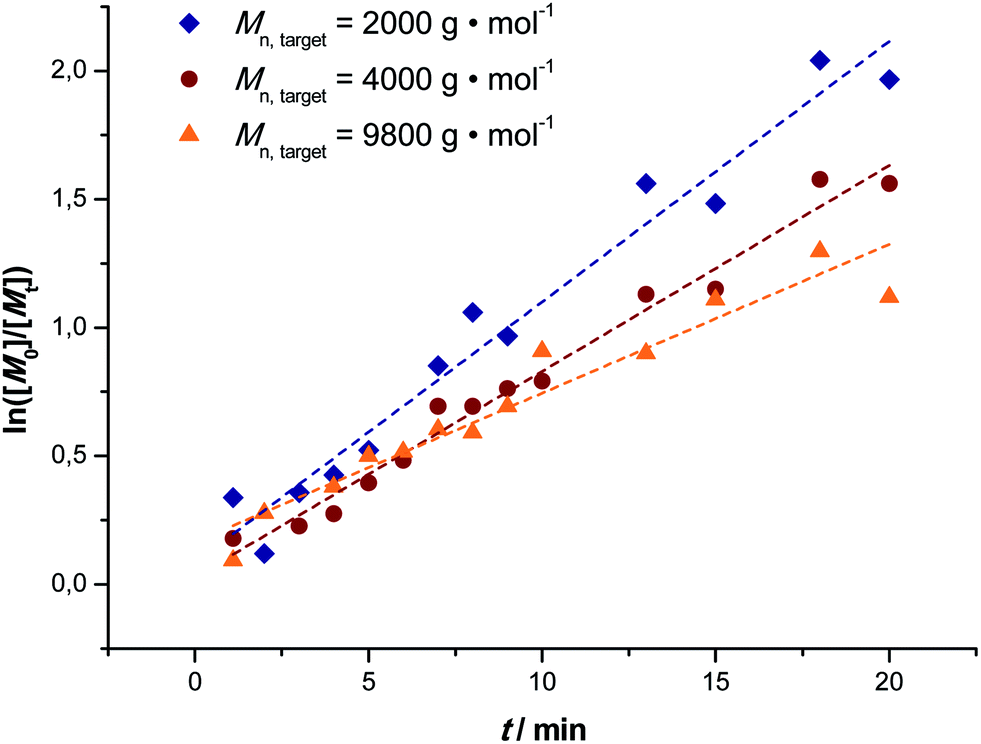 | ||
| Fig. 2 First order kinetic plots of the MA polymerization reactions in a milli-flow reactor with targeted Mn = 2000 (◆), 4000 (●) and 9800 (▲) g mol−1. | ||
Polymerization in a chip micro-flow reactor
Besides the milli-flow reactor, also a micro-flow reactor was employed to carry out UV SET-LRP polymerization to demonstrate that the reaction may also be performed in a true microfluidic device. In the micro-flow reactor a reaction with a targeted Mn = 4000 g mol−1 was performed. A maximum conversion of 80% was reached after a residence time of 20 minutes in this specific reactor and light source combinations, thus relatively comparable yields with the tubular flow reactor. Overall, the polymerization reactions in the micro-flow reactor feature the same characteristics as the in milli-flow reactor. Molecular weight evolution and dispersities are in the same range as in the polymerization reactions discussed above (see ESI† for detailed information on molecular weight and dispersity evolution and molecular weight distributions of the milliflow reactions), as shown in Fig. 3. Fig. 3 displays the molecular weight distributions as obtained from SEC, scaled to monomer conversion of the respective samples. A clear shift of the distributions – also on the low-molecular weight side – is observed, which nicely underpins that the reaction is easily applicable to different reactor setups and is thus highly interesting also for larger flow reactor systems in which kilogram (or higher) production of polymers can be achieved.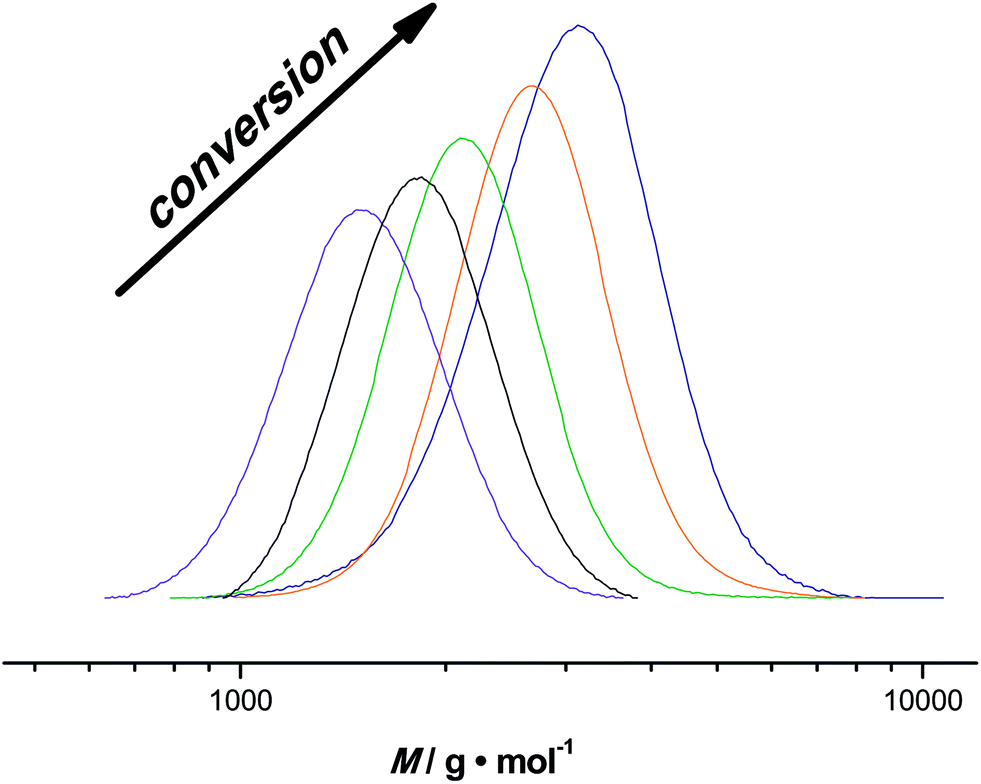 | ||
| Fig. 3 Molecular weight distributions of pMA with increasing residence time in the microflow reactor synthesized via UV-initiated copper-mediated polymerization and with a targeted Mn of 4000 g mol−1. All distributions are scaled to monomer conversion. | ||
The first order kinetic plot in micro- and milliflow reactions shows good linearity, as seen in Fig. 4. However, there is a somewhat lower slope, indicating that radical concentrations in both reactions are slightly different. Since the reactor volume and reactor type had been changed and a light source of different intensity was used, this is not surprising. It must be noted that the fit of the microreactor data indicates that at zero minutes of residence time some polymerization had taken place already (the reactor is fed by light-transparent syringes, thus polymerization could already occur in the feed). This again demonstrates the sensitivity of the reaction.
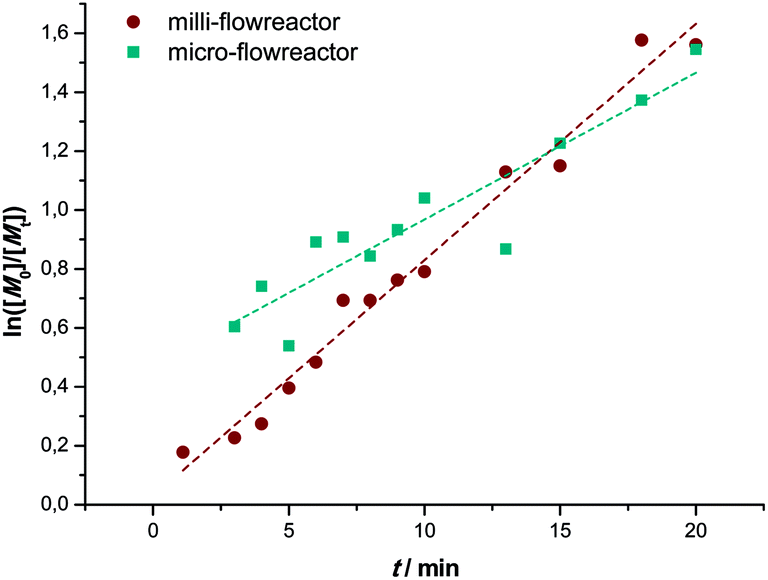 | ||
| Fig. 4 Comparison of the kinetic first-order plots of UV initiated copper-mediated polymerization of MA in a milli- (●) and a micro- (■) flow reactor. | ||
End-group fidelity
Finally, the quality of the polymers were analyzed by mapping the end group fidelity via electrospray ionization mass spectrometry (ESI-MS). Samples of polymers from the polymerization with a target Mn = 2000 g mol−1 after reaction times of 10, 15 and 20 minutes were taken to check for the presence of termination products, which may decrease the livingness of the process. The polymer samples were associated with monomer conversion between 70 and 87%. All show a very high degree of end-group fidelity (see Fig. 5 and Table 1). All ESI-MS spectra display only one single-charged product species, this being the sodium adduct of the expected structure with the initiator group in the α position and a bromine atom at the ω site, Fig. 5. With increasing reaction time, more double-charged polymers can be observed, which is associated with the increase in overall chain length of the samples. Even though traces of side products may be seen in the baseline of the spectra, the absence of any significant amount of termination products (as given in the table for one of the disproportionation species) is clearly seen. The mass-spectrometric analysis thus nicely confirms the above made observation of excellent livingness of the process. While at this stage not unexpected, this is nevertheless a very satisfying result. UV-irradiation may result in several side products, ranging from self-initiated chains over crosslinking to polymer degradation. The practical absence of all such product species confirms that the flow conditions only favor the desired product, but do not cause other processes as are often observed for UV-initiated batch reactions at similar high light intensities. It should thereby be noted that also some samples taken at even higher conversions were analyzed with virtually the same result.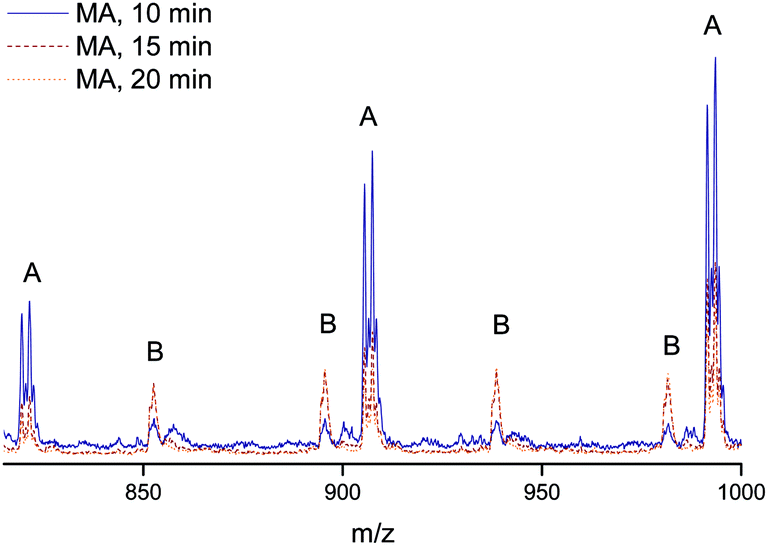 | ||
| Fig. 5 Zoomed in ESI-MS spectra of poly(methyl acrylate) obtained by UV-copper-mediated polymerization in flow reactors. The 15 and 20 min samples show higher relative intensities for double charged species due to their higher Mn which lies above the scan range of 2000 g mol−1. | ||
| Name | α end group | ω end group | Units MA | Ion | m/zexp | m/ztheo | δ/Da |
|---|---|---|---|---|---|---|---|
| A | EBiB | Br | 8 | Na+ | 905.50 | 905.28 | 0.22 |
| B | EBiB | Br | 18 | Na2+ | 894.67 | 894.32 | 0.35 |
| C | EBiB | H | 9 | Na+ | — | 913.46 | — |
Block copolymer formation
Based on pMA obtained from the tubular reactor, block copolymers were synthesized in the micro-flow reactor. For chain extensions, butyl acrylate (BA) was polymerized with a maximum reaction time of 20 minutes. Reaction conditions were chosen analogous to the homopolymerization reactions. pMA with an Mn = 3100 g mol−1 and a dispersity of 1.10 was chosen as the starting material. The targeted Mn of the block copolymer was 7700 g mol−1 (at full BA conversion) and good control over the second block is also achieved, see Fig. 6. Polymerization reactions proceed to high conversions and feature low dispersities. For example, at a BA conversion of 51%, a pMA-b-pBA polymer with an Mn = 4990 g mol−1 and a dispersity index of 1.16 is obtained (theoretical Mn at this conversion is 5400 g mol−1). Block copolymerization reactions can thus also easily be addressed in flow reactions, again underpinning the versatility of the process, as well as the high livingness of the polymerization reactions.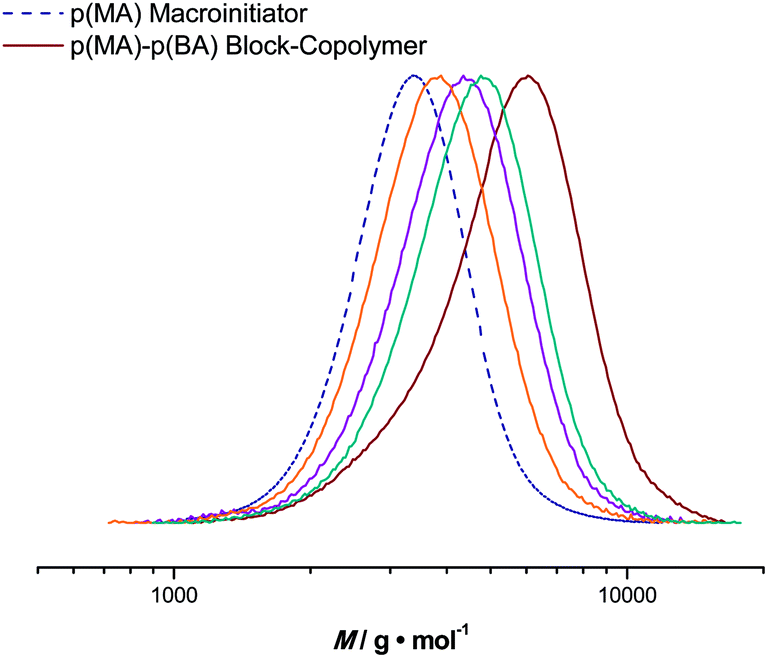 | ||
| Fig. 6 Evolution of molecular weight distributions for the p(MA)-b-p(BA) block copolymers (solid line) obtained in the flow microreactor and the distribution of the p(MA) macroinitiator (dashed line). | ||
In that respect, it is important to test that other monomers can also be polymerized in flow using the same protocol. Batch reactions had shown that a relatively large variety of monomers can be controlled by photo-copper-mediated polymerization. However, in practically all cases, fast polymerizing acrylates were used to stay within reasonable polymerization times to reach high conversions. In flow, several other monomers were also tested with various success. Styrene does not show significant polymerization of the timescale of the flow reactions (we chose 20 minutes as the highest residence time as for the other reactions described in this work). In contrast, methyl methacrylate could well be polymerized by the protocol, reached, however, only ∼35% conversion after 20 minutes (Mn = 2100 g mol−1, Đ = 1.45). Thus, polymerization of such monomers might not be most favorable to produce homopolymers in an efficient manner in the current flow setups. However, for block copolymerization reactions, where reactions are often stopped prematurely to preserve end group fidelity, such reactions could for the future also be of significant interest.
Conclusion
Photo-initiated copper-mediated radical polymerization offers intriguing features for the design and synthesis of complex materials. The translation of the batch process to flow chemistry offers to scale up this reaction for the production of significant amounts of complex materials, which in a batch process is not directly possible due to light absorption profiles and insufficient penetration of light at increased optical path lengths. The good applicability of photo-copper-mediated polymerization of MA is demonstrated for both a commercial glass-chip microreactor (volume 19.5 μL) as well as a simple tubular milli-flow reactor (11 mL). Excellent control over polymerization reactions are observed in both cases. Reactions follow first order kinetics, number average molecular weight increases linearly with monomer conversion and low dispersities are reached for all polymers obtained. Reactions are particularly fast and conversions in the range of 90% are reached within 20 minutes of reactor residence time. All polymers feature excellent endgroup fidelity and allow for efficient block copolymerization reactions, as demonstrated by the synthesis of a series of pMA-b-pBA materials in the microreactor.Overall, the described reactions give a further example for the increasing number of polymer reactions that benefit from microreactor application and flow chemistry in general. The methods described herein make use of comparatively simple and versatile flow reactors and thus do not require sophisticated instrumentation. Both the microflow and the tubular reactors are comparatively cheap and easy to set-up and are thus of potential interest to the whole polymer community as a novel way to produce materials for a broad range of research projects. At the same time the novel photo-copper-mediated polymerization process solves the general problem of inhomogeneities that are commonly observed in thermal copper-mediated polymerization reactions in flow devices, thus making the photo-initiated process also from that point of view the first choice for flow synthesis of materials from controlled polymerization.
Acknowledgements
The authors are grateful for funding in the framework of the European Science Foundation – Precision Polymer Materials (P2M) program. Additional support by the Belgian Science Policy (Belspo) via the Interuniversity Attraction Poles Program IAP P7/05 “Functional Supramolecular Systems” is also kindly acknowledged. The authors are also grateful for support of the Hercules foundation. A. D. C. is grateful for funding from USP Agency for Innovation during the research stay in Belgium. D. M. H. is a Royal Society Wolfson Fellow.References
- J. Vandenbergh, T. de Moraes Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- Z. Nie, S. Xu, M. Seo, P. C. Lewis and E. Kumacheva, J. Am. Chem. Soc., 2005, 127, 8058–8063 CrossRef CAS PubMed.
- C. H. Hornung, C. Guerrero-Sanchez, M. Brasholz, S. Saubern, J. Chiefari, G. Moad, E. Rizzardo and S. H. Thang, Org. Process Res. Dev., 2011, 15, 593–601 CrossRef CAS.
- T. Noda, A. J. Grice, M. E. Levere and D. M. Haddleton, Eur. Polym. J., 2007, 43, 2321–2330 CrossRef CAS PubMed.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- F. Bally, C. A. Serra, V. Hessel and G. Hadziioannou, Chem. Eng. Sci., 2011, 66, 1449–1462 CrossRef CAS PubMed.
- D. Wilms, J. Klos and H. Frey, Macromol. Chem. Phys., 2008, 209, 343–356 CrossRef CAS.
- V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS.
- A. R. Bogdan, S. L. Poe, D. C. Kubis, S. J. Broadwater and D. T. McQuade, Angew. Chem., Int. Ed., 2009, 48, 8547–8550 CrossRef CAS PubMed.
- J. Vandenbergh and T. Junkers, Polym. Chem., 2012, 3, 2739–2742 RSC.
- J. Vandenbergh, T. Tura, E. Baeten and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, DOI:10.1002/pola.27112, in press.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- K. Matyjaszewski, T. E. Patten and J. Xia, J. Am. Chem. Soc., 1997, 119, 674–680 CrossRef CAS.
- D. M. Haddleton, M. C. Crossman, K. H. Hunt, C. Topping, C. Waterson and K. G. Suddaby, Macromolecules, 1997, 30, 3992–3998 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3283–3299 CrossRef CAS.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- D. Konkolewicz, Y. Wang, M. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 CrossRef CAS.
- A. P. Haehnel, S. Fleischmann, P. Hesse, K.-D. Hungenberg and C. Barner-Kowollik, Macromol. React. Eng., 2013, 7, 8–23 CrossRef CAS.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672–2675 RSC.
- J. A. Burns, C. Houben, A. Anastasaki, C. Waldron, A. A. Lapkin and D. M. Haddleton, Polym. Chem., 2013, 4, 4809–4813 RSC.
- Y. Shen and S. Zhu, AIChE J., 2002, 48, 2609–2619 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3081–3096 CrossRef CAS.
- L. D. Arvanitopoulos, M. P. Greuel, B. M. King, A. K. Shim and J. H. Harwood, in Controlled Radical Polymerization, American Chemical Society, 1998, vol. 685, ch. 20, pp. 316–331 Search PubMed.
- C. Detrembleur, D.-L. Versace, Y. Piette, M. Hurtgen, C. Jerome, J. Lalevee and A. Debuigne, Polym. Chem., 2012, 3, 1856–1866 RSC.
- X. Miao, W. Zhu, Z. Zhang, W. Zhang, X. Zhu and J. Zhu, Polym. Chem., 2014, 5, 551–557 RSC.
- L. D. Arvanitopoulos, M. P. Greuel and H. J. Harwood, Polym. Prepr., 1994, 35, 549–550 CAS.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2013, 136, 1141–1149 CrossRef PubMed.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- S. Muthukrishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978–2980 CrossRef CAS.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J.-P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- D.-L. Versace, J. Lalevée, J.-P. Fouassier, Y. Guillaneuf, D. Bertin and D. Gigmes, Macromol. Rapid Commun., 2010, 31, 1383–1388 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS PubMed.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Astolfi, L. Greci, P. Tordo and D. Bertin, Macromolecules, 2007, 40, 3108–3114 CrossRef CAS.
- T. Fukuyama, Y. Kajihara, I. Ryu and A. Studer, Synthesis, 2012, 44, 2555–2559 CrossRef CAS PubMed.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2011, 212, 2036–2042 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 2271 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2011, 212, 2036 CrossRef CAS.
- M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391 CrossRef CAS.
- O. S. Taskin, G. Yilmaz, M. A. Tasdelen and Y. Yagci, Polym. Int., 2013 DOI:10.1002/pi.4573.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- A. A. Alzahrani, A. H. Erbse and C. N. Bowman, Polym. Chem., 2014, 5, 1874–1882 RSC.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- S. A. Piletsky, H. Matuschewski, U. Schedler, A. Wilpert, E. V. Piletska, T. A. Thiele and M. Ulbricht, Macromolecules, 2000, 33, 3092–3098 CrossRef CAS.
- T. Schwalbe, V. Autze, M. Hohmann and W. Stirner, Org. Process Res. Dev., 2004, 8, 440–454 CrossRef CAS.
- F. Lévesque and P. H. Seeberger, Org. Lett., 2011, 13, 5008–5011 CrossRef PubMed.
- B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed.
- M. Conradi and T. Junkers, J. Photochem. Photobiol., A, 2013, 259, 41–46 CrossRef CAS PubMed.
- L. Feng, J. Hu, Z. Liu, F. Zhao and G. Liu, Polymer, 2007, 48, 3616–3623 CrossRef CAS PubMed.
- C. Barner-Kowollik, S. Beuermann, M. Buback, P. Castignolles, B. Charleux, M. L. Coote, R. A. Hutchinson, T. Junkers, I. Lacik, G. T. Russell, M. Stach and A. M. van Herk, Polym. Chem., 2014, 5, 204–212 RSC.
- T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7585–7605 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3py01762a |
| ‡ Equal contribution of both authors. |
| This journal is © The Royal Society of Chemistry 2014 |