
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of highly functionalized C60 fullerene derivatives and their applications in material and life sciences
Weibo
Yan
*ab,
Stefan M.
Seifermann
c,
Philippe
Pierrat
d and
Stefan
Bräse
*ae
aInstitute of Organic Chemistry, KIT-Campus South. Fritz-Haber Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu; Fax: (+49) 721-608-48581; Tel: (+49) 721-608-42902
bBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, China
cCYNORA GmbH, Bruchsal, Germany
dLaboratoire de Conception et Application de Molécules Bioactives CNRS – Université de Strasbourg, Faculté de Pharmacie, 74 route du Rhin 67401 Illkirch, France
eInstitute of Toxicology and Genetics, KIT, Campus North, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 10th October 2014
Abstract
Highly functionalized fullerenes can be efficiently constructed by various techniques. However, the challenge is to synthesize highly symmetrical fullerenes. Recently, a number of X-ray structures have been disclosed showing the high symmetry of substituted fullerenes. By reviewing the major types of multi functionalized fullerenes through selected examples with a link to the structural assignments, the authors intend to give a concise overview to the specialist in the field and to provide the non-specialist with a tool box of possibilities.
 Weibo Yan | Weibo Yan started studying organic chemistry and organic functional materials at the Nankai University in Tianjin, China, in 2006 and obtained his PhD in organic chemistry from the institute of elemento-organic chemistry in 2011. After that, he worked as a postdoctoral fellow in organic nanostructures at Karlsruhe Institute of Technology (KIT) in Germany from 2011 to 2012. Since January 2013, Weibo worked as a postdoctoral fellow at the Institute of Inorganic Chemistry, College of Chemistry and Molecular Engineering, Peking University in Beijing on the synthesis of OLED, solar cell materials and the fabrication of their devices. |
 Stefan M. Seifermann | Stefan Seifermann studied chemistry at Karlsruhe Institute of Technology (KIT). He worked in the group of Stefan Bräse as a member of the Center for Functional Nanostructures (CFN) in the field of fullerene chemistry. After receiving his PhD in 2013, he joined CYNORA GmbH in Bruchsal, Germany, where he is currently studying the development of new materials for Organic Light Emitting Devices (OLEDs). |
 Philippe Pierrat | Philippe Pierrat studied chemistry at Nancy University in France. After 3 years of research in the field of organometallic chemistry in the lab headed by Pr. Yves Fort, he obtained his PhD in 2006. Next, he joined the research group of Pr. Stefan Bräse with an Alexander von Humboldt post-doctoral fellowship to work on fullerene chemistry. In 2009, he was appointed assistant Professor of Organic chemistry at the Strasbourg University in the research group of Dr Luc Lebeau, where he is currently developing new bio-responsive cationic vectors for gene delivery applications. |
 Stefan Bräse | Stefan Bräse studied in Göttingen (Germany), Bangor (UK) and Marseille (France) and received his Ph.D. in 1995, after working with Armin de Meijere in Göttingen. After post-doctoral appointments at Uppsala University (Jan E. Bäckvall) and The Scripps Research Institute (K. C. Nicolaou), he began his independent research career at the RWTH Aachen in 1997 (associated to Dieter Enders). In 2001, he finished his habilitation and moved to the University of Bonn as professor for organic chemistry. Since 2003, he is full professor at the Karlsruhe Institute of Technology and since 2012 director of the Institute of Toxicology and Genetics at the KIT. His research interests include methods in drug discovery (including drug-delivery), combinatorial chemistry towards the synthesis of biologically active compounds, total synthesis of natural products and nanotechnology. |
1. Introduction
Carbon rich nanostructures have been a major topic in chemical research over the past four decades.1–6 In particular, fullerenes that combine three-dimensionality with unique photoelectric and electrochemical properties are extremely promising nanostructures for the preparation of new advanced materials7,8 and biologically active molecules.9–12At present, the most widely used commercial fullerene derivative is a mono-functional [6,6]-phenyl-C61-butyric acid methyl ester (PCBM). It is one of the most important electron acceptor materials presently used in organic bulk-heterojunction solar cells.13
Hexakis-substituted C60 adducts with a Th-symmetrical octahedral addition pattern are unique organic molecules with an appealing compact spherical scaffold for the construction of multifunctional nanomaterials. In addition, many bis-, tris-, tetrakis- and pentakis-functionalized C60 derivatives with special electronic and structural properties have been investigated. They are potentially useful for applications in 2D/3D metal–organic frameworks,14 sensitizers for photodynamic therapy,15 solar energy conversion13 as photoactive molecular devices,16 liquid crystals materials,17 thermoresponsive materials,18 C60–polymer hybrid materials,19 or inhibitors of bacterial growth,10 and so on.
In the last two decades, well-optimized and widely used synthetic methods such as the Bingel cyclopropanation,20 1,3-dipolar additions,21,22 and Diels–Alder reactions,23–28 provided readily accessible building blocks for higher molecular architectures and optoelectronic devices. Several groups have provided in-depth insights into the fascinating field of functionalized fullerenes.
Thus, in this non-comprehensive review, new synthetic strategies of more higher-functionalized fullerene derivatives are summarized. Their properties, application or potential application value will be briefly described and discussed.
The addition of atoms (H, F, Cl, Br, O) to the fullerene core or the addition of radicals as long as they are not stereoselective are not presented in this overview.
1.1. Stereochemical considerations
The addition of functional groups to the C60-fullerene core leads to various substitution patterns.1 In most cases, the electrophilic and reactive [6.6]-bonds (Fig. 1, bold) are the starting points. | ||
| Fig. 1 Fullerene C60 and its Schlegel diagram. | ||
Besides regiochemical issues, stereochemistry plays an important role as well.1 While monosubstituted fullerenes without chiral substituents are in principal achiral, most of the higher substituted fullerenes exhibit – depending on their substitution pattern – chirality.1 As these issues have been discussed in excellent reviews, we will just give a short overview.
Starting from fullerene with Ih symmetry, a number of other symmetric entities can be derived:29C1, Cs, C2, C2v, C2h, C3v, C3h, D2, D3, D5, D2h, D6h, D2d, D5d, S4, Ih, Th, T. However, not all of them have been observed experimentally (Table 1).
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
2. Multiple functionalized fullerenes
2.1. Bis-functional fullerenes
Most of the functionalized fullerenes known to date are disubstituted derivatives because they are promising materials for optoelectronic devices and are easily accessible by a variety of synthetic protocols.In that context e.g. He et al. synthesized ICBA (2) (indene-C60-bis-adduct) by using a simple one-pot reaction.13 Starting from indene (1) and C60 in 1,2,4-trichlorobenzene as the solvent they obtained ICBA (2) in 34% yield (Scheme 1). This bis-adduct exhibits stronger absorption of visible light (400–800 nm) and a 0.17 eV higher LUMO energy level than PCBM, together with an improved solubility in common organic solvents (higher than 90 mg mL−1 in chloroform).
 | ||
| Scheme 1 Synthetic route to ICBA (indene-C60 bis-adduct). | ||
Furthermore, compared to P3HT/bisPCBM devices, PSCs based on the P3HT/ICBA system show better performance (4.5% vs. 5.4% PCE). Thus, ICBA is a promising candidate to be used as acceptor material in high performance PSCs.
A multiphoton absorbing fullerene derivative was published by Figueira-Duarte et al.15 They reported fullerene bis-adduct 4 bearing a hexaphenyl benzene based MPA (multi photon absorption) dye 3, connected to C60via two malonate units. Its synthesis was carried out in a Bingel cyclopropanation of the corresponding bis-malonate with C60, I2, and 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) in toluene at room temperature in 32% yield (Scheme 2). The authors found that the star-shaped system with increased dimensionality and branched structures, lead to the highly effective multiphoton absorption of compound 4.
 | ||
| Scheme 2 Synthetic route to a bis-functional fullerene with star-shaped chromophore. | ||
The authors showed that fluorescence of the π-conjugated system was quenched by the fullerene core due to singlet–singlet energy transfer to the fullerene fragment, followed by intersystem crossing to generate a fullerene-centered triplet. Therefore, in C60-MPA dye dyads the optical limiting properties of C60 are improved which makes such compounds interesting candidates for the protection of optical devices. Furthermore, an application in photodynamic therapy could be possible.
Garg et al. prepared a macrocyclic diporphyrin-fullerene triad 6 that could be used as an artificial photosynthetic reaction centre.16 That compound was synthesized in 20% yield via an unusual double 1,3-dipolar cycloaddition of C60 with the azomethine ylides formed from N-methylglycine (sarcosine) and a hexaphenylbenzene derivative bearing two aldehyde functionalized porphyrin units 5 (Scheme 3). The hexaphenylbenzene architecture was used to establish the required rigidity of the macrocyclic diporphyrin-fullerene. Thus, enforced by the strained 42-atom macrocycle, trans-2 disubstitution pattern and therefore C2-symmetry of the resulting fullerene derivative were achieved. The rigidity of this system results in a very fast photoinduced electron transfer (2000 times faster than charge recombination) and a relatively long-lived charge separated state. In addition, compared to corresponding compounds bearing only one porphyrin unit per fullerene core, the absorption cross section for photoinduced electron transfer is doubled, which leads to a high quantum yield of charge separation. Furthermore the hexaphenylbenzene unit allows for additional functionalization such as with antenna moieties. This way an antenna-reaction centre complex can be obtained.
 | ||
| Scheme 3 Synthesis of a macrocyclic diporphyrin-fullerene with C2 symmetry and trans-2 disubstitution pattern. | ||
Besides the above mentioned charge transfers between C60 and its substituents, there can also be π–π interactions. In this context Figueira-Duarte et al. presented fullerene bis-adducts 8 and 10 that consist of a π-conjugated oligomer unit showing π–π interaction with the C60 core (Scheme 4).40 Compounds 8 and 10 were synthesized from C60 and the corresponding bismalonates 7 and 8, respectively, under the previously mentioned reaction conditions developed by Diederich. The yields were 19% for the cis-2-bis-adduct 8 and 38% for the trans-4 bis-adduct 10.
 | ||
| Scheme 4 Synthesis of bis-adducts with π-conjugated substituents. | ||
Due to ring strain effects in these two macrocyclic dyads, the fullerene moiety and the π-conjugated oligomer substituent come into van der Waals contact. Thus, π–π interactions between these conjugated systems are enabled. The authors proved these π–π interactions by a significant red-shift in the UV/VIS spectra of compounds 8 (24 nm) and 10 (34 nm), as compared to their non-fullerene-bound analogues. They assigned this effect to the lowering of the oligomers’ HOMO–LUMO gaps by through-space interactions with the electron withdrawing C60 core. These intramolecular interactions have a significant influence on the electronic coupling of the two aniline redox units in the dyad.
The above mentioned fullerene bis-adducts all have high potential for applications, due to their electronic and optoelectronic properties. Additionally, multiple functionalized fullerenes are well-suited as building blocks for nanomaterials due to their structural variety.
Sigwalt et al. reported the synthesis of two fullerene bis-adducts (12, 13) that can be used as precursors for nanomaterials (Scheme 5).41 By reacting C60 with cyclic bis-malonate 11 in a modified version of a procedure by Nierengarten et al.42 they obtained compounds 12 and 13 in 8% and 18% yield, respectively.42 The authors assigned the excellent stereoselectivity of this reaction (no further isomers were obtained) to both the ring size of cyclic bis-malonate 11 and the minimum ring strain in trans-1 bis-adduct 12 and trans-3 bis-adduct 13. The former can especially serve as a highly potent building block. Further functionalization of this compound can give access to all-e-tetrakis-adducts carrying four substituents around the equator belt of their C60 core. Such structures can provide a variety of unprecedented nanostructures.
 | ||
| Scheme 5 Synthetic route to new fullerene building blocks. | ||
Matsuo et al. reported a protocol that gives improved yields in mono-addition reactions of Grignard reagents onto Buckminster-fullerenes.
They found out that the addition of DMF as a polar solvent enhanced the reactions of tri(organo)silylmethylmagnesium chlorides with fullerenes to excellent yields within 5–15 min (Scheme 6).43 Furthermore, the authors established a protocol that allows for regioselective alkylation of the corresponding anions 14 and gives rise to 1,4-di(organo)[60]fullerenes 15 in up to 93% yield. Cyclic voltammetry measurements revealed that almost all of the compounds they synthesized had three reversible reduction states around 1.03 V, 1.65 V, 2.27 V and calculated LUMO levels of about 3.77 eV. As these values are slightly higher than those of PCBM (LUMO: 3.80 eV), 1,4-di(organo)fullerenes are interesting alternatives to PCBM. In addition, the authors were able to tune the bulk properties and solubility of these materials by varying the chain length of the organic rests R1 and R2.
 | ||
| Scheme 6 Synthetic route to 1,4-bis-functional fullerenes.44 | ||
In a second publication, Matsuo et al. took advantage of this structure/morphology relationship for the construction of solution-processed bulk heterojunction (BHJ) solar cells with p–i–n structures.44 Their strategy was to spin coat a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 mixture of the donor CP (17) and the acceptor (16a) in a chloroform–chlorobenzene solution as the i layer. Upon thermal annealing at 180 °C, an interdigitated bulk heterojunction was generated due to a thermal retro-Diels–Alder reaction of 17 to insoluble, crystalline BP (18), and due to crystallization of SIMEF 16a (transition amorphous crystalline at 149 °C). SEM images clearly revealed a column/canyon structure of crystalline BP in the device. Since such structures were not found in PCBM-based devices (crystallization of PCBM at 195 °C), the authors ascribe this effect to SIMEF 16a, which crystallizes at 149 °C and therefore forms a crystallization matrix, forcing BP (18) to build columnar crystals. Furthermore, with a PCE of 5.2%, the BP/SIMEF-based OPV cell was one of the most efficient reported in the literature (2009). Therefore, SIMEF derivatives are promising candidates both as acceptor and morphology determining materials for BHJ solar cells.
:7 mixture of the donor CP (17) and the acceptor (16a) in a chloroform–chlorobenzene solution as the i layer. Upon thermal annealing at 180 °C, an interdigitated bulk heterojunction was generated due to a thermal retro-Diels–Alder reaction of 17 to insoluble, crystalline BP (18), and due to crystallization of SIMEF 16a (transition amorphous crystalline at 149 °C). SEM images clearly revealed a column/canyon structure of crystalline BP in the device. Since such structures were not found in PCBM-based devices (crystallization of PCBM at 195 °C), the authors ascribe this effect to SIMEF 16a, which crystallizes at 149 °C and therefore forms a crystallization matrix, forcing BP (18) to build columnar crystals. Furthermore, with a PCE of 5.2%, the BP/SIMEF-based OPV cell was one of the most efficient reported in the literature (2009). Therefore, SIMEF derivatives are promising candidates both as acceptor and morphology determining materials for BHJ solar cells.
In terms of fullerene derivatives, the Nierengarten group contributed another interesting publication, describing good film-forming properties for OPV applications.45 They prepared C60–Ru(II) hybrid structure 21 in 14% yield by first reacting the fullerene with 2,2′-bipyridine containing bis-malonate 19 with compound 20, followed by complexation with [Ru(bipy)2Cl2] (Scheme 7). Due to its amphiphilic character and the alkyl chain decorated bis-malonate moiety, which prevents aggregation of the fullerene subunits, compound 21 forms stable Langmuir films. These films show reversible behaviour in successive compression expansion cycles. In addition the authors proved the occurrence of a photoinduced electron transfer from the Ru(bipy)2 unit to the fullerene core due to effective quenching of the Ru(bipy)2 MLCT emission by the latter. The aforementioned properties turns amphiphilic C60–Ru(bipy)2 derivatives into very promising candidates for the use in solution processed OPVs.
 | ||
| Scheme 7 Amphiphilic C60–Ru(bipy)3 hybrid material. | ||
A different method towards fullerenes with good film-forming properties was presented by Nava et al.19 They prepared a fullerene-containing polymer 24 through polycondensation of acyl chloride 23, which was synthesized from carboxylic acid 22 and tetraethyleneglycol in 70% yield (Scheme 8).
 | ||
| Scheme 8 Synthetic route to fullerene-based polyesters.19 | ||
Because polyester 24 has good solubility in common organic solvents and good film-forming properties, combined with the electronic properties of C60, it was used in all-polymer donor/acceptor BHJ photovoltaic cells. Although the latter showed low short circuit currents and low open circuit voltages compared to optimized PCBM-based devices, polymers like 24 remain interesting candidates for OPV applications.
In contrast to the aforementioned covalent fullerene-containing polymer, fullero-dendrimers represent a class of monodisperse macromolecular fullerene derivatives, based on non-covalent interactions.46–48 In this context van de Coevering et al. reported a non-covalent octa-fullero-dendrimer 27.49 This compound bears a central anionic tetra[bis(benzoylammonium)aryl]silane unit that interacts with eight anionic fullerene bis-adducts. Its synthesis was carried out by an anion exchange reaction between octa-ammonium salt 26 and 8 equiv. of Na[C60CO2] (25) (Scheme 9). The authors claim that in photoluminescence experiments, the characteristic emission of the dendritic benzyl ether units was fully quenched upon excitation in the UV region. This indicates an intramolecular energy transfer from the lowest dendrite singlet excited state of the dendritic core to the peripheral fullerene singlet excited state. Thus, this kind of core-shell fullerene dendrimers may lead to the preparation of unprecedented photoactive molecular devices like antennas for light-harvesting or fullero-dendrimer-based organic solar cells.
 | ||
| Scheme 9 Synthesis of a self-assembled octa-fullero-dendrimer. | ||
Besides the non-covalent dendrimeric structure 27, fullero-dendrimers based on covalent bonds are known as well. Hahn et al., for instance, reported three generations (Gn) of fullero-dendrimers 30 bearing a central hexaphenyl benzene core and six (compound 30a), 12 (30b) or 24 (30c) peripheral C60 units.50 These structures were synthesized starting from dialcohol 28 and first esterified to give bis-(aryl)alkynes 28a–c. The key step was a cobalt-catalyzed cyclotrimerization of the alkynes to yield the corresponding n-generation dendrimers 30a–c in 93%, 62% and 24% yield, respectively. For clarity, in Fig. 2 the full structure of compound 30b is depicted separately (Scheme 10).
 | ||
| Fig. 2 Structure of 2nd-generation dendrimer 30b. | ||
 | ||
| Scheme 10 Synthesis of fullero-dendrimers 30a–c. | ||
2.2. Tris-functionalized fullerenes
The addition of symmetric malonate units onto C60 can theoretically give 14 different regioisomers. Hirsch et al. discovered that the all-equatorial e,e,e-isomers could be synthesized selectively under specific reaction conditions, explaining why the e,e,e-isomers are the most prominent members in the family of trisubstituted fullerenes.51 In this context, Sigwalt et al.52 reported an expeditious synthesis of fullerene e,e,e-tris adducts through a threefold Bingel reaction between C60 and t-butyl(trialkoxy)silane-based tris-malonates 31a–c (Scheme 11). Cyclopropanation of C60 with these malonates in the presence of I2 and DBU in toluene at −15 °C gave 32a–c with 8%, 26% and 14% yield, respectively. In this case, the silane moiety acts as a tether that forces the three adjacent malonate units into the e,e,e-addition pattern. After treatment of 32a with HF·pyridine in THF or 32b and 32c with BF3·Et2O in CH2Cl2–CH3CN, the tether was removed to give triols 33a–c in 85%, 99% and 67% yield, respectively. Bearing three free hydroxyl groups, the latter can serve as precious building blocks for a plenty of further chemical transformations. In addition, their e,e,e-fullerene addition pattern makes them perfectly suited as precursors for fullerene hexakis-adducts with an octahedral addition pattern.53 | ||
| Scheme 11 C60-e,e,e-tris-adducts bearing free hydroxy groups. | ||
This efficient method for the preparation of optically pure e,e,e-tris-adducts was further developed by Nierengarten et al. (Scheme 12).54 They used Si-tethered tris(malonate) 34, bearing enantiomerically pure dioxolane dimethanol entities, to get two diastereomers 35a (7%) and 35b (5%), evoked by the inherently chiral addition pattern on the fullerene core. The structure of one of the diastereomers was elucidated using X-ray crystal structure analysis allowing also therefore the stereochemistry of the other one. The final hydrolysis of the malonate moieties gave access to optically pure C60-tris(malonic acid) derivatives 36a and 36b in 77% and 87% yield, respectively.
 | ||
| Scheme 12 Optically pure C60-e,e,e-tris(malonic acids). | ||
Besides tris-additions by threefold Bingel reactions, combinations of Bingel reactions and Diels–Alder [4 + 2]-cycloadditions are widely used to synthesize fullerene-tris-adducts. In this respect, the group of Diederich reported e,e-trans-1-tris-adduct 38, which was prepared from C60 and bis(1,3-butadienyl)malonate derivative 37 in a successive nucleophilic cyclopropanation-[4 + 2]-cycloaddition sequence (Scheme 13).55,56 The reaction proceeded diastereoselectively in 60% yield. Furthermore, the authors demonstrated the high synthetic potential of compound 38 as a building block by regioselective conversion into tetrakis-, pentakis-, and hexakis-adducts (structures not shown).
 | ||
| Scheme 13 Tethered synthesis of an e,e,trans-1-tris-adduct. | ||
A tether-based synthetic route towards cis-2,cis-2,trans-3,tris-adducts was published by Nierengarten et al. By treatment of mono-functional fullerene 39, which had been prepared from the corresponding THP-protected diol after deprotection and reaction with ethyl malonyl chloride (conditions not shown), with DBU and I2 in toluene under high dilution conditions, fullerene 40 was synthesized in 32% yield (Scheme 14).56,57 The addition pattern, which is caused by the m-xylene units connecting the three malonate units, was proven by NMR spectroscopy.
 | ||
| Scheme 14 Synthesis of a cis-2, cis-2, trans-3-tris-adduct. | ||
All of the aforementioned regioselective syntheses of fullerene tris-adducts are based on tethers that force the substituents into the desired positions with their structure or length.
Regardless of that, the Hirsch group discovered that successive additions of malonate derivatives onto C60 proceeded with remarkably high regioselectivity.51,58,59 They took advantage of this regioselectivity in order to synthesize C60-tris-malonic acid 42 from tris(diethylmalonate) derivative 41 (Scheme 15).60
 | ||
| Scheme 15 Synthesis and incorporation of C60-tris-malonic acid derivatives in thin films. | ||
Cao et al. processed the abovementioned e,e,e-C63 (COOEt)3 (42), together with a diazonium resin, into a thin film (Scheme 15).61 In the first step they formed a layer-by-layer ultrathin film by self-assembly of tris-carboxylate ion 423− and the diazonium moiety of the resin 43 into a poly(acrylic acid) (PAA) matrix through their electrostatic interaction. In a second step the film was annealed under UV irradiation by decomposition of the diazonium moieties and formation of covalent bonds leading to polymer 44. The morphology of this film was studied by AFM/FFM experiments, which indicated its good load-bearing and friction properties. The film of polymer 44 showed good lubricative properties, due to its layered structure. Thus, it might be used as high performance lubricants in the future.
We recently showed that the CBr4-mediated non-tethered regioselective synthesis of higher fullerene-adducts developed by the Sun group is not limited to malonates.62 In this regard, we reported e,e,e-tris adduct 45 bearing three dibenzoylmethane moieties (Fig. 3).63 This compound was synthesized in 15% yield by reacting C60 and dibenzoylmethane in presence of DBU and 100 equiv. CBr4. Furthermore, its molecular structure was proven by X-ray crystallography (Fig. 3). Tris-adduct 45 is a potential candidate for acceptor material in OPVs and a versatile building block for nano-structures due to its highly symmetric structure.
 | ||
| Fig. 3 e,e,e-Tris adduct 45 and its molecular structure. | ||
2.3. Tetrakis-functional fullerenes
Compared to the aforementioned mono, bis and tris-adducts, fullerene tetrakis-adducts play only a minor role. Nevertheless, they are useful building blocks due to their structural diversity and thus represent a highly interesting research field. As their regioselective synthesis cannot be controlled easily, rather complex synthetic routes are needed to gain access to tetrakis-substituted derivatives.Facing these circumstances, the Diederich group developed a tether-based synthesis protocol that leads to malonate tetrakis-adducts 51 and 52 (Scheme 16).32 First, C60 was reacted with tethering agent 46, followed by addition of three diethyl malonate moieties (X in Scheme 16) to give mixed hexakis-adduct 47. Then, under irradiation with a Hg lamp in the presence of O2, dialcohol 48 was synthesized and further converted to compound 49. Cleavage of the tethering ester groups gave symmetric tetrakis(diethyl malonate)-adduct 50 with all four substituents arranged along its equator belt. Its molecular structure was proven by X-ray crystallography (Fig. 4). Finally, transesterification with either dodecan-1-ol or octan-1-ol gave long-chained derivatives 51 and 52 in 16% and 14% yield, respectively. As already mentioned above, such tetrakis-adducts bearing D2h symmetry can act as potential building blocks, leading to highly functional and at the same time highly symmetric nano-structures with unprecedented properties.
 | ||
| Scheme 16 Synthetic route to tetrakis-functional fullerenes. | ||
 | ||
| Fig. 4 Molecular structure of a tetrakis-substituted fullerene 50. | ||
One further interesting fullerene tetrakis-adduct having a fulvene-like π-system was reported by Murata et al. (Scheme 17).64 Through the reaction of C60 with potassium fluorenide (53) in the presence of fluorene and traces of O2, compound 54 was obtained in 40% yield as black crystals whose structure was elucidated by X-ray crystallography (Fig. 5).
 | ||
| Scheme 17 Synthesis of fulvene-like tetrakis-adduct 54. | ||
 | ||
| Fig. 5 Molecular structure of a tetrakis(9-fluorenyl)fullerene 54.64 | ||
The authors demonstrated the fulvene reactivity of this system by reacting it with nucleophiles such as potassium fluorenide 53, lithium organyls or NaCN to obtain the corresponding pentakis-adducts 55 bearing five substituents arranged in a propeller-like fashion.
2.4. Pentakis-functionalized fullerenes
Penta-functional adducts of Buckminsterfullerene have been reported mainly with a focus on pentahaptofullerene and its metal complexes. The fullerene analogues of cyclopentadiene and its anion are obtained by five-fold addition reaction between organocopper compounds and C60 (Fig. 6). | ||
| Fig. 6 Molecular structure of a pentakis-substituted fullerene. | ||
The field of such pentahapto fullerenes and their metal complexes has been intensely studied by Sawamura and Nakamura's group. They found that unlike organolithium or Grignard reagents, cuprates can undergo a five-fold addition onto C60 yielding Cp-like ligands.34 In that context C60 was reacted with an excess amount of Ph2CuMgBr and C60Ph5H (56) was obtained in 94% yield after quenching with aqueous NH4Cl. Furthermore, η5-metal complexes 57 were formed, proving the applicability of fullerene 56 as a cyclopentadiene analogue (Scheme 18). Although this compound bears a Cp-ring that is separated from the remaining C50-π-system by five sp3 carbon atoms, UV-vis spectroscopy of both compound 56 and the corresponding thallium complex indicates that the electronic properties of parental C60 were not severely affected by the pentakis-functionalization. Thus, such fullerene derivatives combine the ability to stabilize numerous metals in a wide range of oxidation states with the outstanding optoelectronic properties of C60. This may lead to new materials with a remarkable application potential in different fields.
 | ||
| Scheme 18 Synthesis of pentahaptofullerene C60Ph5H (56). | ||
Sawamura et al. combined the aforementioned concept of fullerene-based Cp-derivatives with supramolecular chemistry.17 In a three-step protocol consisting of the initial pentakis-functionalization of C60 with (4-(THPO)C6H4)CuMgBr followed by deprotection of the alcohol moiety and subsequent acylation, they synthesized pentakis-adducts 58a–d, in about 60% overall yield (Scheme 19).
 | ||
| Scheme 19 Synthesis of liquid-crystalline C60-pentakis-adducts. | ||
The five biaryl rests arranged around one five-membered ring of the C60 cage create a deeply conical cavity, perfectly suited for the incorporation of a second fullerene molecule through π–π interactions. The latter leads to one-dimensional stacking of these shuttlecock-shaped molecules to give supramolecular columns, which was proven by small-angle (SAXD) and wide-angle X-ray diffraction (WAXD). Due to the flexible aliphatic side chains, these assemblies show thermotropic and lyotropic liquid crystalline behaviour.
In a related report from the same group, the biaryl moieties were not connected directly to C60 like in compounds 58a–d, but via additional CH2SiMe2Ar-linkers (Scheme 20, compounds 59a–d).65 The authors used a similar synthetic strategy as described above, except with the addition of cuprate (4-(THPO)C6H4SiMe2CH2)2CuMgCl as the key step. Their investigation by DSC, polarizing optical microscopy and X-ray diffraction revealed that the cavities now are larger and more cup-shaped due to the rather long Si–C bond. Thus, in the supramolecular assemblies of compounds 59a–d, the fullerene site of a given molecule perfectly fits into the cavity of its neighbour, which allows for higher fullerene–fullerene interaction. Furthermore intracolumnar distances are shorter than in compounds 58a–d.
 | ||
| Scheme 20 Pentakis-adducts bearing a cup-shaped cavity. | ||
Since in R5C60H structures the optoelectronic properties of parental C60 remain unaffected, compounds 58a–d and 59a–d represent a unique material class that unites the features of fullerenes, Cp-ligands and liquid crystals. For example, by complexing the Cp-moieties with metals, unprecedented redox-active liquid-crystalline fullerene/metal hybrid materials could be created in the future.
This issue was impressively addressed again by Sawamura et al., who reported fullerene/ferrocene hybrid material Fe(C60Me5)Cp (61) (Scheme 21).66 This 18-electron sandwich complex is accessible through direct complexation of C60Me5H (60) with [FeCp(CO)2]2 in 52% yield.
 | ||
| Scheme 21 Synthetic route to a fullerene/ferrocene hybrid. | ||
The molecular structure of complex 61 was elucidated by X-ray diffraction, revealing a staggered conformation of the free (Cp) and the fullerene-bound (FCp) cyclopentadienyl units. As the Cp–Fe–FCp distances are very similar to those of ferrocene, both the Cp and the FCp units must be aromatic. This is additionally confirmed by NMR spectroscopy. Furthermore in the 1H NMR all proton resonances are notably shifted downfield compared to ferrocene, which indicates the highly electron withdrawing character of C60. The EPR spectrum of compound 61 did not show any peak, which indicates a singlet ground state like in ferrocene, whereas the UV/vis spectrum looks similar to that of pristine C60. Finally, CV measurements revealed both a ferrocene-like reversible oxidation potential (+0.22 V) and a fullerene-like reversible reduction potential (1.46 V versus ferrocene). With these findings the authors clearly showed that complex 61 is a fullerene/ferrocene hybrid material, comprised of both the electronic properties of pristine ferrocene and fullerene. Since various protocols exist to either modify the ferrocene or the fullerene unit, these structures can make tailored materials accessible and therefore serve as key compounds in material science.
Matsuo et al. investigated the corresponding pentakis-adducts of the endohedral fullerene H2@C60 (62).67 Following the above mentioned cuprate addition protocol, they synthesized endohedral pentakis-adduct 63 in 92% yield by reacting H2@C60/C60) (4/1) with 15 equiv. of the cuprate prepared in situ from PhMgBr and CuBr·SMe2 (Scheme 22). Compound 63 was then converted by treatment with [FeCp(CO)2]2 to the corresponding ferrocene derivative 64 in 71% yield. X-Ray diffraction experiments revealed that the encapsulated dihydrogen has no influence neither on the molecular nor the crystal structure, when compared to those of the corresponding non-hydrogen-containing derivative. Furthermore the authors demonstrated that in compounds 63 and 64 the 1H NMR resonance of the encapsulated H2 was remarkably upfield-shifted (10.39 ppm and 9.79 ppm, respectively), compared to endohedral fullerene 62 (1.44 ppm). The authors assigned these effects to shielding/deshielding effects inside the fullerene core, due to a disturbance of the parental C60-π-system caused by outer sphere functionalization. In summary, since H2 encapsulated in C60 has only neglectable influence on its physical and chemical properties and its 1H NMR resonance responds sensitively to changes on the outer sphere, it can act as a sensing probe to the inside and outside environment of the cage and therefore provide deep insight into electronic properties of functionalized fullerenes.
 | ||
| Scheme 22 Endohedral pentakis-adduct and fullero-ferrocene.67 | ||
2.5. Hexakis-functionalized fullerenes
The bonds between two fused six-membered rings in C60 display the most pronounced double bond character as they are shorter than the other bonds and thus the most reactive. Such bonds theoretically allow to access to highly substituted products of C60. Many hexa-substituted products were accessible through a wide range of reactions and with different symmetry patterns. For instance, the reaction of fullerene C60 with tert-butyl hydroperoxide in the presence of ceric ammonium nitrate (CAN) yields hexakis-adduct 65 (Fig. 7), which is a starting material to isomerically pure fullerenols.68 Star-like water soluble fullerene derivatives can be synthesized by the reaction of C60 with sodium naphthalide, followed by nucleophilic substitution on 1,4-butane sultone to give hexa(sulfobutyl)fullerene C60[(CH2)4SO3]6−. This compound found many applications such as MALDI matrix,69 free radical scavenger,70 or water-soluble electron-accepting layer for photocurrent generation.71,72 | ||
| Fig. 7 Hexa-substitution of C60 with tert-butylperoxo radicals. | ||
Hexakis-substituted fullerenes with an octahedral addition pattern are highly symmetrical three-dimensional structures that have therefore attracted much attention for a diverse range of applications. Two early X-ray structures for hexa-substituted fullerenes are depicted in Fig. 8.
 | ||
| Fig. 8 Molecular structure of A (66): a hexakis-substituted fullerene (CCDC 145416)35 and B (67): a tethered hexakis-substituted fullerene (CCDC).38 | ||
The first examples of octahedral fullerene synthesis appeared in the literature at the beginning of the 90's. A C60 core bearing six octahedrally-arranged [(C2H5)3P]2Pt groups (each bound in a dihapto manner) and having overall Th point group symmetry was synthesized in 88% yield in a one step process involving C60 and 10 equivalents of [(C2H5)3P]4Pt in benzene.73 The structure of this air sensitive product was unambiguously determined by NMR spectroscopy as well as X-ray crystallography. The high degree of regioselectivity is assumed to occur due to reversible metal complexation, slowly equilibrating in favour of the thermodynamic product. Later, Kräutler et al. reported that the Diels–Alder reaction is also a suitable route to access highly symmetrical octahedral fullerene products, since 2,3-dimethyl-1,3-butadiene was efficiently connected to the fullerene core through six regioselective [4 + 2]-cycloaddition reactions producing the expected compounds in 28% yield.74 The preparation of hexa-substituted C60 derivatives bearing six pyrrolidine peripheral groups has been achieved by 1,3-dipolar cycloaddition between C60 and azomethine ylides.75 This reaction led to the formation of two hexa-substituted products, one with the Th symmetry and one with a D3 symmetry, both displaying high fluorescence quantum yields that suggest their applicability in electroluminescent devices and light-harvesting dendritic systems.
The Bingel cyclopropanation approach for C60 functionalization has also be considered as a potential way to access hexa-substituted derivatives, since it deals with irreversible reactions that deliver stable derivatives. Hirsch et al.76 reported a remarkable stepwise synthesis starting from the chiral C3-symmetric tris-adduct whose synthesis was previously reported.51 Successive equatorial (e) additions of diethylbromomalonates in the presence of sodium hydride afforded first the Cs-symmetric tetrakis-adduct, followed by the C2v-symmetric pentakis-adduct and finally the Th-symmetric hexakis-adduct. It is worth noting that each of the intermediates as well as the final hexa-substituted product were isolated by tedious and scale-limiting HPLC purification. Furthermore, the overall synthetic route afforded the expected hexakis-substituted product with a total yield of 0.2% from C60. In the meantime, Diederich et al. reported the syntheses of other hexakis-adducts that were obtained through an original tether-directed remote functionalization concept.77 Next, Lamparth et al. developed a one-step route with good yields to hexa-substituted C60 derivatives from C60 and malonates. This route is based on template-directed activation of the octahedral reactive positions of the fullerene core.36 In the first step, C60 was reacted in a reversible way with 9,10-dimethylanthracene (DMA, 10 equiv.) to form the corresponding Diels–Alder adducts of the general formula C60(DMA)n (Scheme 23).
 | ||
| Scheme 23 First one-step synthesis of hexa-substituted Th fullerene 68 from diethylbromomalonate. | ||
Thus, subsequent treatment with DBU as a base and diethylbromomalonate allowed the formation of the expected hexakis-substituted product 68 in 37% isolated yield, which is 120 times better than the step-by-step approach previously developed. Interestingly, the authors showed that 68 could be quantitatively transformed into the corresponding highly water-soluble hexamalonic acid derivative by treatment with NaH in methanol. Later on, Hirsch et al. further improved the reaction conditions when they found that bromomalonates, which are difficult to prepare and isolate as pure materials due to their complicated structures, could be simply generated in situ by using 10 equiv. of both the starting malonates and CBr4 as the bromination agent.78
Importantly, the research group of Sun has recently brought further improvements to the above-mentioned reaction conditions, which are now the more appropriate method to generate a wide range of octahedral fullerene derivatives from C60 in one step and high yield.62,79 This was achieved by a simple modification of the reaction conditions involving the use of a large excess of bromination agent (100 equiv. of CBr4) without the need of DMA as templating agent and resulted in a dramatic increase of the final hexakis-substituted product yield.
While very useful and despite its many optimizations, the synthetic approach consisting of a one-pot regioselective installation of six malonates entities onto the fullerene core results in low yield and limited purification when starting with bulky and structurally more complicated malonates. To overcome this general trend, different research groups developed a range of synthetic strategies over the last few years to deal with the synthesis of “simple” C60 hexakis-adduct in one step, bearing peripheral reactive groups, that could be further functionalized. To do so, the later reaction must be easy to perform, tolerant to a wide range of functional groups, high yielding and should not produce many by-products for ease of purification.
The Nierengarten group was the first to report on such an approach and evaluated the potential of the copper-mediated Huisgen 1,3-dipolar cycloaddition click reaction to perform further functionalization of fullerene hexakis-adducts. Iehl et al. reported the synthesis of malonate 69 (Scheme 24) bearing two terminal azide functional groups.80 Briefly, the latter was easily obtained in two steps by the reaction of malonyl dichloride with 3-bromopropan-1-ol in the presence of pyridine and subsequent treatment with sodium azide in DMF at room temperature.80 Hexakis-adduct 70 was then readily synthesized by the reaction of C60 with 69, CBr4 and DBU in o-DCB at room temperature for 72 h. The product was obtained as a pure material in 62% yield after silica-gel chromatography. The reaction conditions for the 1,3-dipolar cycloaddition of hexa-substituted fullerene 70 with various aryl acetylenes 71a–f were optimized, and products 72a–f were obtained in 56–81% isolated yield by using the best conditions (CuSO4·5H2O and sodium ascorbate in a mixture of CH2Cl2–H2O at room temperature). The authors proved the high efficiency of this two-step synthetic approach to easily obtain a wide range of highly functionalized octahedral fullerene hexakis-adducts, even with bulky peripheral groups.
 | ||
| Scheme 24 Synthetic route to highly functionalized[60]fullerene hexakis-adducts by Click chemistry. | ||
Next, Iehl et al. reported the complementary version of the above mentioned twelve click reactions around the C60 core by preparing a C60 hexakis-adduct with twelve trimethylsilyl (TMS)-protected alkyne groups, which is suitable for further click reactions in the presence of various azide derivatives (Scheme 25).81 Malonate 73 was accessible starting from malonyldichloride and the corresponding alcohol in the presence of pyridine. Hexakis-adduct 74 was then readily synthesized in 49% yield by the reaction of C60 with 73, CBr4 and DBU in o-DCB at room temperature. The authors also showed that the Huisgen coupling could be directly conducted from protected-alkyne 73 by in situ desilylation in the presence of TBAF, Gn-N3 (75, n = 0–2), CuSO4·5H2O, and sodium ascorbate in CH2Cl2–H2O at room temperature. Under these conditions, products 76a–c were isolated in excellent yields ranging from 58 to 84%.
 | ||
| Scheme 25 Alternative synthetic approach to access highly functionalized hexakis-adducts by click derivatization. | ||
Furthermore, the authors demonstrated that by using a step-wise combination of these complementary click approaches, [5:1]fullerene hexakis-adducts with various peripheral groups are accessible (Scheme 26). To this end, C60 hexakis-adduct bearing two TMS-protected alkynes and ten azide groups has been described. Fullerene mono-adduct 77 was first prepared in 53% yield and further reacted with an excess of complementary malonates to yield the corresponding [5:1]fullerene hexakis-adducts in 45% yield. Hence, successive Huisgen reactions were conducted to afford the expected derivative 78 in good to excellent yields.
 | ||
| Scheme 26 Synthetic route to [5:1]hexakis-adducts of C60 by a sequential click–click approach. | ||
Iehl et al. further enriched the functionalization possibilities on fullerene hexakis-adducts by using the well-documented radical thiol–ene chemistry (Scheme 27).82 Thus, malonate 79 was efficiently linked to the fullerene core by the modified-Bingel cyclopropanation method allowing access to hexakis-adduct 80 in 41% isolated yield. Next, the desired thioether derivatives 81 were obtained by using a mixture of 80, RSH (20 equiv.) and AIBN (3 equiv.) in degassed-benzene at 80 °C for 1 h. Using this approach, fullerene hexakis-adducts were obtained in 30–50% yields.
 | ||
| Scheme 27 Thiol–ene reactions for hexakis-substituted fullerene derivatization. | ||
Based on the efficiency of the derivatization methods previously described on fullerene hexakis-adducts, the authors were even able to produce building blocks incorporating complementary reactive centres with three different peripheral reactive sites (azide, TMS-protected alkyne and alkenes) and to selectively react each of them stepwise. Since the Click reactions (thiol–ene and Huisgen cycloaddition) used for the functionalization of the fullerene core are tolerant to a wide range of functional groups, this methodology allowed for the easy preparation of a large variety of unprecedented globular multifunctional nanomaterials with a controlled distribution of functional groups on the spherical framework, that could be useful for various applications.
For instance, Nierengarten et al. developed an efficient access to fullerene hexakis-adducts derivatives bearing twelve peripheral carbohydrate moieties, to access a potential multivalent effect from cooperative interactions of the carbohydrate groups with the corresponding receptors, such as proteins.82 Synthetic access to such compounds was accomplished by the copper-mediated Huisgen 1,3-dipolar cycloaddition between the sugar, bearing an azide functional group, and the corresponding hexakis-adduct bearing twelve terminal alkyne units, as shown in Scheme 28. Interestingly, the authors showed that instead of a two-step synthetic process, the conjugates could be even accessible in a one-pot reaction involving in situ desilylation with TBAF, followed by subsequent click reaction with the corresponding azide (13 equiv.) in the presence of CuSO4·5H2O (0.1 equiv.) and sodium ascorbate (0.3 equiv.) in DMSO at room temperature. Following this novel synthetic route, the authors synthesized a series of new fullerene glycoconjugates with high isolated yields ranging from 40% to 90% yield. They further demonstrated that the synthetic access to fullerene sugar balls could be achieved in a reverse approach between a fullerene building block bearing twelve terminal azides as functional groups and various sugar moieties bearing terminal alkynes. Thus, it was shown that fullerene hexakis-adducts bearing twelve peripheral carbohydrate moieties can be synthesized in good yields by grafting unprotected sugar derivatives onto the fullerene core by efficient Huisgen-type click reactions.
 | ||
| Scheme 28 Synthetic route to fullerene (imino)sugar balls. | ||
By this synthetic route, Compain et al. reported the synthesis of a fullerene hexakis-adduct, decorated with twelve iminosugar residues 82 in up to 83% yield, with the intent of studying the potential of multivalency on enzyme inhibition.9 The inhibition profile of this fullerene iminosugar ball has thus been systematically evaluated against various glycosidases and compared with its monovalent analogue. The results indicated a dramatic and unprecedented multivalent effect. Indeed, for most of the glycosidases tested, hexakis-substituted fullerene obtained from iminosugar 82e demonstrated a better binding activity than the corresponding monomer. The stronger multivalent effect was obtained with Jack bean α-mannosidase, which had a Ki value up to three orders of magnitude higher than the monomeric control iminosugar. These results highlighted the impact of the central C60 scaffold on the inhibition potential of the multivalent architecture.
It has also been reported that fullerene glycoconjugates have interesting applications in antibacterial therapy.10 Bacteria adhere in the early stages of infection to the cell membrane by interactions between carbohydrate present at the surface of epithelial cells and some proteins such as adhesin present on the tip of bacterial pili.
The group of Nierengarten studied dodecamannosylated fullerenes and their interactions with Escherichia Coli adhesion FimH, which is a particular lectin for which no multivalent effects have been reported so far, in contrast to other lectins that are mostly multimeric.9,82 The key intermediate bearing an azide functionality was thus prepared by a Sonogashira cross-coupling reaction between mannoside 84 and alkyne 85 in 77% yield (Scheme 29). The latter product was then converted into the desired azide precursor 86a, in three steps involving first a mesylation step, followed by the substitution with sodium azide and finally, the removal of the acetyl groups using the Zemplén deprotection method. The last step consisted in the efficient coupling of azide 86b with a fullerene bearing twelve terminal alkyne functionalities to produce the desired mannosylated hexakis-substituted fullerene product 87 having a benzylic aromatic ring at the anomeric position and an amide linker. The ligand potential of this compound towards bacterial adhesin FimH was assessed by isothermal titration calorimetry (ITC), surface plasmon resonance (SPR) and hemagglutination assays. An unexpected phenomenon occurred during the usual titration of FimH by the dodecamannosylated fullerene. Instead of the typical sigmoidal plot, the authors observed a first binding event followed by a strong exothermic jump that could be attributed to an aggregation of the protein. SPR thus confirmed the ITC findings that dodecamannosylated fullerene 87 can accommodate up to seven lectins. It was further demonstrated that the length and the structure of the anomeric substituent are key factors that dramatically enhance the binding affinity of the carbohydrate subunit. This fullerene derivative also displayed a low micromolar inhibition level: in each case lower than its corresponding monomer. Thus, the authors proved that a globular C60 bearing peripheral mannose moieties can accommodate up to seven FimH molecules and thus have a strong affinity to FimH through multivalent effects. The multivalency has been recently corroborated by the Nierengarten group.83,84
 | ||
| Scheme 29 Synthetic access to dodecamannosylated fullerenes. | ||
As an extension of the above-cited results, Cecioni et al.12 reported a preparation of dodecavalent glycoclusters bearing galactose and peripheral glucose residues (Fig. 9) and studied their interactions with lectin PA-IL (LecA), a bacterial lectin from Pseudomonas Aeruginosa. This bacterium is a target of choice as it is responsible for nosocomial infections and is displaying increasing resistance to antibiotics. Interactions between PA-IL and carbohydrates, particularly galactose, are at the origin of this bacterial infection. It was thus anticipated that strong ligands of lectin could be valuable drugs against bacterial infections.
 | ||
| Fig. 9 Structures of hexakis-substituted fullerene-based glycoclusters. | ||
Following the same synthetic pathways described previously, the synthesis of compounds 88, 89 was achieved from the hexakis-substituted fullerene derivative bearing twelve silylated alkyne moieties. This derivative was first treated with TBAF, yielding terminal alkyne moieties, available to undergo Click reaction with the galactoside counterparts containing terminal N3 groups catalysed by CuSO4·5H2O and sodium ascorbate to obtain compound 88 in 74% and 89 in 72% yield. A reverse approach was used to synthesize compound 90 in 73% yield by conjugating the dodecaazide derivative with the corresponding carbohydrate precursor bearing a terminal alkyne functional group. These compounds were then analysed as inhibitors of the bacterial lectin PA-IL by using hemagglutination inhibition assays, enzyme-linked lectin assays and surface plasmon resonance assays. The level of inhibition or binding was found to be highly dependent on the nature of the peripheral sugar residues (galactose vs. glucose) and the nature of the spacer separating the galactose residues from the fullerene core. Importantly, the peculiar spherical distribution of the twelve sugar subunits around the carbon sphere led to a dramatic multivalent effect. Galactosylated fullerene derivatives 90 display up to a 12000-fold increase in binding affinity compared to the monovalent carbohydrate reference. These fullerene derivatives have the highest reported binding affinity in terms of ligand of lectin to date and thus represent potential new anti-adhesive agents against bacterial infection. A recent review written by Nierengarten et al. gives further reference to the biological potential of fullerene sugar balls.85
The multivalent potency of hexakis-substituted fullerene derivatives have also been evaluated as a potential precursor to non-viral gene delivery vehicles. To do so, Sigwalt et al.11 synthesized different polycationic fullerene hexakis-adduct derivatives bearing cationic NH3+ terminal groups with the general structure Gn-3NH3+, as depicted in Scheme 30.11 Precursors Gn-3N3 were obtained from the corresponding benzylic bromides treated with NaN3 in DMF. The subsequent grafting of azides Gn-3N3 (n = 1, 2 or 3) onto the hexa-substituted fullerene core was achieved under copper-catalyzed alkyne-azide 1,3-dipolar cycloaddition conditions. Treatment of fullerene hexakis-substituted building block 91 with a slight excess of azides GnN3 in the presence of TBAF, CuSO4·5H2O and sodium ascorbate gave the corresponding benzyloxycarbonyl (Boc)-protected dendrimers GnNHBoc (n = 1, 2 or 3) in remarkable isolated yields. Finally, treatment of G1–3NHBoc with a large excess of trifluoroacetic acid (TFA) gave the corresponding deprotected derivatives G1-3NH3+ as their trifluoroacetate salts in quantitative yields.
 | ||
| Scheme 30 Synthetic route to polycationic fullerene hexakis-adducts.12 | ||
As a first set of evaluation, the authors showed that polycationic fullerene G1-3NH3+ complexes efficiently plasmidic DNA at N/P ratio of 2, forming “donut-like” polyplex structures with diameters smaller than 100 nm. Next, pCMV-Luc gene delivery experiments were conducted with G1-3NH3+ polyplexes on cultured HeLa cells. While G1-NH3+ is only slightly active in delivering DNA to the cells, G2-NH3+ and G3-NH3+ display approximately the same gene delivery capacity as commercially available JetSI-ENDO, with comparable luciferase expression level, at N/P ratio of 2 (i.e. between 1011 and 1012 RLU per mg of protein). Moreover, the most active derivative G2NH3+ had minimal toxicity effects as revealed by the amount of total proteins that only diminished from 10% of its initial amount in untreated cells at the optimal N/P ratio for transfection. These results showed for the first time that polycationic fullerene hexakis-adducts could be efficient synthetic gene transfection vectors for DNA delivery with low toxicity despite a high density of cationic charges around the C60 core. No siRNA delivery has been reported with this system to date, or in vivo delivery experiments.
Based on the unique Th octahedral symmetry of hexa-substituted fullerene derivatives, researchers have been interested in studying whether hexakis-substituted fullerene could be a suitable scaffold to graft redox entities on the periphery to identify charge hopping from one redox centre to another by ultrafast cyclic voltammetry. With this idea in mind, Fortgang et al. synthesized the desired[5:1]fullerene hexakis-adduct by a stepwise functionalization of the hexa-substituted fullerene scaffold with sequential copper(I)-catalyzed alkyne–azide cycloaddition reactions, which allowed the successive grafting of two kinds of peripheral groups: ferrocene redox subunits and 1,2-dithiolane moieties available for grafting onto the gold electrode (Scheme 31).86
 | ||
| Scheme 31 Synthetic route to [5:1] fullerene hexakis-adduct.86 | ||
Reaction of starting block 93 with ethynylferrocene in the presence of CuSO4·5H2O and sodium ascorbate gave compound 94. Subsequent treatment with azide 95 in the presence of TBAF, CuSO4·5H2O, and sodium ascorbate in a CH2Cl2–H2O mixture afforded the fully reacted [5:1]fullerene hexakis-adduct 96. By usual techniques, this mixed [5:1]fullerene hexakis-adduct was immobilized onto gold ultramicroelectrode due to its dithiolane groups, which allow the ten peripheral ferrocene redox subunits to spatially organize on the surface of the electrode with a certain degree of control by the rigidity of the C60 core. The first oxidation wave of the compound 96, related to oxidation of the ferrocene entities, has been mainly studied. Indeed, at low scan rates, ferrocene entities that lie too far away from the surface cannot communicate directly with the electrode since the electronic coupling is too weak. These ferrocene moieties can however be oxidized in a multistep hopping mechanism, which results from redox centres that interact with one another. At high scan rate, the waves broaden and the peak potentials shift a little because the voltammetric rate compares to the one related to electron transfer between the electrode and the redox sites in its proximity. The results gave an indication that only a fraction of the ferrocene subunits of 96 are oxidized during the forward scan. This results from that some redox centres nearby the electrode remain independent, with pure adsorption behaviour, no electron hopping.86
By the same synthetic approach described above involving sequential CuAAC reactions on a [5:1]fullerene hexakis-adduct precursor bearing ten azide functional groups and two TMS-protected alkyne units, Iehl et al. prepared the fullerene based artificial light-harvesting arrays Y10B2C60 (Fig. 10) in excellent isolated yields with the objective to evaluate and characterize the energy transfer processes within polymer films doped with Y10B2C60.87 The advantage of this synthetic route is that it generates asymmetric fullerene derivatives bearing ten yellow and two blue boron dipyrromethene (Bodipy) dyes, thus displaying a gradient of energy levels around the C60 core that is known to exhibit only low absorption of visible light spectrum. The authors showed that in dry films, energy-transfer occurs such that photons absorbed by the yellow dyes are efficiently emitted by the blue dye. As a first proof of concept, a single layer PMMA-based luminescent solar concentrator (LSC) containing a low density of Y10B2C60 was built and used to sensitize usual amorphous silicon photocell. The obtained photocell was found to display an average light-to-electricity conversion of ca. 5% under ambient light conditions, proving that these decorated nanoparticles sensitize amorphous silicon photocells with increased absorption potential. The biggest challenge lies with self-assembling the loaded C60 particles into a loose network where inter-particle energy transfer is optimized.
 | ||
| Fig. 10 Structure of C60 hexakis-adduct Y10B2C60 decorated with yellow and blue Bodipy-based dyes. | ||
Molecular switches and nanomachines have attracted considerable attention within the last years to build responsive materials for many applications. For instance, many efforts have been made to generate molecules in which electric stimulus results in molecular motions. Iehl et al. reported a copper-mediated Huisgen 1,3-dipolar cycloaddition starting from easily accessible fullerene hexakis-adduct 70 (1 equiv.) bearing twelve peripheral azide functional groups in the presence of 97·2PF6 (13 equiv.), Cu(MeCN)4PF6 (0.1 equiv.), and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (0.1 equiv.) in DMF for 24 h at room temperature (Scheme 32).88 Compound 98 was thus obtained in 78% yield. Viologens (V2+) are usually easily converted to radical cations V˙+ upon a one electron reduction process that further leads to π-dimerization at high concentrations of viologen units.
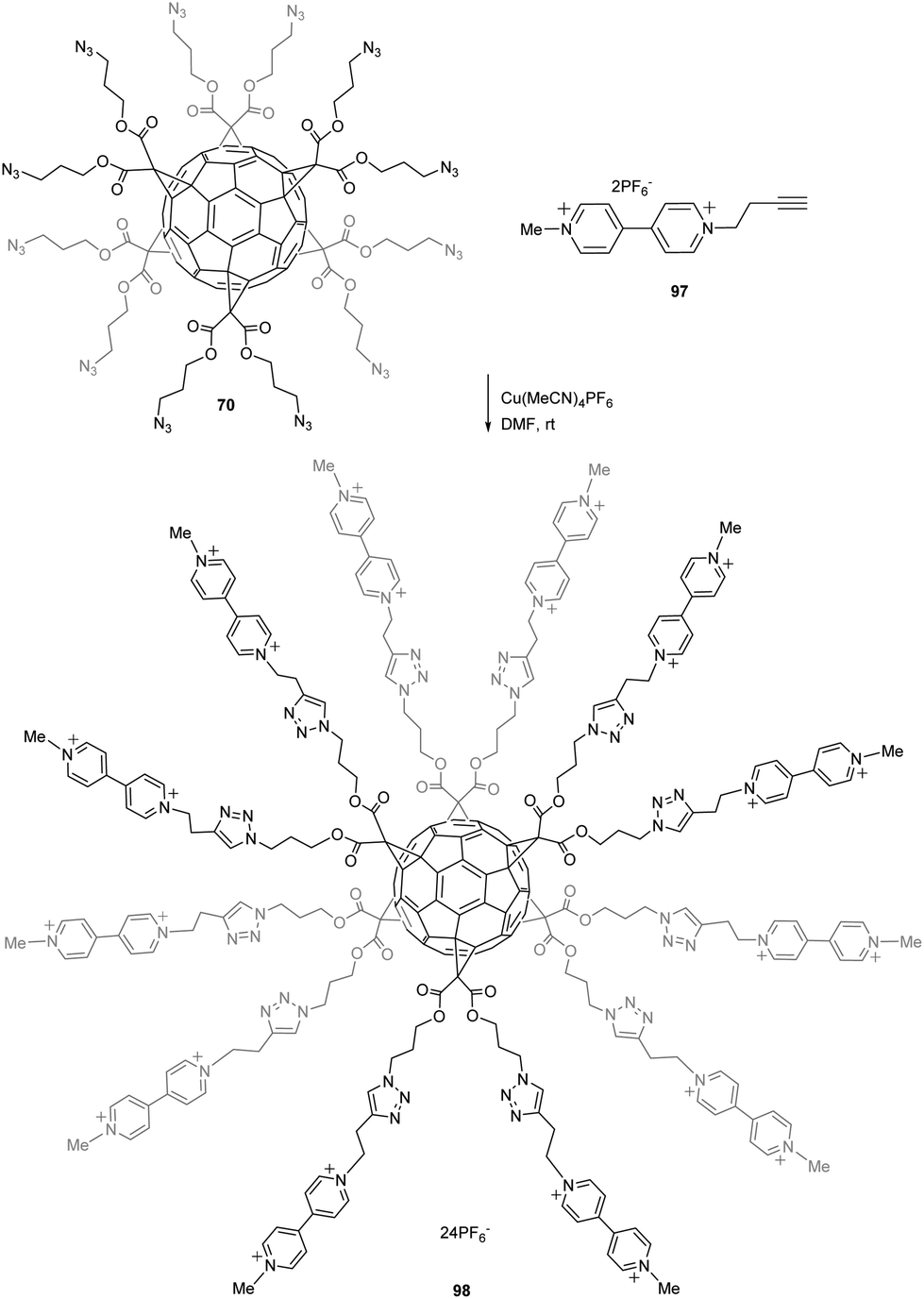 | ||
| Scheme 32 Synthetic route to hexakis-adduct C60 with viologen subunits around the core.88 | ||
The authors evaluated this feature of the viologen terminal units within fullerene 98, demonstrating that upon reduction, six intramolecular π-dimers are formed leading to fullerene molecules able to electrochemically and reversibly switch from an “open” charge-repelled state (twelve arms) to a “close” state (six arms). These multiple “open/close” molecular movements around the C60 core, triggered by electron-transfers centered on the π-dimerizable bipyridinium units, have been shown to occur in organic solvents and in aqueous media. This system opens the way towards more elaborate redox switchable structures where mechanically interlocked molecules could be incorporated around the all-carbon sphere using a similar synthetic protocol.
We have discussed the fact that the C60 core can be transformed to fullerene hexakis-adducts of C2v symmetry by sequential Bingel reactions with two different malonates. Using this approach, Brettreich et al. prepared globular amphiphiles described in Scheme 33.89 First, malonate 99 (obtained from malonyl dichloride and two equivalents of the corresponding alcohol) was reacted with C60 in a nucleophilic Bingel cyclopropanation reaction to afford the expected monoadduct 100. Next, the five remaining octahedral positions on C60 were functionalized in a DMA-templated cyclopropanation involving didodecenyl malonate to give the expected mixed hexakis-adduct 101 in 23% isolated yield. tert-Butyl groups were then removed with TFA, unmasking the terminal carboxylic acids that were then reacted with amide dendron 102 in presence DCC and HOBt to give the intermediate in 30% yield. A final treatment of the latter product with TFA gave access to fullerene amphiphile 103 bearing 18 carboxylic acid groups on the hydrophilic tail and ten dodecyl chains in the hydrophobic region. Aggregation of 103 was studied by UV spectroscopy, TEM and light scattering measurements, showing that it forms spontaneously vesicles of approximately 100–400 nm, which is similar to those obtained from common phospholipids. However, it was observed that compounds 103 tend to form unilamellar aggregates, as well as a variety of different bilayered structures.
 | ||
| Scheme 33 Synthetic route to globular amphiphiles based on hexakis-adducts of C60.89 | ||
The influence of polyaddition of malonates to C60 was also explored with the intent of forming chiral liquid-crystalline phases and appeared to be a suitable approach to generate a wide variety of mesomorphic materials.90–92
The direct introduction of six bulky malonates around the fullerene C60 through Bingel cyclopropanation reactions is rapidly slowed down by the large malonates, and results in the formation of the expected hexakis-adduct with low to negligible yield. To overcome this problem, several groups have developed methods for post-functionalization of the methanofullerene adducts. For instance, Nierengarten et al. adapted the copper-mediated Huisgen 1,3-dipolar cycloaddition reaction and applied it to the preparation of complex hexa-substituted fullerenes from a easily accessible dodecaazide derivative, as discussed before. Additionally, Pierrat et al. developed a synthetic strategy using a Mitsunobu post-functionalization of a hexa-substituted fullerene derivative bearing twelve hydroxyl groups (Scheme 34).93 The silyl-protected malonate 104 was conjugated to C60 using usual Bingel conditions to form the expected hexakis-adduct that was further deprotected to give polyol 105 in 28% over two steps. The latter was then successfully functionalized by a Mitsunobu reaction in the presence of a range of activated carboxylic acids. The reaction was optimized with a combination of diethyl azodicarboxylate (DEAD) as oxidizing azo-reagent and triphenylphosphine (TPP) as reducing agent, which gave the desired dodeca-esters in moderate to good yields (Scheme 34). Moreover, the product 106d bearing twelve aromatic iodide units was further transformed by efficient Sonogashira cross-coupling reactions to afford product 107 in 52% yield, that corresponds to 95% per iodide. Although results obtained so far suggest that this approach is limited to activated Mitsunobu substrates, this approach enables for third generation derivatization of the fullerene substrates.
 | ||
| Scheme 34 Mitsunobu and Sonogashira cross-coupling reactions to post-functionalize fullerene hexakis-adducts.93 | ||
Seifermann et al. reported on the first enantiomerically pure[6:0]hexakis[(bisoxazolinyl)methano]fullerene derivatives shown in Scheme 35, which were obtained via cyclopropanation of C60 with the corresponding C2-symmetrical enantiomerically pure bis(oxazolines) 108 and adaptation of the conditions developed by the Sun group (100-fold excess CBr4).37 In these conditions, both enantiomers of the phenyl derivatives all-S-109 and all-R-109 were obtained in 32% and 31% yield, respectively. These compounds provide an all-organic rigid scaffold bearing six orthogonally directed metal-chelation sites. Preliminary experiments indicate that the generation of six-fold metal-complexes is possible. Thus, this new material holds great promise in terms of potential applications in asymmetric catalysis as well as tectons for the construction of enantiomerically pure supramolecular 3D structures.
 | ||
| Scheme 35 Synthetic route to enantiomerically pure novel [6:0]hexakis[(bisoxazolinyl)methano]fullerenes.37 | ||
Since the discovery of the Bingel reaction, malonates and β-keto esters are starting materials of choice to functionalize the fullerene core and to access various fullerene adducts. Recently, our group reported the synthesis of hexakis-diphenylmethane adduct that could have applications in the construction of 3D metal organic frameworks owing to the coordination properties of 1,3-diketones.63 The reaction conditions involved the use of α-bromo dibenzoylmethane (6 equiv.) with the non-nucleophilic base P(tBu)3 in toluene. After column chromatography, the expected product 110 (Fig. 11) was clearly identified by 1H NMR and MALDI-TOF-MS. Unfortunately, the title compound contained some aliphatic impurities that could not be removed by any purification methods tested so far.
 | ||
| Fig. 11 Structure of hexakis-diphenylmethane adduct 110 based on DFT calculations.63 | ||
The method of choice to synthesize hexakis-methanofullerenes deals with the Bingel reaction involving malonates. This route leads to the formation of dodeca-substituted products with only limited applications as octahedral building blocks in material science. To circumvent this problem, Pierrat et al. reported on the synthesis of hexakis-substituted fullerene adducts 112a–d bearing only six peripheral functional groups (halides, pyridine and azide) from the corresponding malonate-isophthalate oxyethylene macrocycles that were anchored onto the C60 by the usual Bingel reaction (Scheme 36).94 Derivative 112a bearing six peripheral pyridine sticky sides is a useful starting point to generate octahedral metal–organic frameworks through metal–ligand recognition processes.
 | ||
| Scheme 36 Synthetic access to hexakis-substituted fullerenes bearing six sticky sides. | ||
Interestingly, compounds 112b (bearing six peripheral azides) and 112c (bearing six aromatic iodides) could be further functionalized by Click chemistry and various cross-coupling reactions (Scheme 37). The cross-coupling reactions presented are modular, tolerant to a wide range of functional groups and high-yielding. This method easily opens synthetic access to a large number of functionalized Th symmetric fullerene derivatives.
 | ||
| Scheme 37 Six-fold Huisgen, Sonogashira, Suzuki and Heck reactions on fullerene hexakis-adducts. | ||
Using this synthetic approach, Inglis et al. reported the first examples of well-defined C60-based star polymers, whose structures have been confirmed by gel permeation chromatography and 1H NMR spectroscopy.18 These C60-based star polymers were prepared by the CuAAC of alkyne-terminated polymers 113–116 with C60-N3 in the presence of copper(II)sulfate and sodium ascorbate in DMF (Scheme 38). It was shown that compound 116, bearing six peripheral poly(n-isopropylacrylamide) (PNIPAM) showed quick reversible, thermally-induced flocculation in water. This contribution has thus shown that hexakisazido fullerene derivative 112b may be efficiently used in the synthesis of well-defined, star-shaped polymers via click chemistry. The authors demonstrated that the use of a large, sterically non-demanding core such as C60 allows for a near flawless star construction even at high molecular weights. In addition, thermoresponsive star-shaped fullerene derivatives are easily accessible by this synthetic route.
 | ||
| Scheme 38 Synthetic route to star-shaped polymer-fullerene hybrids. | ||
MOFs containing C60 linkers in well-defined geometries would be expected to exhibit unique structures and properties.
Habicher et al. reported on the synthesis and X-ray crystal structure of fullerene-containing rigid dinuclear cyclophane which was obtained by self-assembly of the novel fullerene ligand with Pt(II) centres.95 Dipyridylchloromethane 118 was treated under the usual Bingel cyclopropanation method to afford the fullerene monoadduct 119 in 32% yield (Scheme 39). Any attempt to form metal complexes from fullerene 119, was unsuccessful because of the low solubility of the fullerene monoadduct.
 | ||
| Scheme 39 Synthetic route to a Pt(II)-central dinuclear cyclophane-containing two fullerene units. | ||
In order to overcome this problem, the authors prepared the yellow C2v-symmetrical fullerene hexakis-adduct 120 by DMA-templated addition of diethyl 2-bromomalonate on fullerene 119. Next, by mixing an equimolar amount of fullerene hexakis-adduct and [cis-Pt(PEt3)2(OTf)2] in CD2Cl2 at room temperature, the tetracationic cyclophane 121 was obtained quantitatively as its tetrakistriflate salt. The structure of the complex was fully characterized by means of NMR spectroscopy (1H, 13C, 19F and 31P) and by X-ray crystal structure analysis. This structure highlights the potential of metal-directed self-assembly to build complex supermolecular fullerene arrays. For instance, by replacing further diethyl malonate substituents with dipyridylmethano groups, larger assemblies such as rods or two- and three-dimensional fullerene networks were accessible.
Peng et al. reported the synthesis of a new fullerene linker and its use in the formation of a linear coordination polymer by silver complexation (Scheme 40).96 A new hexakis-fullerene adduct with two 4,5-diazafluorene groups strategically located at trans-1 positions to allow linear polymerization was selectively synthesized in very high yield. A one dimensional metal–organic coordination polymer was obtained by reaction with silver triflate. The 4,5-diazafluorene group was selected since it can efficiently react in the Bingel method and is known to be an excellent ligand for metal coordination. The synthesis of mono-unit 125 for coordination was accomplished starting with the bis-adduct of C60 with anthracene protecting groups located in the trans positions. The remaining four equatorial positions around the C60 were then decorated by symmetric addition of four diethyl malonate groups. The anthracene groups were removed by a heat-mediated retro-Diels–Alder reaction to afford tetrakis-functional fullerene 124, which was subsequently reacted with 4,5-diazafluorene to produce the hexakis-trans-1 adduct 125 in 91% yield. Light yellow-green crystals of the polymeric silver complex were obtained by slow diffusion of silver triflate in methanol into a solution of mono-unit 125 solubilized in dichloromethane followed by the addition of toluene. The structure of the crystal was characterized by X-ray crystal structure analysis. No intermolecular interactions between fullerene units were observed. This was explained by the presence of four bulky equatorial ethyl malonate groups on the C60 cage. Interestingly, diazafluorene rings of adjacent fullerenes interact clearly by π–π interactions. Furthermore, the coordination geometry of the silver centre can be considered as distorted tetrahedral. Thus, hexakis-substituted fullerene adducts with coordination sites are suitable building blocks for the generation of linear polymers through coordination with metal ions.
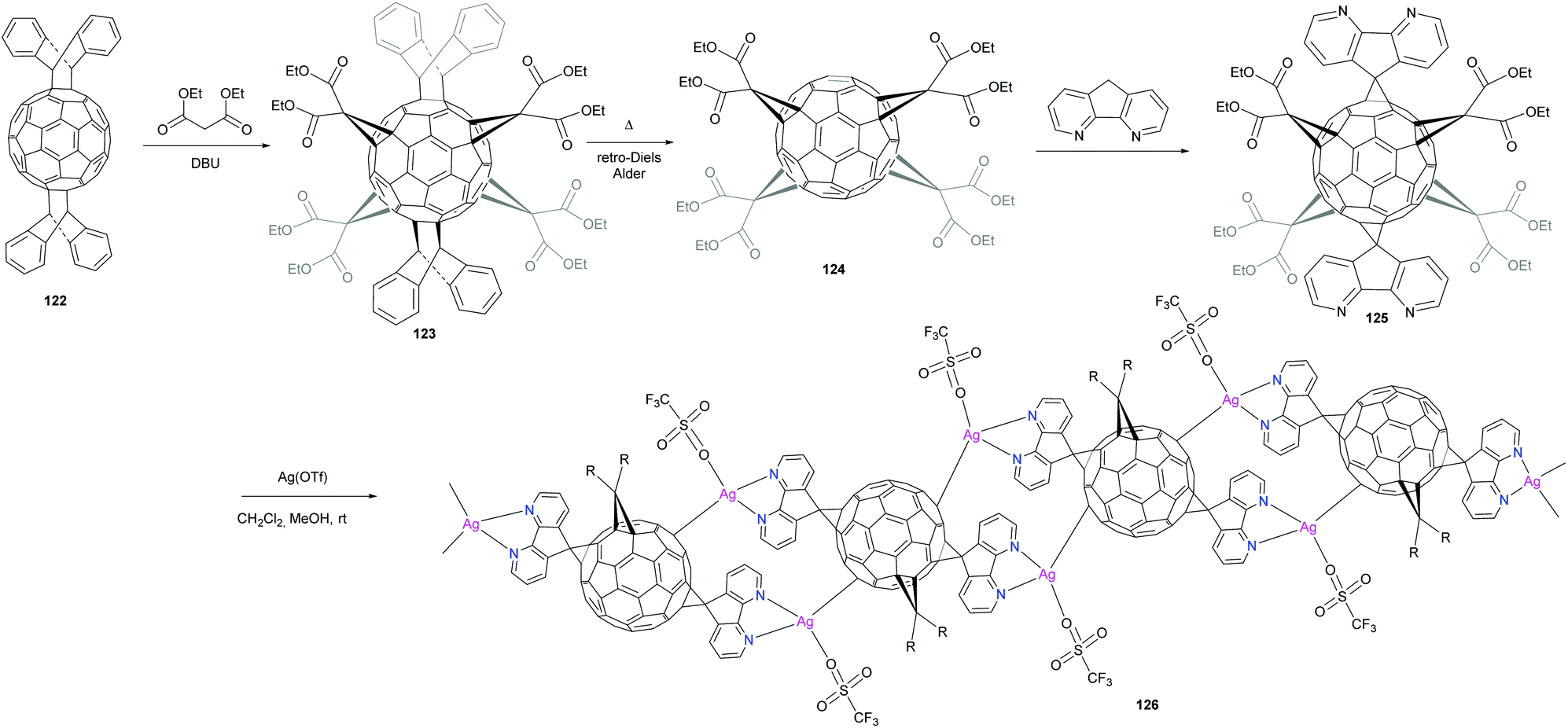 | ||
| Scheme 40 Synthetic route to a new fullerene linker and linear silver-coordination polymer. | ||
Recently, Peng et al. reported the design and synthesis of a hexakisfullerene adduct with four pyridyl groups whose nitrogen atoms reside in a rectangle.14 This adduct was employed as a linker to build the first two-dimensional (2D) fullerene-based MOF. The 2D layers packed in the crystal with interlayers composed of isolated hexakisfullerene molecules form an intricate three-dimensional (3D) structure (Scheme 41). The authors used tetrakis[di(ethoxycarbonyl)methano]-C60130 as a starting material and reacted it under photoirradiation at 365 nm with 4,4′-(4,4′-(diazomethylene)bis(4,1-phenylene))dipyridine 129 to access the hexakis-adduct 131, which displayed a D2h symmetry according to 1H NMR spectroscopy. Compound 131 was designed to have four pyridyl nitrogen atoms, to define a perfectly planar rectangle. Crystals of complex 131[(130)2·Cd(NO3)2]∞ were obtained by the reaction of 5 with Cd(NO3)2·4H2O in DMF at 100 °C, followed by careful addition of methanol to the cooled solution. Single-crystal X-ray diffraction analysis clearly showed the single layer formed upon coordination of 131 with cadmium cations, in which each pyridine group coordinates with four different cadmium ions. Furthermore, the fullerene pillars exhibit interactions with the layers surrounding them. On the basis of these results, it is anticipated that by connecting metal centres of the 2D fullerene layers with dipyridine-based pillars, 3D MOF can be built.
 | ||
| Scheme 41 Synthetic route to the first fullerene-linked 2D metal–organic framework. | ||
Hörmann et al. have reported the preparation of C2v-symmetrical pentakis-adducts of C60 as versatile building blocks for fullerene architectures that involve a mixed octahedral addition pattern.97 To efficiently synthesize such structures, the authors developed a strategy involving isoxazolinofullerenes 133 (Scheme 42) in which the isoxazoline moiety can be considered a protecting group as it can be easily removed by efficient retro-cycloaddition by refluxing in o-dichlorobenzene in the presence of an excess of a dienophile. Fullerene 133 was subjected to five-fold cyclopropanation at octahedral positions in the presence of CBr4, DBU and a range of malonates to afford the expected product 134 in 29–53% yields. The completion of the octahedral addition pattern was accomplished by means of five-fold cyclopropanation with malonate and CBr4 in the presence of DBU. Deprotection of the protected[5:1]hexakis-adducts was achieved under irradiation and in the presence of maleic anhydride in 44–79% yields. Cyclopropanation of 135 with cyclic bismalonate 136 afforded adduct 137 in 86% yield, that could be further functionalized in the presence of 135, CBr4 and DBU to give 136 in 67% yield. Thus, the authors have introduced a new protection–deprotection sequence for the selective and efficient synthesis of a variety of C2v-symmetrical fullerene pentakis-adducts with an incomplete octahedral addition pattern. Through the use of this protecting group, the hexakis-adduct synthesis that often represents the yield-limiting step, was done at the beginning of the synthesis, which reduced the quantities of malonate needed and made the purification process easier.
 | ||
| Scheme 42 Synthetic route to C2v-symmetrical pentakis-adducts of C60 to synthesize [5:1]hexakis-adducts. | ||
Moreover, the use of C2v-symmetrical fullerene pentakis-adducts 135 for the synthesis of [5:1]hexakis-adducts afforded a convergent synthesis. The isoxazoline protection–deprotection strategy was successfully applied to the synthesis of a heptakis-fullerene 139 (Fig. 12).98 The success of this synthesis relied on the mono-activation of the bis-malonate cyclo-[2]-octylmalonate 140 (Scheme 43). The authors demonstrated that the introduction of a bromide atom is not suitable for this purpose due to unexpected scrambling of the bromide atom between both malonate sides in the presence of DBU. This was not observed with a chloride atom at this position. Nevertheless, it was shown that chlorinated malonates are less reactive than the brominated ones for the substitution of C60 in the Bingel reaction. The monochlorination of 140 was performed in the presence of a stoichiometric amount of sulfuryl chloride to give the expected compound 141 in 36% isolated yield. A 25-fold excess of substituents 141 was reacted with C60 by templating with 9,10-dimethylanthracene to get the Th-symmetric hexakis-adduct with DBU as a base in toluene as solvent. After purification by silica gel column chromatography, the expected hexakis-adduct 142 was isolated in 3.3% yield. In the final step, pentakis-substituted fullerene 143 (7 equiv.) prepared as described previously was conjugated to hexakis-adduct 142 in toluene in the presence of CBr4 and tert-butylimino-tri(pyrrolidino)phosphorane as a base.97 Owing to solubility problems, the reaction was stirred for one month at room temperature, and the expected heptafullerene was obtained after purification in 43% yield. Authors have thus developed an efficient approach for the sequential fullerenylation of bis-malonates with different fullerene units. This method for the sequential and orthogonal oligofunctionalization of oligomalonates bears many possibilities for the design of highly functional oligofullerene architectures with tunable properties.
 | ||
| Fig. 12 Structure of the heptafullerene 139. | ||
 | ||
| Scheme 43 Synthetic route to oligocluster heptafullerene. | ||
2.6. Heptakis-substituted fullerenes
There are only a few reports dealing with the synthesis of hepta-substituted fullerenes. Most of them were synthesized by diazomethane addition and characterized by mass-spectroscopy.99–104 As there no structural details corroborated by X-ray crystallography are known, these systems were not further discussed.2.7. Octakis-substituted fullerenes
Octakis-substituted fullerenes are quite rare and were only produced by dipolar cycloaddition reactions.99 As for these no structural details are known, structures are not shown herein.2.8. Higher substituted fullerenes
Highly reactive species also lead to higher-substituted fullerenes. Some of them, such as octakis(trifluoromethyl)octakishydrido fullerene were characterized by X-ray crystallography.105,1063. Conclusions
It is remarkable that different C60-derivatives can be obtained by controlling the accessible extent of functionalization of C60. Most functionalized fullerenes described in this review were obtained in good to excellent yields and provide a number of useful and unique fullerene-based materials such as indene-C60 bis-adduct and silylmethyl[60]fullerene for high-performance solar cells; self-assembly films; penta-haptofullerene metal complexes; liquid crystals, glycosidase inhibition with fullerene iminosugar balls; bacterial anti-adhesives, gene delivery with polycationic fullerene; artificial light-harvesting arrays; star shaped polymer-fullerene hybrids; fullerene-linked metal–organic frameworks. Some derivatives hold the promise of a number of applications; however, the application of these derivatives has not been deeply studied.Given the number of developed methods to synthesize flexible structures, we anticipate that the key challenge is the construction of defined nanostructures according to specific needs in functional fullerene materials and biologically useful molecules, and so on. And it is anticipated this will be extensively investigated in the near future.
Acknowledgements
We appreciate the stimulating atmosphere of the KIT fullerene group (mentioned in the publications) and also the reviewers for very useful comments. Our own work has been supported by the DFG (CFN).Notes and references
- C. Thilgen and F. Diederich, Chem. Rev., 2006, 106, 5049–5135 CrossRef CAS PubMed.
- F. Giacalone and N. Martin, Chem. Rev., 2006, 106, 5136–5190 CrossRef CAS PubMed.
- O. Vostrowsky and A. Hirsch, Chem. Rev., 2006, 106, 5191–5207 CrossRef CAS PubMed.
- Y. Matsuo and E. Nakamura, Chem. Rev., 2008, 108, 3016–3028 CrossRef CAS PubMed.
- P. A. Troshin, A. S. Peregudov, S. I. Troyanov and R. N. Lyubovskaya, Russ. Chem. Bull., 2008, 57, 887–912 CrossRef CAS PubMed.
- U. Hahn, F. Vögtle and J. F. Nierengarten, Polymers, 2012, 4, 501–538 CrossRef PubMed.
- M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC.
- S. S. Babu, H. Mohwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC.
- P. Compain, C. Decroocq, J. Iehl, M. Holler, D. Hazelard, T. M. Barragan, C. O. Mellet and J. F. Nierengarten, Angew. Chem., Int. Ed., 2010, 49, 5753–5756 CrossRef CAS PubMed.
- M. Durka, K. Buffet, J. Iehl, M. Holler, J. F. Nierengarten, J. Taganna, J. Bouckaert and S. P. Vincent, Chem. Commun., 2011, 47, 1321–1323 RSC.
- D. Sigwalt, M. Holler, J. Iehl, J. F. Nierengarten, M. Nothisen, E. Morin and J. S. Remy, Chem. Commun., 2011, 47, 4640–4642 RSC.
- S. Cecioni, V. Oerthel, J. Iehl, M. Holler, D. Goyard, J. P. Praly, A. Imberty, J. F. Nierengarten and S. Vidal, Chem. – Eur. J., 2011, 17, 3252–3261 CrossRef CAS PubMed.
- Y. J. He, H. Y. Chen, J. H. Hou and Y. F. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- P. Peng, F. F. Li, V. S. P. K. Neti, A. J. Metta-Magana and L. Echegoyen, Angew. Chem., Int. Ed., 2014, 53, 160–163 CrossRef CAS PubMed.
- T. M. Figueira-Duarte, J. Clifford, V. Amendola, A. Gegout, J. Olivier, F. Cardinal, M. Meneghetti, N. Armaroli and J. F. Nierengarten, Chem. Commun., 2006, 2054–2056 RSC.
- V. Garg, G. Kodis, M. Chachisvilis, M. Hambourger, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2011, 133, 2944–2954 CrossRef CAS PubMed.
- M. Sawamura, K. Kawai, Y. Matsuo, K. Kanie, T. Kato and E. Nakamura, Nature, 2002, 419, 702–705 CrossRef CAS PubMed.
- A. J. Inglis, P. Pierrat, T. Muller, S. Bräse and C. Barner-Kowollik, Soft Matter, 2010, 6, 82–84 RSC.
- M. G. Nava, S. Setayesh, A. Rameau, P. Masson and J. F. Nierengarten, New J. Chem., 2002, 26, 1584–589 RSC.
- C. Bingel, Chem. Ber./Recl., 1993, 126, 1957–1959 CrossRef CAS.
- M. Prato, T. Suzuki, H. Foroudian, Q. Li, K. Khemani, F. Wudl, J. Leonetti, R. D. Little, T. White, B. Rickborn, S. Yamago and E. Nakamura, J. Am. Chem. Soc., 1993, 115, 1594–1595 CrossRef CAS.
- M. Prato, Q. C. Li, F. Wudl and V. Lucchini, J. Am. Chem. Soc., 1993, 115, 1148–1150 CrossRef CAS.
- T. Tago, T. Minowa, Y. Okada and J. Nishimura, Tetrahedron Lett., 1993, 34, 8461–8464 CrossRef CAS.
- H. H. Wang, J. A. Schlueter, A. C. Cooper, J. L. Smart, M. E. Whitten, U. Geiser, K. D. Carlson, J. M. Williams, U. Welp, J. D. Dudek and M. A. Caleca, J. Phys. Chem. Solids, 1993, 54, 1655–1666 CrossRef CAS.
- P. Belik, A. Gugel, A. Kraus, J. Spickermann, V. Enkelmann, G. Frank and K. Mullen, Adv. Mater., 1993, 5, 854–856 CrossRef CAS.
- V. M. Rotello, J. B. Howard, T. Yadav, M. M. Conn, E. Viani, L. M. Giovane and A. L. Lafleur, Tetrahedron Lett., 1993, 34, 1561–1562 CrossRef CAS.
- F. Diederich, U. Jonas, V. Gramlich, A. Herrmann, H. Ringsdorf and C. Thilgen, Helv. Chim. Acta, 1993, 76, 2445–2453 CrossRef CAS.
- B. Krautler and M. Puchberger, Helv. Chim. Acta, 1993, 76, 1626–1631 CrossRef.
- D. Babic, D. J. Klein and C. H. Sah, Chem. Phys. Lett., 1993, 211, 235–241 CrossRef CAS.
- F. Cardullo, P. Seiler, L. Isaacs, J. F. Nierengarten, R. F. Haldimann, F. Diederich, T. MordasiniDenti, W. Thiel, C. Boudon, J. P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 1997, 80, 343–371 CrossRef CAS.
- T. Kusukawa and W. Ando, Angew. Chem., Int. Ed. Engl., 1996, 35, 1315–1317 CrossRef CAS.
- F. Cardullo, P. Seiler, L. Isaacs, J. F. Nierengarten, R. F. Haldimann, F. Diederich, T. Mordasini-Denti, W. Thiel, C. Boudon, J. P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 1997, 80, 343–371 CrossRef CAS.
- F. Hormann, M. Brettreich, W. Donaubauer, F. Hampel and A. Hirsch, Chem. – Eur. J., 2013, 19, 2814–2825 CrossRef PubMed.
- M. Sawamura, H. Iikura and E. Nakamura, J. Am. Chem. Soc., 1996, 118, 12850–12851 CrossRef CAS.
- C. R. Woods, J. P. Bourgeois, P. Seiler and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 3813–3816 CrossRef CAS.
- I. Lamparth, C. Maichle-Mössmer and A. Hirsch, Angew. Chem., Int. Ed. Engl., 1995, 34, 1607–1609 CrossRef CAS.
- S. M. Seifermann, C. Rethore, T. Muller and S. Bräse, Sci. Rep., 2013, 3 Search PubMed , 2817.
- P. Seiler, L. Isaacs and F. Diederich, Helv. Chim. Acta, 1996, 79, 1047–1058 CrossRef CAS.
- Y. Z. Tan, Z. J. Liao, Z. Z. Qian, R. T. Chen, X. Wu, H. Liang, X. Han, F. Zhu, S. J. Zhou, Z. P. Zheng, X. Lu, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Mater., 2008, 7, 790–794 CrossRef CAS PubMed.
- T. M. Figueira-Duarte, V. Lloveras, J. Vidal-Gancedo, B. Delavaux-Nicot, C. Duhayon, J. Veciana, C. Rovira and J. F. Nierengarten, Eur. J. Org. Chem., 2009, 5779–5787 CrossRef CAS.
- D. Sigwalt, M. Holler and J. F. Nierengarten, Tetrahedron Lett., 2013, 54, 3160–3163 CrossRef CAS PubMed.
- J.-F. Nierengarten, V. Gramlich, F. Cardullo and F. Diederich, Angew. Chem., Int. Ed. Engl., 1996, 35, 2101–2103 CrossRef CAS.
- Y. Matsuo, A. Iwashita, Y. Abe, C. Z. Li, K. Matsuo, M. Hashiguchi and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15429–15436 CrossRef CAS PubMed.
- Y. Matsuo, Y. Sato, T. Niinomi, I. Soga, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 16048–16050 CrossRef CAS PubMed.
- F. Cardinali, J. L. Gallani, S. Schergna, M. Maggini and J. F. Nierengarten, Tetrahedron Lett., 2005, 46, 2969–2972 CrossRef CAS PubMed.
- J. Nierengarten, in Dendrimers V, ed. C. Schalley and F. Vögtle, Springer, Berlin, Heidelberg, 2003, vol. 228, ch. 4, pp. 87–110 Search PubMed.
- A. Hirsch and O. Vostrowsky, in Dendrimers IV, ed. F. Vögtle and C. Schalley, Springer, Berlin, Heidelberg, 2001, vol. 217, ch. 2, pp. 51–93 Search PubMed.
- J.-F. Nierengarten, Chem. – Eur. J., 2000, 6, 3667–3670 CrossRef CAS.
- R. van de Coevering, R. Kreiter, F. Cardinali, G. van Koten, J. F. Nierengarten and R. J. M. K. Gebbink, Tetrahedron Lett., 2005, 46, 3353–3356 CrossRef CAS PubMed.
- U. Hahn, E. Maisonhaute, C. Amatore and J. F. Nierengarten, Angew. Chem., Int. Ed., 2007, 46, 951–954 CrossRef CAS PubMed.
- A. Hirsch, I. Lamparth and H. R. Karfunkel, Angew. Chem., Int. Ed. Engl., 1994, 33, 437–438 CrossRef.
- D. Sigwalt, F. Schillinger, S. Guerra, M. Holler, M. Berville and J. F. Nierengarten, Tetrahedron Lett., 2013, 54, 4241–4244 CrossRef CAS PubMed.
- F. Beuerle and A. Hirsch, Chem. – Eur. J., 2009, 15, 7447–7455 CrossRef CAS PubMed.
- S. Guerra, F. Schillinger, D. Sigwalt, M. Holler and J. F. Nierengarten, Chem. Commun., 2013, 49, 4752–4754 RSC.
- L. Isaacs, F. Diederich and R. F. Haldimann, Helv. Chim. Acta, 1997, 80, 317–342 CrossRef CAS.
- C. Thilgen, S. Sergeyev and F. Diederich, Top. Curr. Chem., 2004, 248, 1–61 CrossRef.
- J. F. Nierengarten, T. Habicher, R. Kessinger, F. Cardullo, F. Diederich, V. Gramlich, J. P. Gisselbrecht, C. Boudon and M. Gross, Helv. Chim. Acta, 1997, 80, 2238–2276 CrossRef CAS.
- F. Djojo and A. Hirsch, Chem. – Eur. J., 1998, 4, 344–356 CrossRef CAS.
- F. Djojo, A. Hirsch and S. Grimme, Eur. J. Org. Chem., 1999, 3027–3039 CrossRef CAS.
- I. Lamparth and A. Hirsch, J. Chem. Soc., Chem. Commun., 1994, 1727–1728 RSC.
- T. B. Cao, F. Wei, Y. L. Yang, L. Huang, X. S. Zhao and W. X. Cao, Langmuir, 2002, 18, 5186–5189 CrossRef CAS.
- H. Li, S. A. Haque, A. Kitaygorodskiy, M. J. Meziani, M. Torres-Castillo and Y.-P. Sun, Org. Lett., 2006, 8, 5641–5643 CrossRef CAS PubMed.
- A. Giovannitti, S. M. Seifermann, A. Bihlmeier, T. Muller, F. Topic, K. Rissanen, M. Nieger, W. Klopper and S. Bräse, Eur. J. Org. Chem., 2013, 2013, 7907–7913 CrossRef CAS.
- Y. Murata, M. Shiro and K. Komatsu, J. Am. Chem. Soc., 1997, 119, 8117–8118 CrossRef CAS.
- Y. Matsuo, A. Muramatsu, R. Hamasaki, N. Mizoshita, T. Kato and E. Nakamura, J. Am. Chem. Soc., 2004, 126, 432–433 CrossRef CAS PubMed.
- M. Sawamura, Y. Kuninobu, M. Toganoh, Y. Matsuo, M. Yamanaka and E. Nakamura, J. Am. Chem. Soc., 2002, 124, 9354–9355 CrossRef CAS PubMed.
- Y. Matsuo, H. Isobe, T. Tanaka, Y. Murata, M. Murata, K. Komatsu and E. Nakamura, J. Am. Chem. Soc., 2005, 127, 17148–17149 CrossRef CAS PubMed.
- F. Wang, Z. Xiao, Z. Yao, Z. Jia, S. Huang, L. Gan, J. Zhou, G. Yuan and S. Zhang, J. Org. Chem., 2006, 71, 4374–4382 CrossRef CAS PubMed.
- J. Shiea, J.-P. Huang, C.-F. Teng, J. Jeng, L. Y. Wang and L. Y. Chiang, Anal. Chem., 2003, 75, 3587–3595 CrossRef CAS.
- S. C. Chueh, M. K. Lai, M. S. Lee, L. Y. Chiang, T. I. Ho and S. C. Chen, Transplant. Proc., 1999, 31, 1976–1977 CrossRef CAS.
- H. Li, Y. Li, J. Zhai, G. Cui, H. Liu, S. Xiao, Y. Liu, F. Lu, L. Jiang and D. Zhu, Chem. – Eur. J., 2003, 9, 6031–6038 CrossRef CAS PubMed.
- H. Li, Y. Zhou, Y. Li, Y. Song, H. Fang, S. Xiao, H. Liu, H. Gan, T. Jiu and D. Zhu, Chem. Phys. Lett., 2004, 383, 230–234 CrossRef CAS PubMed.
- P. J. Fagan, J. C. Calabrese and B. Malone, J. Am. Chem. Soc., 1991, 113, 9408–9409 CrossRef CAS.
- B. Kräutler and J. Maynollo, Angew. Chem., Int. Ed. Engl., 1995, 34, 87–88 CrossRef.
- G. Schick, M. Levitus, L. Kvetko, B. A. Johnson, I. Lamparth, R. Lunkwitz, B. Ma, S. I. Khan, M. A. Garcia-Garibay and Y. Rubin, J. Am. Chem. Soc., 1999, 121, 3246–3247 CrossRef.
- A. Hirsch, I. Lamparth, T. Groesser and H. R. Karfunkel, J. Am. Chem. Soc., 1994, 116, 9385–9386 CrossRef CAS.
- L. Isaacs, R. F. Haldimann and F. Diederich, Angew. Chem., Int. Ed. Engl., 1994, 33, 2339–2342 CrossRef.
- X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997, 1595–1596 RSC.
- H. Li, A. Kitaygorodskiy, R. A. Carino and Y.-P. Sun, Org. Lett., 2005, 7, 859–861 CrossRef CAS PubMed.
- J. Iehl, R. P. de Freitas, B. Delavaux-Nicot and J. F. Nierengarten, Chem. Commun., 2008, 2450–2452 RSC.
- J. Iehl and J. F. Nierengarten, Chem. – Eur. J., 2009, 15, 7306–7309 CrossRef CAS PubMed.
- J. F. Nierengarten, J. Iehl, V. Oerthel, M. Holler, B. M. Illescas, A. Munoz, N. Martin, J. Rojo, M. Sanchez-Navarro, S. Cecioni, S. Vidal, K. Buffet, M. Durka and S. P. Vincent, Chem. Commun., 2010, 46, 3860–3862 RSC.
- I. Nierengarten, K. Buffet, M. Holler, S. P. Vincent and J.-F. Nierengarten, Tetrahedron Lett., 2013, 54, 2398–2402 CrossRef CAS PubMed.
- R. Risquez-Cuadro, J. M. Garcia Fernandez, J.-F. Nierengarten and C. Ortiz Mellet, Chem. – Eur. J., 2013, 19, 16791–16803 CrossRef CAS PubMed.
- I. Nierengarten and J.-F. Nierengarten, Chem. – Asian J., 2014, 9, 1436–1444 CrossRef CAS PubMed.
- P. Fortgang, E. Maisonhaute, C. Amatore, B. Delavaux-Nicot, J. Iehl and J. F. Nierengarten, Angew. Chem., Int. Ed., 2011, 50, 2364–2367 CrossRef CAS PubMed.
- J. Iehl, J. F. Nierengarten, A. Harriman, T. Bura and R. Ziessel, J. Am. Chem. Soc., 2012, 134, 988–998 CrossRef CAS PubMed.
- J. Iehl, M. Frasconi, H. P. J. de Rouville, N. Renaud, S. M. Dyar, N. L. Strutt, R. Carmieli, M. R. Wasielewski, M. A. Ratner, J. F. Nierengarten and J. F. Stoddart, Chem. Sci., 2013, 4, 1462–1469 RSC.
- M. Brettreich, S. Burghardt, C. Bottcher, T. Bayerl, S. Bayerl and A. Hirsch, Angew. Chem., Int. Ed., 2000, 39, 1845–1848 CrossRef CAS.
- T. Chuard, R. Deschenaux, A. Hirsch and H. Schonberger, Chem. Commun., 1999, 2103–2104 RSC.
- S. Campidelli, T. Brandmueller, A. Hirsch, I. M. Saez, J. W. Goodby and R. Deschenaux, Chem. Commun., 2006, 4282–4284 RSC.
- S. Gottis, C. Kopp, E. Allard and R. Deschenaux, Helv. Chim. Acta, 2007, 90, 957–962 CrossRef CAS.
- P. Pierrat, C. Rethore, T. Muller and S. Bräse, Chem. – Eur. J., 2009, 15, 11458–11460 CrossRef CAS PubMed.
- P. Pierrat, S. Vanderheiden, T. Muller and S. Bräse, Chem. Commun., 2009, 1748–1750 RSC.
- T. Habicher, J. F. Nierengarten, V. Gramlich and F. Diederich, Angew. Chem., Int. Ed., 1998, 37, 1916–1919 CrossRef CAS.
- P. Peng, F. F. Li, F. L. Bowles, V. S. P. K. Neti, A. J. Metta-Magana, M. M. Olmstead, A. L. Balch and L. Echegoyen, Chem. Commun., 2013, 49, 3209–3211 RSC.
- F. Hörmann, W. Donaubauer, F. Hampel and A. Hirsch, Chem. – Eur. J., 2012, 18, 3329–3337 CrossRef PubMed.
- L. K. Wasserthal, A. Kratzer and A. Hirsch, Eur. J. Org. Chem., 2013, 2355–2361 CrossRef CAS.
- R. F. Haldimann, F. G. Klarner and F. Diederich, Chem. Commun., 1997, 237–238 RSC.
- A. Herrmann, M. W. Ruttimann, T. Gibtner, C. Thilgen, F. Diederich, T. Mordasini and W. Thiel, Helv. Chim. Acta, 1999, 82, 261–289 CrossRef CAS.
- J. P. Bourgeois, C. R. Woods, F. Cardullo, T. Habicher, J. F. Nierengarten, R. Gehrig and F. Diederich, Helv. Chim. Acta, 2001, 84, 1207–1226 CrossRef CAS.
- R. F. Haldimann, C. Fokas and F. Diederich, Helv. Chim. Acta, 2001, 84, 1635–1660 CrossRef CAS.
- M. J. van Eis, P. Seiler, L. A. Muslinkina, M. Badertscher, E. Pretsch, F. Diederich, R. J. Alvarado, L. Echegoyen and I. P. Nunez, Helv. Chim. Acta, 2002, 85, 2009–2055 CrossRef CAS.
- Y. Chen, Y. L. Wang, A. Rassat, P. Sinay, Y. Zhao and Y. M. Zhang, Tetrahedron, 2006, 62, 2045–2049 CrossRef CAS PubMed.
- I. E. Kareev, N. B. Shustova, B. S. Newell, S. M. Miller, O. P. Anderson, S. H. Strauss and O. V. Boltalina, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, O3154–O3156 CAS.
- N. B. Shustova, D. V. Peryshkov, I. E. Kareev, O. V. Boltalina and S. H. Strauss, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, O3398–U2441 CAS.
| This journal is © The Royal Society of Chemistry 2015 |