
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cross-catalytic peptide nucleic acid (PNA) replication based on templated ligation†
Abhishek
Singhal‡
and
Peter E.
Nielsen
*
Department of Cellular and Molecular Medicine, Faculty of Health Sciences, University of Copenhagen, The Panum Institute, Blegdamsvej 3c, DK-2200 Copenhagen N, Denmark. E-mail: ptrn@sund.ku.dk; Fax: +45 35396042; Tel: +45 35327762
First published on 17th July 2014
Abstract
We report the first PNA self-replicating system based on template directed cross-catalytic ligation, a process analogous to biological replication. Using two template PNAs and four pentameric precursor PNAs, all four possible carbodiimide assisted amide ligation products were detected and identified by HPLC and MALDI-TOF analysis. We conclude that the two template complementary reaction products are generated via cross-catalysis, while the other two self-complementary (and in principle auto-catalytic) products are formed via intra-complex coupling between the two sets of complementary PNA precursors. Cross-catalytic product formation followed product inhibited kinetics, but approximately two replication rounds were observed. Analogous but less efficient replication was found for a similar tetrameric system. These results demonstrate that simpler nucleobase replication systems than natural oligonucleotides are feasible, thereby strengthening the foundation for the discussion of a possible role for PNA (like) genetic material in the prebiotic evolution of life and lay the ground for further studies into evolution of such potentially prebiotic systems.
Introduction
Replication of self is the key feature of all (cellular) life. Although an RNA world is generally accepted as a likely predecessor of contemporary “central dogma life”, there are also valid arguments for a possible pre-RNA “proto-life”, and it is still unclear how biological replicating systems developed and evolved during an ancestral pre-RNA world. In principle, self replicating and template directed self assembling systems represent potential models for exploring the molecular basis of evolution of synthetic chemical systems from simple pre-biotic precursors. A wide range of self-replicating molecules including nucleotide-based oligomers1–6 peptides scaffolds7–11 and small molecules without natural homologues12–16 have previously been described. The pseudopeptide nucleic acid mimic PNA (peptide nucleic acid) has been proposed as a (model for a) robust prebiotic evolutionary predecessor of RNA17–20 being capable of chemical sequence information transfer from one PNA oligomer to another (a replicative process) as well as from a PNA oligomer to an RNA oligomer (a PNA to RNA transition). It has been demonstrated that a PNA G-oligomer can be synthesized on a PNA homocytosine decameric template using PNA G-dimers as substrates and water-soluble carbodiimide (EDC) as condensing/activating agent in an imidazole buffer18 (it is necessary to use G-dimers rather than G-monomers as precursors because of the ease by which the monomer cyclizes to the piperazinone). Longer PNA oligomers may also be assembled by DNA directed PNA–PNA ligation21,22 but a PNA replicator has not been described. In the evaluation and discussion of the possible involvement of non sugar-phosphate nucleobase oligomers in the prebiotic emergence of life and also in terms of the emergence of “self-replication” processes in general, we have designed and characterized a PNA assisted and directed cross-catalytic self-replicating system, capable of performing PNA sequence (hybridization) directed cross-catalytic ligation reactions. Characterization of the properties of such model systems is important for understanding the fundamental mechanistic behavior of PNA replication systems and for the discussion of any role such systems may (or may not) have played during prebiotic evolution. Also such systems may eventually provide essential components for future “artificial life” approaches, and may be used as models for studying in vitro evolution of chemical replicator systems.The underlying principle of biological replication is a cross-catalytic reaction in which one nucleic acid strand acts as a catalyst for the formation of the other strand and vice versa. A minimal implementation of such a cross-catalytic self-replicating (oligonucleobase) system can be represented by simple template ligation reactions consisting of two sequence complementary templates and four precursors which in pairs are complementary to each of the templates. Thus two interconnected template directed ligation reactions are required, where the product of the one ligation acts as the template for the other (see also Fig. 1A and 1E). This type of simple replication system has previously been realized using hexameric DNA templates and trimer ligation precursors3,6 and we now report an analogous and longer, non-nucleic acid PNA system.
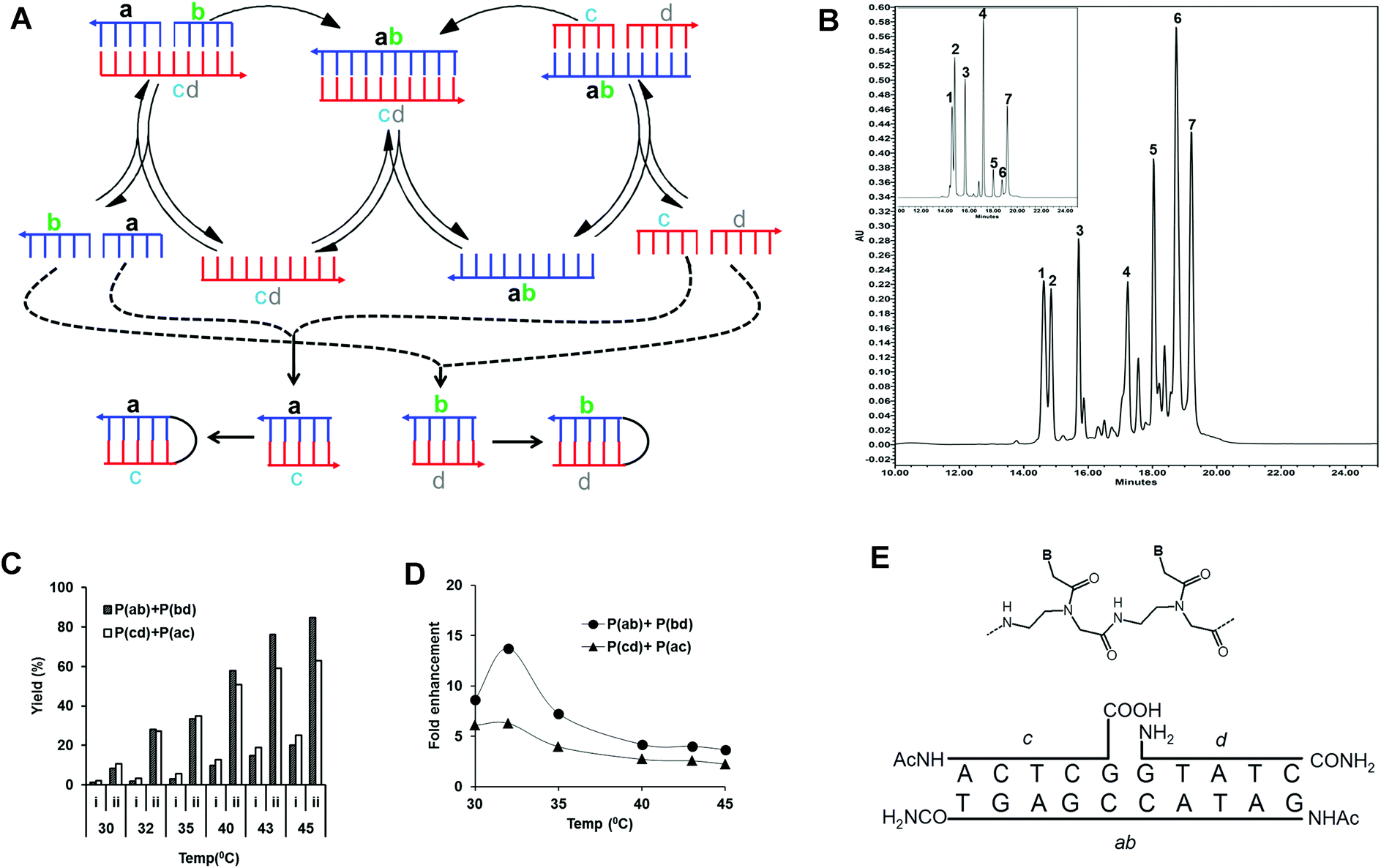 | ||
| Fig. 1 (A) Scheme of the pentameric PNA, cross-catalytic self-replication system. In this scheme, two parallel pathways exist. As a result of cross-catalytic ligation reactions, one template strand catalyzes the formation of the other template strand and vice versa. ab and cd denote templates, and a, b, c, and d denote PNA precursors complementary to the templates. As a side reaction to cross-catalytic formation of the complementary products ab and cd, the precursors may also generate the self-complementary products ac and bd which in principle can engage in autocatalysis reactions. (B) HPLC analysis of the reaction mixture. Reactions were carried out with an equimolar mixture of all four pentameric PNA precursors (a, b, c and d; concentration 100 μM) in the presence of chemically synthesized complementary decameric templates (Tab and Tcd; concentration 10 μM) (see Table 1) and 150 mM EDC in 50 mM imidazole buffer (pH 7) at 45 °C for 1 h. The reactions were followed by HPLC analysis and all indicated peaks were assigned by retention time (compared to authentic products) and MALDI-TOF mass spectrometry analysis. Peaks 1–4 correspond to pentameric PNA precursors. Peak 7 corresponds to the internal standard. Peak 5 and 6 are each heterogeneous mixtures of two amide ligation products: Peak 5 consists of products Pcd (m/z = 2743.4) and Pac (m/z = 2768.9) while peak 6 contains Pab (m/z = 2937.2) and Pbd (m/z = 2911.4). Insert shows a control reaction without EDC. (C) Effect of temperature on the yield (%) and the relative fold enhancement of formed products (Pab, Pcd, Pac and Pbd) in the pentameric PNA, cross-catalytic self-replicating reaction system. Reactions were carried out with an equimolar mixture of all four pentameric PNA precursors (a, b, c and d; concentration 100 μM) in the absence or presence of chemically synthesized complementary templates (Tab and Tcd; concentration 10 μM) in 50 mM imidazole buffer (pH 7), 150 mM EDC at different temperatures (30–45 °C) for 1 h. Reaction mixtures were analyzed by HPLC and the product yields (%) were calculated related to the internal standard. Due to co-migration by HPLC the sum of the Pab, Pbd and Pcd, Pac are reported (see also (B)). Columns “i” Correspond to non-templated reactions, while columns “ii” correspond to templated reactions after subtraction of initial template amounts. (D) Relative fold enhancement of formed products by addition of initial templates calculated from the data set of (C). Tm values of complementary PNA precursors were shown in Table S2.† (E) Chemical structure of the PNA units (B signifies the nucleobase) and schematic drawing of the ternary complex leading to PNA ab catalyzed ligation of PNAs c and d. | ||
Results
Four PNA precursors and two templates were designed in accordance with a cross catalytic replication scheme (Fig. 1A, Tables 1 and S1†). In this scheme ab and cd denote templates, and a, b, c, and d denote PNA precursors complementary to the templates. In order to favor cross-catalysis and avoid side products, the amino-end of two carboxyl PNA precursors b and c were N-terminally acetylated, while the carboxy-end of the two amine PNA precursors a and d was C-terminally amidated (Fig. 1E).| PNA no. | PNA sequence | Bases | Designation |
|---|---|---|---|
| PNA precursors | |||
| 3603 | Ac-eg1-GATAC-OH | 5 | b |
| 3605 | H-CGAGT-NH2 | 5 | a |
| 3791 | H-GTATC-NH2 | 5 | d |
| 3792 | Ac-ACTCG-OH | 5 | c |
| 3947 | H-CGAG-NH2 | 4 | k |
| 3948 | Ac-eg1-ATAC-OH | 4 | l |
| 3950 | H-GTAT-NH2 | 4 | n |
| 3951 | Ac-CTCG-OH | 4 | m |
| PNA templates | |||
| 3737 | Ac-ACTCGGTATC-NH2 | 10 | cd |
| 3111 | Ac-eg1-GATACCGAGT-NH2 | 10 | ab |
| 3956 | Ac-ACTCGCGAGT-NH2 | 10 | ac |
| 3957 | Ac-eg1-GATACGTATC-NH2 | 10 | bd |
| 3946 | Ac-CTCGGTAT-NH2 | 8 | mn |
| 3949 | Ac-eg1-ATACCGAG-NH2 | 8 | kl |
| 4334 | Ac-CTCGCGAG-NH2 | 8 | km |
| 4333 | Ac-eg1-ATACGTAT-NH2 | 8 | ln |
In the reaction precursors a, b, c, and d hybridize to the templates ab and cd forming the ter-molecular complexes a·b·cd and c·d·ab, respectively (Fig. 1E). In these complexes the reactive ends of the respective PNA precursors are set up for ligation by amide bond formation via EDC activation. Furthermore, the product of the one ligation reaction can serve as the template for the other, and vice versa, thereby enabling cross-catalysis. As a side reaction to cross-catalytic formation of the complementary products ab and cd, the precursors may also generate the self-complementary products ac and bd, which in principle comprise two independent autocatalytic systems.
In order to identify reaction conditions allowing for self-replication, an equimolar mixture of all four pentameric PNA precursors (a, b, c and d) both in the absence or presence (10 fold excess of precursors) of chemically synthesized cross-catalytic decameric templates (Tab and Tcd) were incubated for 1 h in the presence of EDC as the condensing agent at different temperatures (30–45 °C) and analyzed by HPLC. All four precursor PNAs, internal standard and formed reaction products were separated by HPLC and identified on the basis of retention times (compared to authentic products), and additionally, individual peaks were isolated and characterized by MALDI-TOF mass spectrometry as exemplified in Fig. 1B. However, MALDI mass spectrometry analysis showed that the ligation products each consisted of a mixture of a cross-catalytic and an autocatalytic product (Pcd and Pac for peak 5 and Pab and Pbd for peak 6, respectively) showing that both types of products were formed in the reaction.
The ligation reaction is clearly catalyzed by the initial presence of cross-catalytic ab and cd templates yielding several fold more ligation product (Fig. 1C), and the yields also increase with increasing temperature, showing an optimum in terms of template yield enhancement around 32 °C (Fig. 1D). This optimum most likely reflects the relative stability of the ternary pre-ligation complex versus that of the product duplex (see Discussion).
In order to resolve the problem associated with HPLC overlap of the cross-catalytic and auto-catalytic products, and also to simplify the analysis, we turned to a method based on MALDI-TOF mass spectrometry as exemplified in Fig. 2H. This allowed separation of all reaction products, and using an internal standard combined with calibration using authentic compounds, quantitative analysis is feasible.23 Applying this analysis we measured the time course of the reaction as a function of the amount of initial templates added (Fig. 2A–E). These data show that while the rate of formation of the cross-catalytic products Pab and Pcd increase with increasing template amount (and tend to exhibit saturation-like kinetics) (Fig. 2A,B), the formation of the auto-catalytic products Pac and Pbd decrease with increasing concentration of the cross catalytic templates (and seem to follow linear kinetics (at least up to 20% yield)) (Fig. 2C,D). Furthermore, the rate shows a square root dependence on the initial template concentration (Fig. 2E). These results are fully consistent with cross catalytic and product inhibited formation of Pab and Pcd.
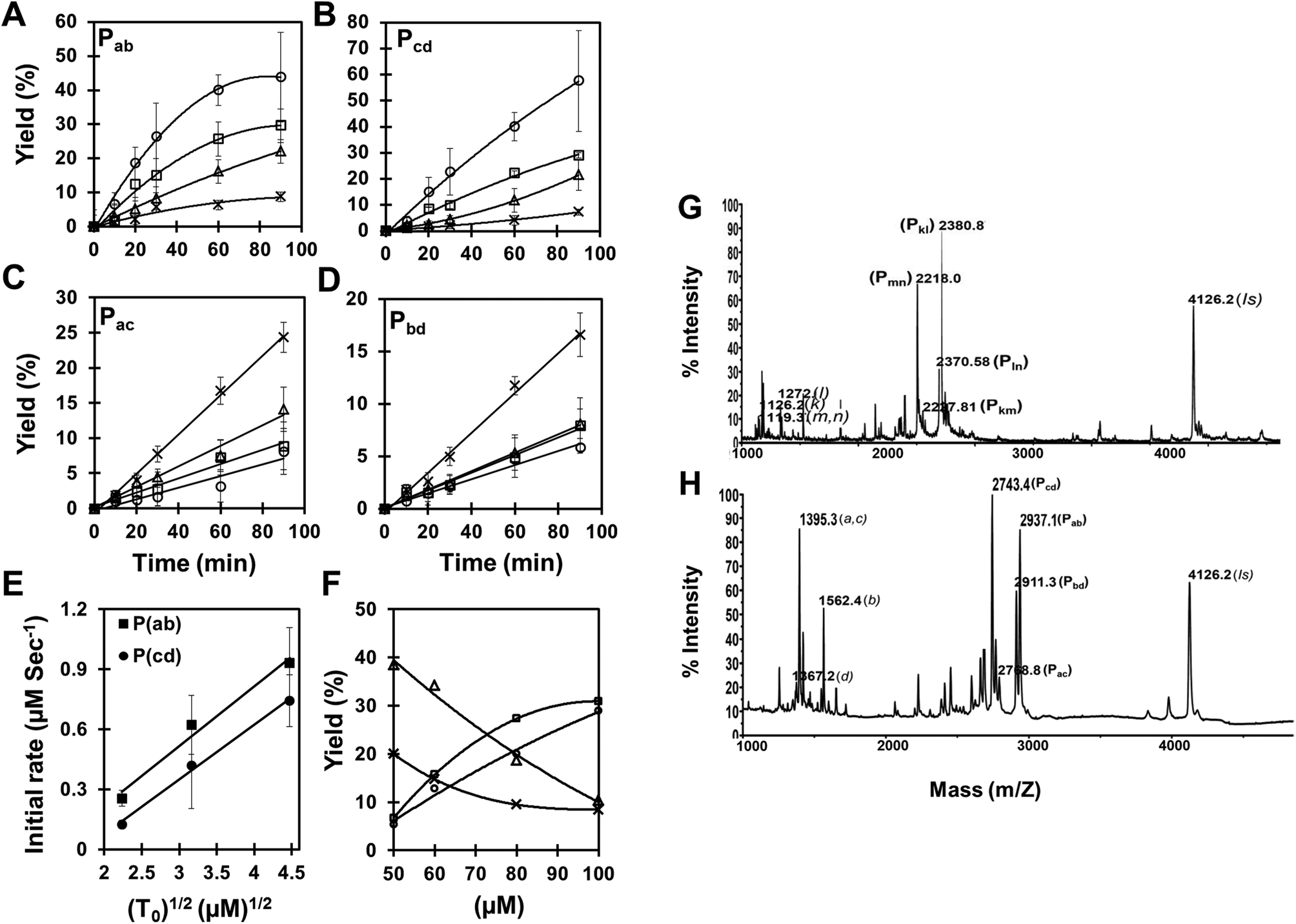 | ||
Fig. 2 (A–E) Kinetics of complementary (Pab and Pcd) and self-complementary (Pac and Pbd) products formation. Reactions were carried out with increasing amount of both chemically synthesized complementary templates (Tab and Tcd), (cross) 0 μM; (triangle) 5 μM; (square) 10 μM; (circle) 20 μM, while fixing the concentration of all four pentameric PNA precursors (a, b, c and d; concentration 100 μM) under the same experimental conditions (see Fig. 1B). Reaction mixture was analyzed through MALDI mass spectrometry and the product yields (%) relative to initial precursor concentration (100 μM) were calculated using the internal standard (see supplementary material Fig. S1 and S4 and Tables S2 and S3†). (A) Pab, (B) Pcd, (C) Pac, (D) Pbd and (E) Initial rate of product formation (Pab and Pcd) as a function of the square root of the initial template concentration (T0). SEM represents 4 independent experiments. (F) Concentration dependence of the product (Pab, Pcd, Pac and Pbd) yields in the pentameric PNA, cross-catalytic self-replicating reaction system. Reactions were carried out by increasing the total concentration of reacting PNA species (50–100 μM) in a fix ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) between complementary templates (Tab and Tcd) and PNA precursors (a, b, c and d) in the presence of chemically synthesized complementary decameric templates under the same experimental conditions (see Fig. 1B). Reaction mixture was analyzed through MALDI mass spectrometry and the product yields (%) (square) Pab; (circle) Pcd; (triangle) Pac; (cross) Pbd were calculated related to the internal standard. (G–H) Raw data of MALDI mass spectrometry analysis of tetrameric, and pentameric PNA cross-catalytic self-replicating reaction system. Reactions were carried out with an equimolar mixture of all four precursors (100 μM each) (G) tetrameric: k, l, m and n; (H) pentameric: a, b, c and d; in the absence or presence of chemically synthesized complementary templates (octameric: Tkl and Tmn; decameric: Tab and Tcd) under the same experimental conditions (see Fig. 1B). The reaction products were quantified by MALDI-TOF mass spectrometry. IS corresponds to the internal standard. :10) between complementary templates (Tab and Tcd) and PNA precursors (a, b, c and d) in the presence of chemically synthesized complementary decameric templates under the same experimental conditions (see Fig. 1B). Reaction mixture was analyzed through MALDI mass spectrometry and the product yields (%) (square) Pab; (circle) Pcd; (triangle) Pac; (cross) Pbd were calculated related to the internal standard. (G–H) Raw data of MALDI mass spectrometry analysis of tetrameric, and pentameric PNA cross-catalytic self-replicating reaction system. Reactions were carried out with an equimolar mixture of all four precursors (100 μM each) (G) tetrameric: k, l, m and n; (H) pentameric: a, b, c and d; in the absence or presence of chemically synthesized complementary templates (octameric: Tkl and Tmn; decameric: Tab and Tcd) under the same experimental conditions (see Fig. 1B). The reaction products were quantified by MALDI-TOF mass spectrometry. IS corresponds to the internal standard. | ||
We further examined whether the products Pac and Pbd were indeed the outcome of an auto-catalytic process or merely due to a simple second order reaction, possibly enhanced via duplex hybridization of complementary oligomers. The results (Table 2) clearly show that formation of Pac and Pbd is not enhanced (catalyzed) by initial presence the product itself, thereby strongly arguing against efficient auto-catalysis. From the present data we cannot distinguish whether Pac and Pbd are formed via simple single strand collision reaction or by ac bd duplex facilitated ligation, either by direct end ligation or via end stacking ligation (see Fig. 1A). The observation that the Pac product is formed in significantly higher yield than the Pbd product could be taken as argument for a duplex mediated ligation because of the higher stability of the ac duplex compared to the bd duplex (Tm = 30 °C and 23 °C, respectively) (Table S2†). In addition, a direct end ligation mechanism is also consistent with the concentration dependence of the replication reaction (Fig. 2F), showing that increased concentration of all components leads to a significantly higher yield of the cross-catalytic Pab and Pcd products (formed in a 3rd order, ter-molecular reaction) relative to the Pac and Pbd products (thus believed to form in a 2nd order, bi-molecular reaction, while an end stacking ligation should be 4th order and thus be relatively more efficient at higher total oligomer concentration). The cross catalytic products Pab and Pcd observed in the absence of any initially added template (Fig. 2A,B, crosses) may likewise be formed by a duplex assisted end ligation mechanism as discussed above. Once formed these should act as catalysts for further replication, and a more exponential like kinetic behavior could have been anticipated, at least in the initial phase. However, the present data do not allow such detailed analysis.
| Self-complementary product | Temp. (°C) | (−) T | (+) T |
|---|---|---|---|
| a Reactions were carried out with an equimolar mixture of two complementary pentameric PNA precursors (a and c) or (b and d); concentration 100 μM both in the absence or presence of one of the chemically synthesized self-complementary decameric template (Tac or Tbd; concentration 10 μM) under the same experimental conditions (see Fig. 1B) at the designated temperature. Reaction mixture was analyzed through MALDI mass spectrometry and the product yields (% relative to initial amount of precursor) were calculated related to the internal standard. | |||
| Pac | 35 | 16.8% | 17.0% |
| 45 | 15.4% | 16.0% | |
| Pbd | 35 | 10.5% | 11.7% |
| 45 | 6.0% | 6.4% | |
In a further characterization of the replication reaction we studied 12 individual reaction setups and controls (Fig. S1†). The results of these experiments corroborate the conclusion drawn, by demonstrating that each of the two ligation reactions of the cross-catalytic system is catalyzed by the template (while the other template has no additional effect) (compare reactions I, II & III and IV, V and VI), and the full system is catalyzed equally well by each of the two templates and a combination of the two (compare reactions IX, X, XI & XII), since of course in the first ligation round of reactions X and XI an equal amount of the non-added template is formed.
Finally, we investigated the effect of oligomer length using a shorter tetrameric (octameric product) cross-catalytic system. Together with the data from the pentameric system these results (Table 3, Fig. 2G,H) quite clearly show that the catalytic effect of initial templates is much more pronounced for the shorter oligomers. Furthermore, the “auto-catalytic” products are more prevalent for the longer oligomers, which is consistent with the higher stability of the longer duplex intermediates.
| Product/species | (−) T | (+) Tmn,Tkl | |
|---|---|---|---|
| (A) Tetrameric precursors | |||
| Complementary | Pmn | 1.6 ± 0.2% | 10.8 ± 1.1% |
| Pkl | 1.1 ± 0.2% | 10.9 ± 0.8% | |
| Self-complementary | Pkm | 1.2 ± 0.1% | 1.2 ± 0.1% |
| Pln | 2.6 ± 0.4% | 1.4 ± 0.1% | |
| Product/species | (−) T | (+)Tab,Tcd | |
|---|---|---|---|
| (B) Pentameric precursors | |||
| a Reactions were carried out with an equimolar mixture of all four precursors (100 μM each) (A) tetrameric: k, l, m and n; (B) pentameric: a, b, c and d in the absence or presence of chemically synthesized complementary templates (octameric: Tkl and Tmn; decameric: Tab and Tcd) under the same experimental conditions (see Fig. 1B). Four independent experiments were performed and the reaction products were quantified by MALDI-TOF mass spectrometry and the product yields (% relative to initial amount of precursor) ± SEM values were calculated related to the internal standard via standard curves. | |||
| Complementary | Pab | 8.8 ± 1.2% | 29.6 ± 8.7% |
| Pcd | 7.3 ± 1.2% | 29.0 ± 10.3% | |
| Self-complementary | Pac | 24.3 ± 3.3% | 8.9 ± 3.2% |
| Pbd | 16.6 ± 0.3% | 8.0 ± 1.4% | |
Discussion
Several examples of non-enzymatic, template-directed chemical self-replicating systems based on oligonucleotides,1–6 peptides scaffolds7–11 have previously been reported. However, it still remains unclear how contemporary biological replicating systems evolved from primitive prebiotic systems. The present peptide nucleic acid based cross catalytic template ligation directed replication system is relevant for this discussion. This closed system composed of four pentameric precursors and two decameric templates (and thus also products) yields approximately two replication rounds (starting with 10 fold excess of the precursors) despite showing product inhibited kinetics. Such kinetics would be expected from the behavior of analogous oligonucleotide replication systems,2,3 especially considering the higher stability of the duplex product (Tm = 76 °C) compared to the ternary pre-ligation complex (Tm ∼ 48 °C). Furthermore, the replication yield decreased as the total PNA concentration was decreased as would be expected from the effect on the corresponding equilibrium constants (the product duplex formation is bi-molecular while formation of the ternary pre-ligation complex is a tri-molecular reaction). However, the cross-catalytic pentameric PNA replicator should be able to perform continuous replication in an open flow system, which could allow for in vitro evolution type experiments.Two self complementary products which may engage in auto catalytic amplification were also formed in the reaction, and these are unavoidable side products in this type of auto catalytic oligo nucleobase systems, as also observed previously (at similar rates) with oligonucleotides.2,3 However, in the present PNA system, the products are not formed via auto-catalysis, but most probably catalyzed by the duplex formed between the two sequence complementary precursors (a–c and b–d). This behavior distinguishes the pentameric PNA system from the previously described trimer oligonucleotide system.2,3 First of all the longer template (10 versus 6 bases) allows the formation of a more stable hairpin which would also compete with templating activity in an auto-catalytic reaction, and in addition PNA–PNA duplexes have higher stability than DNA–DNA duplexes. Furthermore, the higher flexibility and dynamics of the PNA backbone may facilitate the ligation of the ends of the a–c and b–d precursor duplexes. It is also important to note that formation of the “auto-catalytic” products in the PNA system is very significantly subdued relative to the cross-catalytic products by increased oligomer concentration, as well as by the presence of the cross catalytic templates. The cross-catalytic replication reaction shows a temperature optimum around 30 °C. This optimum must reflect the very delicate balance between the thermal stabilities (i.e., the temperature dependence of the equilibrium constant) of the ter-molecular pre-ligation complex and the (product inhibitory) duplex product, and most likely also the temperature dependence of the rate constants for the formation of these complexes.
Although our findings demonstrate the feasibility of simple chemical PNA replication systems of comparable properties to oligonucleotide replicators, and thereby can support models of PNA as a prebiotic genetic material, much further work is required for validation of such a hypothetical scenario. In particular it is important to consider if replicators may function with shorter precursors, such as dimers, and also to explore how they behave in terms of fidelity, evolution and selection when presented with a mixture of precursors. The present results now allow pursuing such studies, which could elucidate general properties of in vitro evolution of simple chemical nucleobase replicators.
Furthermore, we find it worth considering that the quest for genuine exponential replication systems may not necessarily be crucial for the discussion of the origin of prebiotic evolution. This is particularly true for DNA like nucleobase oligomer systems as these are inherently product inhibited. Thus although biological cells can show exponential growth, their DNA replication process as such is not kinetically exponential, and is crucially dependent on DNA template denaturing processes, e.g., in the form of helicases, and of course is taking place in a complex, thermodynamically open environment. Likewise, simple replicators like the present, could depend on auxiliary factors as exemplified by the surface promoted replication developed by the Kiedrowski group.6 For instance it may be very plausible that circadian temperature changes in geological microenvironments (e.g. shallow puddles) of the primordial world could support day rhythm replication, analogously to the temperature cycles of the polymerase chain reaction (PCR). Additionally, it should be remembered that evolutionary selection only operates on the species available at a specific point in time, and per definition always operates on suboptimal solutions. Thus although chemical replicator systems are far less sophisticated than self-replicative systems based on ribonucleic acid polymers with RNA polymerase like ribozyme activity (which eventually may be able to replicate themselves24–26), more primitive replicators based on prebiotic nucleic acids or nucleic acid mimics such as PNA may still have played a crucial role in the early evolution of life. Therefore, obtaining more detailed characterization of such systems and their behavior and properties will allow us to validate and compare different prebiotic scenarios, and it might even guide us towards the development of artificial “living systems”, e.g. in combination with proto cell approaches.27
Experimental section
All solvents were of analytical quality (p.a.). PNA oligomers were synthesized by conventional solid phase Boc-chemistry.28 PNA concentrations were determined spectrophotometrically at 65 °C using molar extinction coefficients: ε260 for adenine = 15400 M−1 cm−1, ε260 for guanine = 11700 M−1 cm−1, ε260 for thymine = 8800 M−1 cm−1 and ε260 for cytosine = 7400 M−1 cm−1.
HPLC analysis
Analytical HPLC (Waters Alliance 2690 (pump, autosampler, and degasser) with a PDA UV absorbance diode array detector Model 996 (195 nm–600 nm), and Millennium 32 Chromatography software version 3.2) was run on a Waters Symmetry 300 C18, 3.9 × 150 mm (5 μm particles with 100 Å pore size) analytical column equipped with a Zorbax Eclipse XDBC18 (5 μm particles with 80 Å pore size) guard column (Agilent) with a linear gradient of 0–50% of solvent B (0.1% TFA in acetonitrile) (0–35 min) in solvent A (0.1% TFA in water) at 50 °C with a flow rate of 1 mL min−1. Each individual HPLC peak was purified, dried under vacuum and characterized as an individual fraction by mass spectrometry analysis.MALDI-TOF/MS analysis
Each HPLC fraction was dissolved in 20 μL of solvent A. A saturated solution of sinapinic acid in MeCN was used for generating the probe-matrix solution. An aliquot of each sample (1 μL) was mixed with the sinapinic acid matrix aqueous solution in 1:5 ratios. 1 μL of each mixture was deposited onto a MALDI target plate and allowed to dry on air. MALDI-TOF mass spectra were recorded on a Voyager-DE Pro bio spectrometry workstation of PerSeptive Biosystems operating in reflector mode. Mass data were obtained by accumulating spectra from 200 laser shots with an accelerating voltage of 20 kV.
MALDI-TOF mass spectrometry quantification of the reaction kinetics (Fig. 2) was done by calculating the ion current (% intensity) of each product in the MALDI mass spectrum relative to the internal standard (Table S1†). In parallel standard calibration curves using the authentic product compounds were made (Fig. S3†), and from these the relative values (Tables S3 and S4†) were converted to absolute concentrations (Tables S5 and S6†) and plotted (Fig. S4†). The data of four independent experiments were used (Fig. 2).
T M measurements
Thermal melting experiments were run at 260 nm on a Cary 300 Bio UV-visible spectrophotometer (Varian, Cary, NC, USA) connected to a temperature controller. Main stock solutions of PNAs were prepared by dissolution in deionized, double distilled water. Thermal melting profiles were obtained using heating–cooling cycles in the range of +5 to 95 °C. The PNA samples (1:1 stoichiometry in single strands) in 50 mM imidazole buffer (pH 7) were heated from 5 °C to 95 °C at a rate of 0.5 °C min−1. The melting temperature (Tm) was determined from the peak of the first derivative of the heating curve. Cuvettes of 1.0 cm path length and 1.0 ml volume were used for all experiments.
Acknowledgements
This work was supported by the European Commission in the 6th framework SYNTCELLS project, contract no. 043359. Annette Jørgensen and Jolanta Ludvigsen are acknowledged for technical assistance.References
- G. von Kiedrowski, Angew. Chem., Int. Ed. Engl., 1986, 25, 932–935 CrossRef.
- D. Sievers and G. von Kiedrowski, Nature, 1994, 369, 221–224 CrossRef CAS PubMed.
- D. Sievers and G. von Kiedrowski, Chem. – Eur. J., 1998, 4, 629–641 CrossRef CAS.
- T. Li and K. C. Nicolaou, Nature, 1994, 369, 218–221 CrossRef CAS PubMed.
- W. S. Zielinski and L. E. Orgel, Nature, 1987, 327, 346–347 CrossRef CAS PubMed.
- A. Luther, R. Brandsch and G. von Kiedrowski, Nature, 1998, 396, 245–248 CrossRef CAS PubMed.
- K. Severin, D. H. Lee, J. A. Martinez and M. R. Ghadiri, Chem. – Eur. J., 1997, 3, 1017–1024 CrossRef CAS.
- D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin and M. R. Ghadri, Nature, 1996, 382, 525–528 CrossRef CAS PubMed.
- J. Chmielewski, S. Yao, I. Ghosh and R. Zutshi, Nature, 1998, 396, 447–450 CrossRef PubMed.
- A. Saghatelian, Y. Yokobayashi, K. Soltani and M. R. Ghadiri, Nature, 2001, 409, 797–801 CrossRef CAS PubMed.
- R. Issac and J. Chmielewski, J. Am. Chem. Soc., 2002, 124, 6808–6809 CrossRef CAS PubMed.
- J. Rebek, Angew. Chem., Int. Ed. Engl., 1990, 29, 245–255 CrossRef.
- T. Tjivikua, P. Ballester and J. Rebek, J. Am. Chem. Soc., 1990, 112, 1249–1250 CrossRef CAS.
- R. J. Pieters, I. Huc and J. Rebek, Angew. Chem., Int. Ed. Engl., 1994, 33, 1579–1581 CrossRef.
- E. A. Wintner and J. Rebek, Acta Chem. Scand., 1996, 50, 469–485 CrossRef CAS PubMed.
- D. N. Reinhoudt, D. M. Rudkevich and F. deJong, J. Am. Chem. Soc., 1996, 118, 6880–6889 CrossRef CAS.
- P. E. Nielsen, Origins Life Evol. Biospheres, 1993, 23, 323–327 CrossRef CAS.
- C. Bohler, P. E. Nielsen and L. E. Orgel, Nature, 1995, 376, 578–581 CrossRef CAS PubMed.
- K. E. Nelson, M. Levy and S. L. Miller, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3868–3871 CrossRef CAS.
- U. J. Meierhenrich, G. M. Munoz Caro, J. H. Bredehoft, E. K. Jessberger and W. H. Thiemann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9182–9186 CrossRef CAS PubMed.
- A. Mattes and O. Seitz, Chem. Commun., 2001, 2050–2051 RSC.
- S. Ficht, A. Mattes and O. Seitz, J. Am. Chem. Soc., 2004, 126, 9970–9981 CrossRef CAS PubMed.
- T. J. Griffin, W. Tang and L. M. Smith, Nat. Biotechnol., 1997, 15, 1368–1372 CrossRef CAS PubMed.
- T. A. Lincoln and G. F. Joyce, Science, 2009, 323, 1229–1232 CrossRef CAS PubMed.
- C. Olea Jr., D. P. Horning and G. F. Joyce, J. Am. Chem. Soc., 2012, 134, 8050–8053 CrossRef PubMed.
- P. Holliger, A. W. Wochner, A. J. Attwater and A. Coulson, Science, 2011, 332, 209–212 CrossRef PubMed.
- P. Stano, E. D'Aguanno, J. Bolz, A. Fahr and P. L. Luisi, Angew. Chem., Int. Ed., 2013, 52, 13397–13400 CrossRef CAS PubMed.
- L. Christensen, R. Fitzpatrick, B. Gildea, K. H. Petersen, H. F. Hansen, T. Koch, M. Egholm, O. Buchardt, P. E. Nielsen, J. Coull and R. H. Berg, J. Pept. Sci., 1995, 1, 175–183 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01158a |
| ‡ Present address: Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, Michigan, USA. |
| This journal is © The Royal Society of Chemistry 2014 |