Synthesis of aromatic 13C/2H-α-ketoacid precursors to be used in selective phenylalanine and tyrosine protein labelling†
Received
2nd June 2014
, Accepted 30th July 2014
First published on 31st July 2014
Abstract
Recent progress in protein NMR spectroscopy revealed aromatic residues to be valuable information sources for performing structure and motion analysis of high molecular weight proteins. However, the applied NMR experiments require tailored isotope labelling patterns in order to regulate spin-relaxation pathways and optimize magnetization transfer. We introduced a methodology to use α-ketoacids as metabolic amino acid precursors in cell-based overexpression of phenylalanine and/or tyrosine labelled proteins in a recent publication, which we have now developed further by providing synthetic routes to access the corresponding side-chain labelled precursors. The target compounds allow for selective introduction of 13C–1H spin systems in a highly deuterated chemical environment and feature alternating 12C–13C–12C ring-patterns. The resulting isotope distribution is especially suited to render straightforward 13C spin relaxation experiments possible, which provide insight into the dynamic properties of the corresponding labelled proteins.
Introduction
Aromatic amino-acids represent a sensitive source of structural and dynamic parameters in high-molecular weight protein NMR spectroscopy.1 Phenylalanines and tyrosines are substantially overrepresented at protein binding interfaces due to their ability to contribute to hydrophobic, as well as to electrostatic interactions.2 Examples from the literature have proven the importance of aromatic residue based NOE data to complement the set of methyl-group derived distance restraints for structure calculation.3 Moreover, aromatic side chains display a remarkable flexibility in dynamic motion, which can be sensitively probed by 13C–1H spin pair relaxation.4 Insufficient chemical shift dispersion, extensive 13C–13C spin coupling and retarded side-chain motion strongly affect the signal assignment and analysis in the aromatic spectral region.
Selective stable-isotope patterns are required to enable effective magnetisation transfer and well defined spin relaxation, which is both necessary to decrypt the structural information buried in these residues. Alternating 12C–13C–12C and/or 2H–1H–2H arrangements in the aromatic ring systems have been shown to result in well resolved NMR signals due to significant reduction of scalar and dipolar couplings.5 Isolated 13C–1H spin systems in an otherwise 2H-containing aromatic ring have additionally been used as valuable tools to elucidate the aromatic side chain motion by erasing unwanted relaxation pathways.6 Reports on labelling phenylalanine and tyrosine residues with stable isotopes include cell-free (CF) protein synthesis,7 as well as cell-based expression systems.6,8 CF-approaches require the sophisticated synthesis of 15N-labelled amino acids, but display highly selective isotope composition in the target proteins. Cell-based overexpression, on the other hand, makes use of amino acid precursor compounds, which are introduced to the metabolism of a protein expressing organism.9
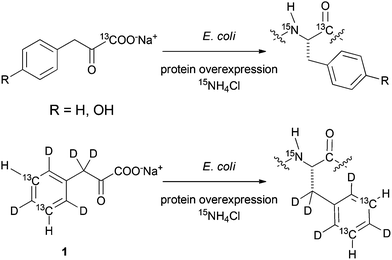
Although economically preferred, cell-based methods often suffer from low incorporation rates and selectivity due to the loss of heavy isotopes at metabolic crossroads. In order to expand the methodology of introducing stable isotopes at distinct positions of a target protein, we recently presented highly selective phenylalanine- and tyrosine-residue labelling based on the corresponding metabolic α-ketoacid precursors sodium phenylpyruvate and sodium 4-hydroxyphenylpyruvate (Scheme 1).10
 |
| | Scheme 1
E. coli based overexpression of a model protein in the presence of labelled aromatic precursor compounds of phenylalanine and tyrosine results in selective protein isotope labelling as shown in previous studies. | |
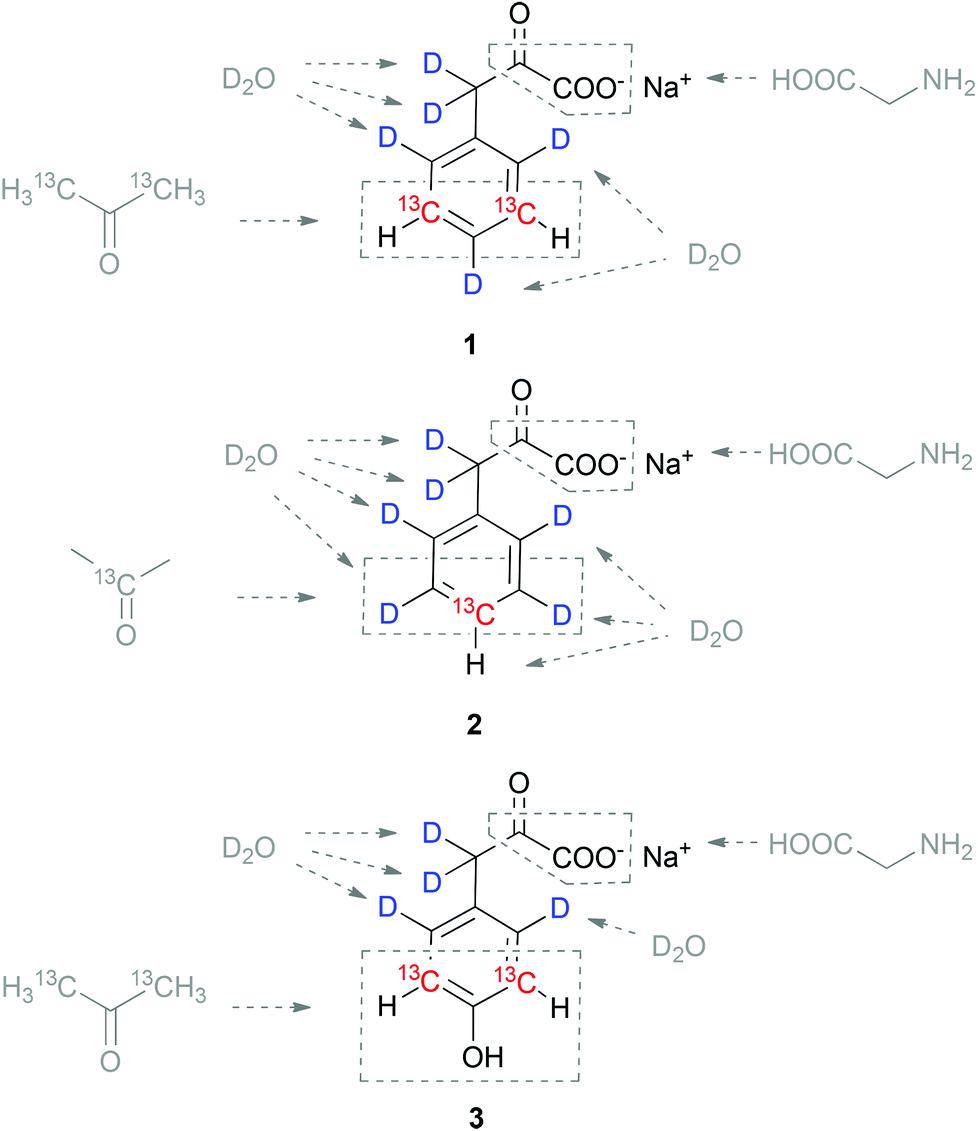
Protein synthesis using an E. coli overexpression host in the presence of the labelled aromatic α-ketoacids thus resulted in the incorporation of 13C without any cross-labelling to other residues. This new methodology combines the robustness and versatility of in-cell overexpression with high incorporation selectivity, which is usually the domain of cell-free protein synthesis. In order to further develop our α-ketoacid precursor based approaches towards selective side-chain labelling,11 we developed a synthetic route to sodium phenylpyruvate 1 containing 13C–1H at meta-positions in an otherwise perdeuterated chemical environment. We could already demonstrate that this side-chain labelled precursor is selectively converted to Phe-residues in an E. coli expression medium.10 This article describes the synthetic details to obtain the 13C/2H aromatic α-ketoacids illustrated in Scheme 2. In addition to the already mentioned precursor 1, synthetic approaches to access para13C–1H labelled phenylalanine precursor 2, as well as the meta13C–1H tyrosine precursor 3 are presented. The routes feature acetone and heavy water as 13C and 2H sources, respectively. Labelling of backbone positions is feasible by application of 13C-glycine as shown previously.10
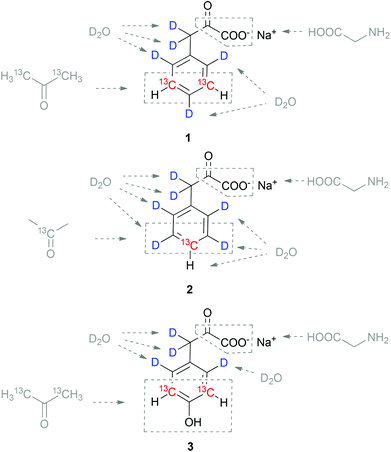 |
| | Scheme 2 Target compounds for the selective labelling of Phe- (1 and 2) and Tyr- (3) residues in cell-based protein overexpression systems. Isotope sources labelled acetone and D2O are shown in grey, as well as the potential source for backbone labelling glycine. | |
Results and discussion
The approach to access the target compounds 1–3 (Scheme 2) is based on the synthesis of the aromatic ring by the reaction of labelled acetone with nitromalonaldehyde in basic aqueous solution.12 Selective deuteration at activated ring-positions was planned in acidic D2O using aniline or 4-aminophenol as electron rich substrates at elevated temperatures.13 On the one hand, this synthetic concept was designed as an economically practicable way of synthesizing enough material to be used in cell-based protein overexpression (quantitative isotope incorporation at 100–200 mg L−1 minimal medium) due to the relatively cheap sources of stable isotopes and robust reaction steps. On the other hand, the routes should be flexible enough to access alternative isotope patterns by simply switching to commercially available starting compounds with different stable isotope composition (e.g. various patterns of labelled acetone for side-chain-, or glycine as a 13C-source for backbone labelling).
The synthesis of sodium 3,3-dideuterio([3,5-13C2]2,4,6-trideuteriophenyl)pyruvate 1 was performed as outlined in Scheme 3. Initially, a straightforward way to access the aromatic ring system in one step was applied by the reaction of commercially available [1,3-13C2]acetone 4 with sodium nitromalonaldehyde 5. Compound 5 can be prepared from mucobromic acid as a stable solid.14 Subsequent deoxygenation of [2,6-13C2]4-nitrophenol 6 was performed in a two-step reaction sequence via the 1-phenyl-1H-tetrazolylether 7.15 Compound 7 was prepared by reaction of the phenolic hydroxy group with 5-chloro-1-phenyl-1H-tetrazole in the presence of KOtBu. Hydrogenation using palladium on charcoal at room temperature and a pressure of 4 bar removed the oxygen from the aromatic ring, while at the same time the nitro-group was reduced yielding [3,5-13C2]aniline 8.16 At this stage, the deuterium pattern at the aromatic ring was installed, as compound 8 shows highly selective 1H/2H exchange at the electron-rich ortho/para positions in the presence of D2O and HCl under microwave irradiation.13 Subsequent formation of [3,5-13C2]2,4,6-trideuteriobenzonitrile 10 was achieved by using potassium tetracyanonickelate in ammonium chloride buffer.17 Reduction of compound 10 using diisopropylaluminium hydride yielded [3,5-13C2]2,4,6-trideuteriobenzaldehyde 11 which was then used in the subsequent condensation step with hydantoin.18 The preparation of labelled benzalhydantoin 12 was done in the presence of ammonium acetate, which provided higher and more reproducible yields than the use of sodium acetate reported in the literature.18a Finally, the hydantoin ring of compound 12 was hydrolysed using 20% NaOD solution, which simultaneously introduced 2H at the C3-position. Labelled sodium phenylpyruvate 1 was obtained by lyophilisation from aqueous solution as a stable white powder in an overall yield of ∼16% in 8 steps from [1,3-13C]acetone 4.
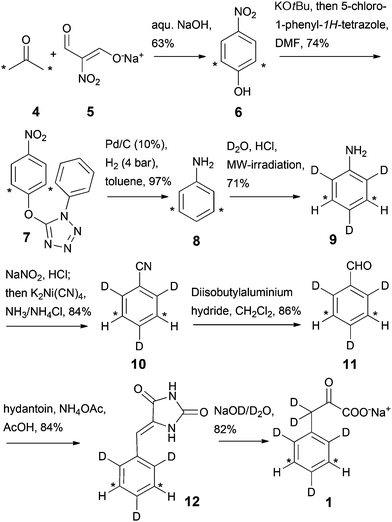 |
| | Scheme 3 Synthesis of labelled phenylpyruvate 1. Asterisks denote 13C labelling. | |
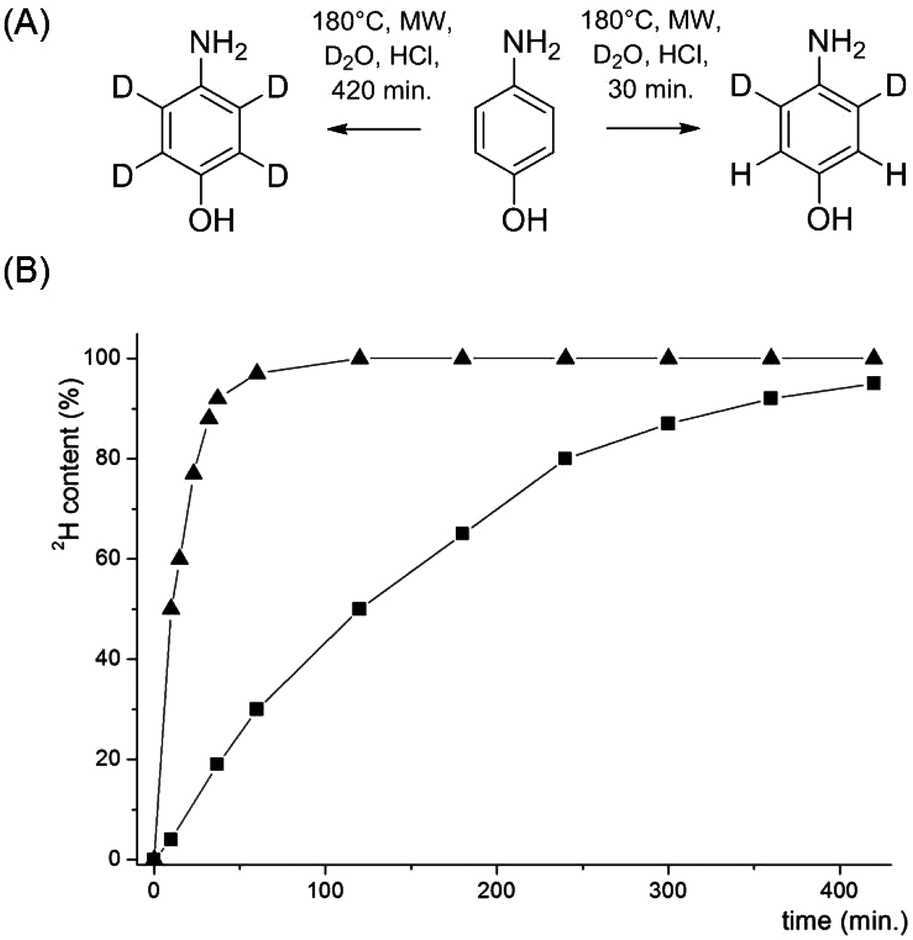
In order to access compounds 2 and 3, the deuteration of 4-aminophenol upon microwave irradiation was thoroughly studied (Scheme 4). A nearly quantitative deuteration at positions 3 and 5 was achieved within 30 minutes at 180 °C in the presence of D2O and HClconc. (1.25% v/v). Additional incorporation of 2H at positions 2 and 6 was performed at a much slower rate with >95% deuteration after 8 hours and only minimal aminophenol degradation. The side-chain deuteration patterns for compounds 2 and 3 could thus be installed by varying the reaction time of the microwave mediated deuteration. Sodium 3,3-dideuterio([4-13C]2,3,5,6-tetradeuterio phenyl)pyruvate 2 was prepared by reducing [1-13C]4-nitrophenol 14 to [1-13C]4-aminophenol 15 using the continuous-flow hydrogenation reactor H-cube® (Scheme 5). After microwave induced deuteration at positions 2, 3, 5 and 6, deoxygenation was again performed via the corresponding 1-phenyl-1H-tetrazolylether 17.
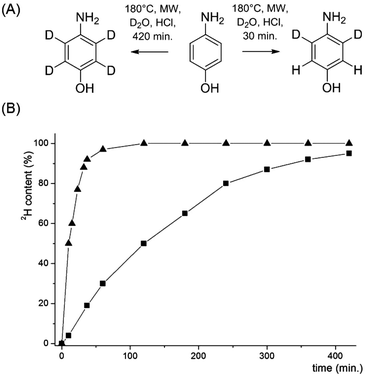 |
| | Scheme 4 (A) Selective deuteration of 4-aminophenol. Reagents and conditions: 4-aminophenol (400 mg), D2O (4 mL), HClconc. (50 μL), 180 °C, microwave irradiation; (B) time dependent progress of 4-aminophenol deuteration in positions 3 and 5 (▲) and additional deuteration in positions 2 and 6 (■). The solvents were evaporated after 180 min and replaced by fresh D2O. 2H content was analysed by integration of the corresponding 1H-NMR signals. | |
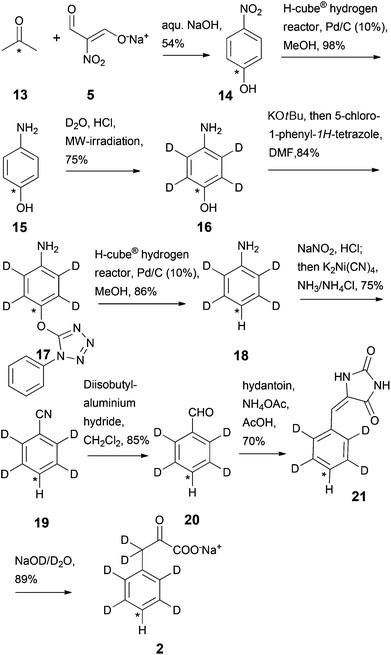 |
| | Scheme 5 Synthesis of labelled phenylpyruvate 2. Asterisks denote 13C labelling. | |
In this case, the Pd/C-mediated hydrogenation was again conducted in the continuous-flow hydrogenation reactor, leading to an isolated hydrogen atom in the para position of the resulting labelled aniline 18. The following reaction steps were performed analogously to the reaction sequence reported for the preparation of the labelled sodium phenylpyruvate 1 leading to the target compound sodium 3,3-dideuterio([4-13C]2,3,5,6-tetradeuterio-phenyl)pyruvate 2 in 9 steps and an overall yield of ∼11%.
To achieve straightforward labelling at the aromatic side chain of tyrosine residues, a route to sodium 3,3-dideuterio([3,5-13C2]2,6-dideuterio-4-hydroxyphenyl)pyruvate 3 was developed as outlined in Scheme 6. After formation of the aromatic system, [2,6-13C2]4-nitrophenol 6 was converted to [2,6-13C2]4-aminophenol 22 as described in the synthesis of compound 2. Deuteration in the ring positions 3 and 5 was then conducted in D2O–HCl at 180 °C for 37 minutes, followed by formation of labelled 4-hydroxybenzonitrile 24 using K2Ni(CN)4. Diisobutylaluminium hydride reduction gave 4-hydroxybenzaldehyde 25, which subsequently underwent condensation with hydantoin in the presence of piperidine.19 Hydrolysis of the hydantoin ring in NaOD–D2O finally gave sodium 3,3-dideuterio([3,5-13C2]2,6-dideuterio-4-hydroxy-phenyl)pyruvate 3 as a stable white solid. This 7-step sequence yielded the target compound 3 in a total yield of ∼28%, which contains ∼23% deuterium in positions 3 and 5 of the aromatic ring (determined by NMR signal integration).
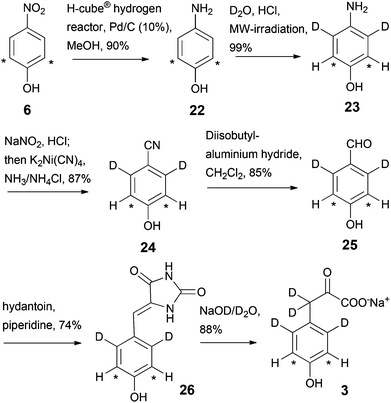 |
| | Scheme 6 Synthesis of labelled hydroxyphenylpyruvate 3. Asterisks denote 13C labelling. | |
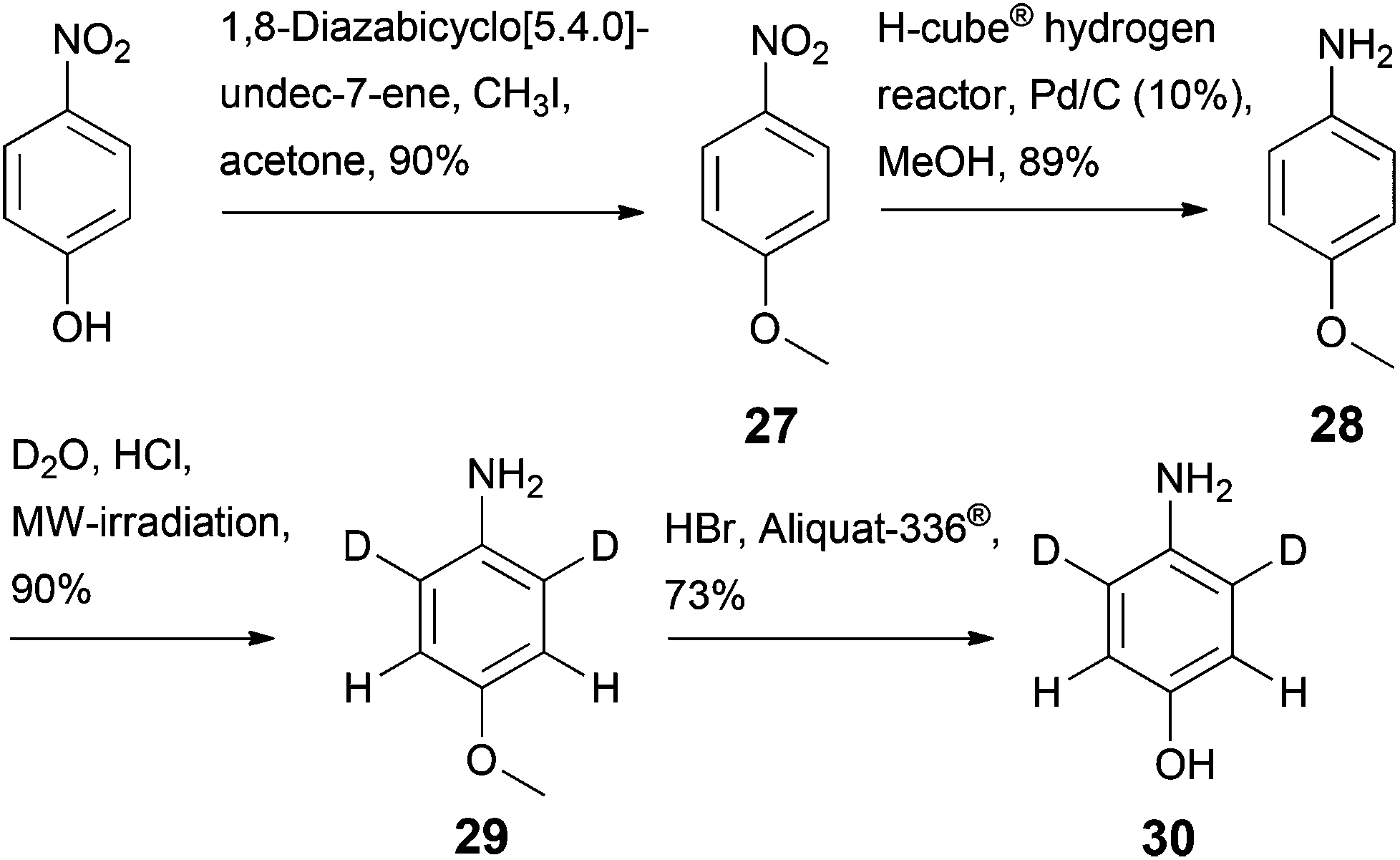
A more selective deuteration pattern can be achieved, if required, as shown in Scheme 7. Methylation of 4-nitrophenol20 and subsequent reduction of the nitro group yielded p-anisidine 28, which showed no reactivity in the deuteration step meta to the amino group (28 → 29).21 Demethylation using HBr in the presence of a phase transfer catalyst (Aliquat-336®) gave selectively deuterated aminophenol 30.22 This sequence, which was verified using unlabelled 4-nitrophenol as a starting material, increases the number of reactions in the route to prepare sodium 3,3-dideuterio([3,5-13C2]2,6-dideuterio-4-hydroxyphenyl)pyruvate 3 by two steps, but represents an effective approach to avoid partial deuteration at the 13C labelled aromatic positions in the target compound 3. The aromatic α-ketoacids 1–3 display high stability in their lyophilized forms as sodium salts, but undergo oxidative degradation in basic solution in the presence of atmospheric oxygen.18d NMR spectra of compounds 1–3 in D2O show mainly the keto forms, whereas in DMSO-d6 the enol forms predominate, which is in accordance with literature data.23
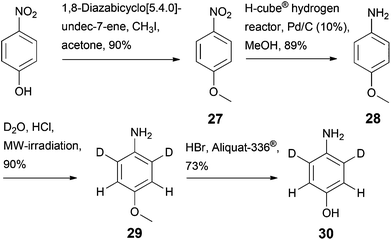 |
| | Scheme 7 Synthesis of selectively deuterated aminophenol 30. | |
Conclusions
An efficient synthetic concept is presented to access labelled metabolic precursor compounds of phenylalanine and tyrosine based on the low-cost isotope sources 13C-acetone and D2O. The routes enable the construction of specific labelling patterns in the aromatic side chains with special focus on alternating 12C–13C–12C ring sequences and isolated 13C–1H spin systems in an otherwise deuterated chemical surrounding. Highly selective aromatic side-chain labelling is thus feasible in cell-based overexpression systems without the need for chiral labelled amino acid additives. The resulting isotope arrangements facilitate the interpretation of Carr–Purcell–Meiboom–Gill (CPMG) based spin-relaxation experiments,6b improve the quality of aromatic proton NOE derived distance restraints3b and enable the unambiguous assignment of aromatic ring signals even in very large proteins. The precursors presented constitute valuable reporters of motional dynamics in complex molecular processes, such as protein folding, allostery and enzymatic catalysis. The straightforward and economic synthetic protocols shown below will further promote the efforts to turn aromatic residue labelling into a routinely used concept and complement the techniques of NMR-based analysis of protein dynamics, which traditionally rely on the interpretation of spin relaxation residing at the backbone or 13C and 2H methyl bearing side-chains.24
Experimental section
General methods
All solvents were distilled prior to use. Anhydrous tetrahydrofuran and dimethylformamide were purchased from commercial suppliers. Dichloromethane was dried by elution over an aluminium oxide column. Isotope labelled reagents were purchased from Sigma-Aldrich ISOTEC with the following purity grades: [1,3-13C2]acetone (99% 13C), [2-13C]acetone (99% 13C) and D2O (99.9% 2H). Column chromatography was performed using silica gel 60 (0.040–0.063 μm, 240–400 mesh) from Merck. Thin layer chromatography (TLC) was done on precoated silica gel (Merck 60 F254) glass plates. TLC detection was carried out using a UVAC-60 neolab ultraviolet lamp, an iodine chamber, or by application of a Mo–Ce(SO4)2 complex solution (48 g (NH4)6Mo7O24·4H2O and 2 g Ce(SO4)2 in 100 mL 10% H2SO4). NMR spectra were recorded on a Bruker AVANCE-DPX 400 spectrometer at 400 MHz. Chemical shifts are given in parts per million (ppm). NMR solvent signals have been calibrated to the following ppm values: 2.5 (DMSO-d6), 4.79 (D2O), 7.26 (CDCl3) and 3.31 (CD3OD). NMR signal multiplicity is abbreviated as singlet (s), doublet (d), multiplet (m), doublet of doublets (dd), doublet of multiplets (dm), etc. Mass spectrometry (MS) and high resolution mass spectrometry (HRMS) experiments were done using electron ionization (EI, 70 eV) or electrospray ionization (ESI, 3 keV). Continuous-flow hydrogenations were performed in an H-Cube® reactor from ThalesNano®. Microwave reactions were conducted in a Biotage Initiator® microwave synthesizer.
Sodium nitromalonaldehyde monohydrate 5.
Sodium nitrite (30 g) was dissolved in water (30 mL) using a three necked round bottomed flask, equipped with a thermometer, a dropping funnel and a tube to drain the evolved gases. The mixture was slightly warmed to dissolve all of the NaNO2. A solution of mucobromic acid (30 g) in ethanol (30 mL) was slowly added for a period of 1 h. After additional stirring for 15 minutes, the reaction mixture was cooled to 0 °C and the precipitate was filtered off. The resulting solid was transferred into a round bottomed flask and stirred under reflux with ethanol (50 mL) and water (10 mL). The hot solution was filtered and the filtrate was subsequently cooled to 0 °C, which led to product precipitation. The solid was filtered off and washed with small portions of cold ethanol. Drying the product under vacuum gave 7.67 g (42%) of sodium nitromalonaldehyde monohydrate 5 as a white solid, which was stored over CaCl2. 1H NMR (400 MHz, DMSO-d6): 9.72 (s, 2H, CHO); 13C NMR (DMSO-d6): 181.20 (CHO), 132.38 (C).
[2,6-13C2]4-nitrophenol 6.
An aqueous NaOH solution (4.4 g in 20 mL) was slowly added to a mixture of sodium nitromalonaldehyde monohydrate 5 (3.25 g) and [1,3-13C2]acetone 4 (1 g) in H2O (200 mL) at 0 °C using a dropping funnel. After the addition was complete, the flask was tightly closed and stirred for 6 days at 4 °C. The resulting brown solution was cooled to 0 °C and 6 N HCl (26 mL) was slowly added. Filtration of the solution resulted in a dark solid, which was taken up in 6 N HCl (26 mL) and boiled gently for 10 minutes. The warm mixture was filtered and the two combined filtrates were extracted with diethyl ether (6 × 100 mL). Subsequent drying of the combined organic phases over MgSO4 and evaporation of the diethyl ether under reduced pressure yielded a yellow solid. The crude product was purified over a silica gel chromatography column by elution with hexane–ethyl acetate (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v). The reaction yielded 1.47 g (63%) of [2,6-13C2]4-nitrophenol 6. 1H NMR (400 MHz, CDCl3): 8.18 (d, J = 8.7 Hz, 2H, CHarom.), 6.90 (dd, J = 8.7 Hz, J = 159.7 Hz, 2H, 13CHarom.), 5.64 (t, J = 4.8 Hz, 1H, OH); 13C NMR (100.6 MHz, CDCl3): 116.10 (13CH). HRMS (ESI): calcd for C413C2H6NO3 [M + H]+ 142.0415; found 142.0430.
:4 v/v). The reaction yielded 1.47 g (63%) of [2,6-13C2]4-nitrophenol 6. 1H NMR (400 MHz, CDCl3): 8.18 (d, J = 8.7 Hz, 2H, CHarom.), 6.90 (dd, J = 8.7 Hz, J = 159.7 Hz, 2H, 13CHarom.), 5.64 (t, J = 4.8 Hz, 1H, OH); 13C NMR (100.6 MHz, CDCl3): 116.10 (13CH). HRMS (ESI): calcd for C413C2H6NO3 [M + H]+ 142.0415; found 142.0430.
5-([2,6-13C2]4-nitrophenoxy)-1-phenyl-1H-tetrazole 7.
A solution of [2,6-13C2]4-nitrophenol 6 (1.4 g) in dry dimethylformamide (18.4 mL) was stirred at room temperature, while potassium tert-butoxide (1.31 g) was added within 5 minutes in small aliquots under a constant stream of argon. After 1 h of vigorous stirring under an argon atmosphere, a solution of 5-chloro-1-phenyl-1H-tetrazole (1.9 g) in dry dimethylformamide (8 mL) was added and the reaction mixture was stirred for further 3 h. The solution was warmed to 65 °C and stirring continued overnight. Precipitation of the crude product was induced by pouring the mixture in ice water (100 mL) and completed at 4 °C in 12 h. The resulting precipitate was separated by filtration and washed with small portions of ice water. The reaction yielded 2.3 g of a crude product, which was further purified by column chromatography. Elution with hexane–ethyl acetate (8:2) gave 5-([2,6-13C2]4-nitrophenoxy)-1-phenyl-1H-tetrazole 7 (2.07 g, 74%) as a white solid. 1H NMR (400 MHz, CDCl3): 8.35 (d, J = 9.3 Hz, 2H, CHarom.), 7.76 (dd, J = 8.2 Hz, J = 1.7 Hz, 2H, CHphenyl.), 7.67–7.54 (m, 3H, CHphenyl.), 7.69 (dm, J = 164.6 Hz, 2H, 13CHarom.); 13C NMR (100.6 MHz, CDCl3): 130.34 (CH), 122.90 (CH), 120.29 (13CH), 116.06 (CH); MS (EI): calcd for C1113C2H9N5O3 [M] 285.08; found 284.9.
[3,5-13C2]aniline 8.
Palladium on charcoal (10%, 1.04 g) was added to a solution of 5-([2,6-13C2]4-nitrophenoxy)-1-phenyl-1H-tetrazole 7 (1.04 g) in dry toluene (150 mL) in a thick walled hydrogenation flask. The flask was mounted on a hydrogenation Parr-apparatus and a pressure of 4 bar of hydrogen was applied. After 12 h of agitation, the pressure was released, the flask flushed with argon and the black solid palladium catalyst separated from the solution by filtration. The catalyst was washed with toluene (30 mL) and the combined filtrates poured on a 0.5 N NaOH solution (150 mL). After separation of the two layers, the aqueous phase was extracted with toluene (3 × 100 mL). The combined organic phases were then extracted using 0.5 N HCl (3 × 100 mL). Addition of concentrated HCl (0.5 mL) to the combined aqueous phases was followed by reducing the volume of the resulting solution by half under reduced pressure at 50 °C. NaOH (1 N) was added until the solution showed a pH of ∼10. The product was extracted from the solution with dichloromethane (3 × 100 mL). Drying of the combined organic phases over MgSO4 and subsequent careful evaporation of the solvents under reduced pressure (>100 mbar) gave 394 mg of a product/dichloromethane mixture which was used for further conversion. The reaction yield was determined by integrating the corresponding NMR signals to be 338 mg (97%). 1H NMR (400 MHz, CDCl3): 7.16 (ddd, J = 8.4 Hz, J = 7.4 Hz, J = 156.7 Hz, 2H, 13CHarom.), 6.80–6.62 (m, 3H, CHarom.), 3.63 (bs, 2H, NH2); 13C NMR (100.6 MHz, CDCl3): 129.69 (13CH); MS (EI): calcd for C413C2H7N [M] 95.06; found 94.9.
[3,5-13C2]2,4,6-trideuterioaniline 9.
A microwave vessel (0.5–2 mL) was charged with [3,5-13C2]aniline 8 (338 mg), D2O (1.5 mL) and 10 drops of HClconc.. After the vessel had been irradiated for 10 minutes (150 °C), the solvents were evaporated and the residue was again dissolved in D2O (1.5 mL). The vessel was tightly closed and again irradiated for 10 minutes (150 °C). The procedure of evaporation, addition of D2O (1.5 mL) and application of microwave irradiation was performed two more times. The solution was then brought to a neutral pH by addition of 1 N NaOH and the product extracted with diethyl ether (3 × 60 mL). Drying of the organic phases over MgSO4 and careful evaporation of the solvents under reduced pressure gave 280 mg of a dark crude product. The same reaction procedure was applied to 250 mg of the substrate, which gave 230 mg of the crude product. The two batches were combined and purified using bulb-to-bulb distillation (50 mbar; up to 120 °C), which yielded [3,5-13C2]2,4,6-trideuterioaniline 9 (430 mg, 71%) as a light yellow liquid. 1H-NMR spectroscopy analysis showed quantitative deuterium incorporation at positions 2, 4 and 6. 1H NMR (400 MHz, CDCl3): 7.15 (dd, J = 8.5 Hz, J = 158.3 Hz, 2H, 13CHarom.), 3.62 (bs, 2H, NH2); 13C NMR (100.6 MHz, CDCl3): 129.46 (13C); MS (EI): calcd for C413C2H4D3N [M] 98.08; found 98.0.
[3,5-13C2]2,4,6-trideuteriobenzonitrile 10.
A solution of sodium nitrite (380 mg) in water (25 mL) was slowly added to a stirred mixture of [3,5-13C2]2,4,6-trideuterioaniline 9 (420 mg) in HCl (0.4%, 160 mL) at 0 °C using a dropping funnel. After 2 h of stirring at 0 °C, the reaction mixture was brought to pH 7 by addition of saturated aqueous Na2CO3. The resulting solution was slowly added to potassium tetracyanonickelate hydrate (1.06 g) in NH3–NH4Cl buffer (60 mL, pH = 10). Stirring was continued for 15 min at 60 °C. The solution was then filtered and the solid residue was washed with small aliquots of water. The combined filtrates were extracted with diethyl ether (4 × 100 mL) and the combined organic phases were dried over MgSO4. Evaporation of the solvents under reduced pressure gave a crude product, which was further purified by bulb-to-bulb distillation (30 mbar; up to 120 °C) to yield [3,5-13C2]2,4,6-trideuteriobenzonitrile 10 (387 mg, 84%) as a slightly yellow liquid. 1H NMR (400 MHz, CDCl3): 7.47 (dd, J = 8.1 Hz, J = 164.0 Hz, 2H, 13CHarom.); 13C NMR (100.6 MHz, CDCl3): 129.29 (13C); MS (EI): calcd for C513C2H2D3N [M] 108.07; found 108.0.
[3,5-13C2]2,4,6-trideuteriobenzaldehyde 11.
A solution of [3,5-13C2]2,4,6-trideuteriobenzonitrile 10 (380 mg) in dry dichloromethane (30 mL) was set under an argon atmosphere and cooled to −78 °C. After the addition of diisobutylaluminium hydride (1 M in dichloromethane, 3.9 mL) was accomplished using a syringe, the mixture was allowed to warm to −40 °C for a period of 2 h. The reaction was quenched by addition of silica gel (5.4 g) and water (3 mL) in small portions. Subsequently, the mixture was stirred at 0 °C for 1 h. The solution was transferred into an Erlenmeyer flask and a spatula of K2CO3 was added. After drying over MgSO4, the solids were separated by filtration and rinsed with ethyl acetate (150 mL). Evaporation of the organic solvents under reduced pressure gave a crude product, which was purified using bulb-to-bulb distillation (20 mbar; up to 110 °C). The reaction yielded [3,5-13C2]2,4,6-trideuteriobenzaldehyde 11 (332 mg; 86%) as a colorless liquid. 1H NMR (400 MHz, CDCl3): 10.04 (s, 1H, CHO), 7.54 (dd, J = 7.8 Hz, J = 162.5 Hz, 2H, 13CHarom.); 13C NMR (100.6 MHz, CDCl3): 129.17 (13C); MS (EI): calcd for C513C2H3D3O [M] 111.07; found 111.0.
5-([3,5-13C2]2,4,6-trideuteriobenzylidene)hydantoin 12.
A solution of [3,5-13C2]2,4,6-trideuteriobenzaldehyde 11 (225 mg), hydantoin (300 mg) and ammonium acetate (226 mg) was stirred in acetic acid (0.7 mL) using a round bottomed flask, equipped with a short reflux condenser. The mixture was heated to 120 °C for 4 h. The hot solution was cooled in an ice bath, leading to the precipitation of a yellow solid, which was separated by filtration. Drying in vacuo yielded 5-([3,5-13C2]2,4,6-trideuteriobenzylidene)hydantoin 12 (488 mg, 84%). 1H NMR (400 MHz, CD3OD): 7.42 (dd, J = 7.9 Hz, J = 160.4 Hz, 2H, 13CHarom.), 6.57 (s, 1H, CH); 13C NMR (100.6 MHz, CD3OD): 128.77 (13C); MS (EI): calcd for C813C2H5D3N2O2 [M] 193.08; found 193.1.
Sodium 3,3-dideuterio([3,5-13C2]2,4,6-trideuteriophenyl)pyruvate 1.
A two necked round bottomed flask, equipped with a reflux condenser, was loaded with 5-([3,5-13C2]2,4,6-trideuteriobenzylidene)hydantoin 12 (217 mg) and set under an argon atmosphere. Addition of NaOD in D2O (20%, 6 mL, prepared by slow addition of Na to D2O) was accomplished via a syringe and the mixture was stirred at 100 °C for 5 hours. After allowing the mixture to reach room temperature, the solution was extracted with diethyl ether (2 × 20 mL). The aqueous phase was brought to pH < 1 by slow addition of HClconc. at 0 °C. This mixture was then extracted with diethyl ether (5 × 30 mL) and the combined organic phases were dried over MgSO4. The solvent was removed in vacuo yielding a white solid. To this residue D2O (30 mL) was added and the pH set to 7 by careful addition of 1 M NaOD. Lyophilization overnight yielded sodium 3,3-dideuterio([3,5-13C2]2,4,6-trideuteriophenyl)pyruvate 1 (176 mg, 82%) as a white powder. NMR analysis showed residual 1H at C3 (<5%). 1H NMR (400 MHz, D2O): 7.45 (dd, J = 8.1 Hz, J = 160.9 Hz, 2H, 13CHarom.), 4.12 (s, 0.06 H, residual CH2); 13C NMR (100.6 MHz, D2O): 129.05 (13C); HRMS (ESI): calcd for C713C2H2D5O3 [M − Na]− 170.0777; found 170.0781.
[1-13C]4-nitrophenol 14.
The synthesis was performed according to the preparation of [2,6-13C2]4-nitrophenol 6 using [2-13C]acetone as a reagent. Purification of the raw product by column chromatography eluting with hexane–ethyl acetate (6:4 v/v) yielded [1-13C]4-nitrophenol 14 (1.28 g, 54%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 11.02 (s, 1 H), 8.12 (dd, J = 9.2 Hz, J = 9.2 Hz, 2H, m-CHarom.), 6.93 (dd, J = 9.2 Hz, J = 2.1 Hz, 2H, o-CHarom.), 13C NMR (100.6 MHz, DMSO-d6): 164.37 (13C), 126.65 (m-CHarom.), 116.24 (d, J = 63.0 Hz, o-CHarom.); HRMS (ESI): calcd for C513CH4NO3 [M − H]− 139.0225; found 139.0234.
[1-13C]4-aminophenol 15.
[1-13C]4-nitrophenol 14 (1.28 g) was dissolved in MeOH (90 mL). This solution was loaded onto a Pd/C (10%) catalyst cartridge in a continuous-flow hydrogen reactor (H-cube® – Thalesnano) at a flow-rate of 1 mL min−1 and at room temperature. The hydrogen generator was set to full hydrogen mode. Evaporation of methanol under reduced pressure gave [1-13C]4-aminophenol 15 (986 mg, 98%). 1H NMR (400 MHz, DMSO-d6): 8.30 (d, J = 2.1 Hz, 1H, OH), 6.47 (dd, J = 8.8 Hz, J = 8.8 Hz, 2H, o-CHarom.), 6.42 (dd, J = 8.8 Hz, J = 2.1 Hz, 2H, m-CHarom.), 4.36 (s, 2H, NH2); 13C NMR (100.6 MHz, DMSO-d6): 148.68 (13C), 141.12 (d, J = 8.8 Hz, CNH2), 115.97 (d, J = 66.0 Hz, o-CHarom.), 115.69 (m-CHarom.); HRMS (ESI): calcd for C513CH8NO [M + H]+ 111.0639; found 111.0636.
[1-13C]2,3,5,6-tetradeuterio-4-aminophenol 16.
[1-13C]4-aminophenol 15 (980 mg) was heated to 180 °C, together with D2O (10 mL) and HClconc. (125 μL) using a microwave reactor. The microwave vessel was purged with argon before the reaction was started. After 2.5 h, the reaction mixture was allowed to cool to room temperature, transferred to a round bottomed flask and the solvents were evaporated in vacuo. After the addition of fresh D2O (10 mL), the mixture was again transferred to a microwave vessel and irradiation was continued for another 5.5 h at 180 °C. The solvents were then removed in vacuo and the residual black solid purified over silica gel column chromatography using ethyl acetate as the eluent. The reaction yielded [1-13C]2,3,5,6-tetradeuterio-4-aminophenol 16 (766 mg, 75%) as a light brown solid. NMR spectroscopy revealed quantitative deuteration at positions 2 and 6 and a deuteration grade of >95% in positions 3 and 5. 1H NMR (400 MHz, DMSO-d6): 8.29 (s, 1H, OH), 6.46 (d, J = 3.0 Hz, 0.07 H, residual o-CHarom.), 4.34 (s, 2H, NH2); 13C NMR (100.6 MHz, DMSO-d6): 148.48 (13C), 140.86 (d, J = 8.9 Hz, CNH2), 115.63 (dt, J = 62.0 Hz, J = 26.2 Hz, o-CHarom.), 115.29 (t, J = 23.0 Hz, m-CHarom.); HRMS (ESI): calcd for C513CH4D4NO [M + H]+ 115.0891; found 115.0884.
5-([1-13C]-2,3,5,6-tetradeuterio-4-aminophenoxy)-1-phenyl-1H-tetrazole 17.
A three necked round bottomed flask was charged with [1-13C]2,3,5,6-tetradeuterio-4-aminophenol 16 (760 mg). Potassium-tert butoxide (1.1 g) was loaded in a slightly-bent round bottomed flask attached to one neck and the apparatus set under an argon atmosphere. The addition of dry DMF (18 mL) was conducted via a syringe through a septum and the KOtBu was slowly added within 15 min. The mixture was then stirred for 90 min before a solution of 5-chloro-1-phenyl-1H-tetrazole (1.27 g in 6 mL dry DMF) was added via a syringe. Stirring was continued at room temperature for another 90 min. The reaction was quenched by pouring the mixture on ice-water (150 mL). The precipitated solid was filtered off and dissolved again in dichloromethane (200 mL). This solution was washed with water until the aqueous phase remained colorless. The organic phase was dried over MgSO4 and the solvents removed under reduced pressure to yield 5-([1-13C]-2,3,5,6-tetradeuterio-4-aminophenoxy)-1-phenyl-1H-tetrazole 17 (1.48 g, 84%). 1H NMR (400 MHz, DMSO-d6): 7.85–7.80 (m, 2H, CHphenyl.), 7.69–7.56 (m, 3H, CHphenyl.), 7.11 (d, J = 4.6 Hz, 0.08 H, residual CHarom.), 5.17 (s, 2H, NH2); 13C NMR (100.6 MHz, DMSO-d6): 161.60, 155.68, 154.78, 147.62 (d, J = 10.2 Hz), 144.35 (13C), 133.14, 130.28 (CH), 130.14 (CH), 123.55 (CH); HRMS (ESI): calcd for C1213CH8D4N5O [M + H]+ 259.1327; found 259.1326.
[4-13C]2,3,5,6-tetradeuterioaniline 18.
5-([1-13C]-2,3,5,6-tetradeuterio-4-aminophenoxy)-1-phenyl-1H-tetrazole 17 (1.48 g) was loaded onto a Pd/C (10%) catalyst cartridge at a pressure of 10 bar and a flow rate of 1 mL min−1. using a continuous-flow hydrogen reactor (H-cube® – Thalesnano). After the solvents have been removed at reduced pressure, the residue was taken up in 0.5 M NaOH (200 mL) and the resulting basic solution extracted with diethylether (4 × 50 mL). The combined organic phases were extracted with 0.5 M HCl (4 × 50 mL) and the acidic solutions pooled and then reduced to half of their volume under reduced pressure after addition of conc. HCl (1 mL). Addition of 1 M NaOH set the pH > 12 and the resulting solution was extracted with dichloromethane (4 × 50 mL). Drying of the combined organic phases over MgSO4 and subsequent careful evaporation of the solvents at reduced pressure (>200 mbar) gave [4-13C]2,3,5,6-tetradeuterioaniline 18 in residual dichloromethane, which was used without further purification. NMR signal integration revealed a yield of 490 mg (86%). 1H NMR (400 MHz, DMSO-d6): 6.99 (d, J = 7.5 Hz, 0.08 H, residual m-CHarom.), 6.47 (d, J = 159.8 Hz, 1H, 13CH), 4.96 (s, 2H); 13C NMR (100.6 Hz, DMSO-d6): 116.23 (13C); HRMS (ESI): calcd for C513CH4D4N [M + H]+ 99.0941; found 99.0940.
[4-13C]2,3,5,6-tetradeuteriobenzonitrile 19.
The synthesis was conducted similar to the conversion of compound 9 to [3,5-13C2]2,4,6-trideuteriobenzonitrile 10. Purification of the crude product using bulb-to-bulb distillation gave [4-13C]2,3,5,6-tetradeuteriobenzonitrile 19 (390 mg, 75%) as a colourless liquid. 1H NMR (400 MHz, CDCl3): 7.61 (d, J = 161.5 Hz, 1H, 13CHarom.), 7.47 (d, J = 7.8 Hz, 0.08 H, residual m-CHarom.); 13C NMR (100.6 MHz, CDCl3): 132.53 (13C); HRMS (EI): calcd for C613CHD4N [M] 108.0707; found 108.0697.
[4-13C]2,3,5,6-tetradeuteriobenzaldehyde 20.
Compound 20 was synthesized according to the procedure described for the preparation of [3,5-13C2]2,4,6-trideuteriobenzaldehyde 11. The reaction yielded [4-13C]2,3,5,6-tetradeuteriobenzaldehyde 20 (290 mg, 85%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3): 10.04 (s, 1H, CHO), 7.63 (d, J = 160.0 Hz, 1H, 13CHarom.), 7.54 (d, J = 7.5 Hz, 0.08 H, residual m-CHarom.); 13C NMR (100.6 MHz, CDCl3): 134.22 (13C); MS (EI): calcd for C613CH2D4O [M] 111.08; found 111.1.
5-([4-13C]2,3,5,6-tetradeuteriobenzylidene)hydantoin 21.
Preparation of compound 21 was accomplished similar to the preparation of 5-([3,5-13C2]2,4,6-trideuteriobenzylidene)hydantoin 12 yielding 363 mg (70%) of the target compound as a slightly green solid. 1H NMR (400 MHz, DMSO-d6): 11.12 (bs, 1H, NH), 10.59 (bs, 1H, NH), 7.33 (d, J = 160.5 Hz, 1H, 13CHarom.), 7.40 (d, J = 7.3 Hz, 0.1 H, residual m-CHarom.), 6.41 (s, 1H, CH); 13C NMR (100.6 MHz, DMSO-d6): 166.12 (CO), 156.28 (CO), 128.57 (13C), 108.57 (CH); HRMS (ESI): calcd for C913CH5D4N2O2 [M + H]+ 194.0949; found 194.0938.
Sodium 3,3-dideuterio([4-13C]2,3,5,6-tetradeuteriophenyl)pyruvate 2.
The synthesis of compound 2 was performed according to the preparation of sodium 3,3-dideuterio([3,5-13C2]2,4,6-trideuteriophenyl)pyruvate 1, but using 5-([4-13C]2,3,5,6-tetradeuteriobenzylidene)hydantoin 21 (85 mg) as a substrate. The reaction yielded sodium 3,3-dideuterio([4-13C]2,3,5,6-tetradeuteriophenyl)pyruvate 2 (76 g, 89%) as a colourless lyophilisate. NMR analysis showed residual 1H at C3 (<6%). 1H NMR (400 MHz, D2O): 7.40 (d, J = 160.9 Hz, 1H, 13CHarom.), 4.13 (s, 0.11 H, residual m-CHarom.); 13C NMR (100.6 MHz, DMSO-d6): 127.48 (13C); HRMS (ESI): calcd for C813CHD6O3 [M − Na]− 170.0805; found 170.0803.
[2,6-13C2]4-aminophenol 22.
The reaction was performed analogously to the synthesis of [1-13C]4-aminophenol 15 using [2,6-13C2]4-nitrophenol 6 (1.4 g) as a substrate. The reaction yielded [2,6-13C2]4-aminophenol 22 (985 mg, 90%) as a brown solid. 1H NMR (400 MHz, DMSO-d6): 8.29 (t, J = 4.1 Hz, 1H, OH), 6.45 (dm, J = 153.2 Hz, 2H, 13CHarom.), 6.41 (d, J = 8.6 Hz, 2H, CHarom.), 4.35 (s, 2H, NH2); 13C NMR (100.6 MHz, DMSO-d6): 116.00 (13C); HRMS (ESI): calcd for C413C2H8NO [M + H]+ 112.0673; found 112.0668.
[2,6-13C2]3,5-dideuterio-4-aminophenol 23.
[2,6-13C2]4-aminophenol 22 (460 mg) was treated with D2O (4.6 mL) and HClconc. (57 μL) at 180 °C for 37 min in a microwave reactor. The solvents were removed in vacuo and the residual black solid dissolved in methanol (10 mL). Evaporation of the solvent gave [2,6-13C2]3,5-dideuterio-4-aminophenol 23 (464 mg, 99%) as a dark solid. NMR spectroscopy showed a deuteration grade of 92% in positions 3 and 5 whereas positions 2 and 6 revealed a deuteration grade of 23%. 1H NMR (400 MHz, DMSO-d6): 8.65 (s, 1H, OH), 6.60–6.56 (m, 0.17 H, residual m-CHarom.), 6.55 (dm, J = 156.2 Hz, 1.55 H, 13CHarom.), 6.08 (bs, 2H, NH2); 13C NMR (100.6 MHz, DMSO-d6): 115.92 (13C); MS (EI): calcd for C413C2H5D2NO [M] 113.07; found 113.1.
[3,5-13C2]2,6-dideuteriohydroxybenzonitrile 24.
A solution of sodium nitrite (796 mg) in water (50 mL) was slowly added to a stirred mixture of [2,6-13C2]3,5-dideuterio-4-aminophenol 23 (900 mg) in HCl (0.4%; 325 mL) at 0 °C using a dropping funnel. After 2 h of stirring at 0 °C, the solution was brought to pH 7 by addition of saturated aqueous Na2CO3. The resulting mixture was slowly added to a stirred solution of potassium tetracyanonickelate in NH3–NH4Cl buffer (112 mL, pH = 10). Stirring was continued for 15 min at 60 °C. The solution was filtered and the solid residue was washed with small aliquots of water. The filtrates were extracted with ethyl acetate (6 × 70 mL) and the combined organic phases were dried over MgSO4. Evaporation of the solvents under reduced pressure gave 915 mg of a brown solid, which was purified by column chromatography (eluent: hexane–ethyl acetate 7:3). The reaction yielded 850 mg (87%) of [3,5-13C2]2,6-dideuteriohydroxybenzonitrile 24 as a yellow solid. 1H NMR (400 MHz, DMSO-d6): 10.58 (s, 1H, OH); 7.65–7.61 (m, 0.14 H, residual o-CHarom.); 6.89 (dm, J = 162.0 Hz, 1.44 H, 13CHarom.); 13C NMR (100.6 MHz, DMSO-d6): 116.53 (13C); HRMS (ESI): calcd for C513C2H3D2NO [M + H]+ 123.0564; found 123.0559.
[3,5-13C2]2,6-dideuteriohydroxybenzaldehyde 25.
A solution of [3,5-13C2]2,6-dideuteriohydroxybenzonitrile 24 in dry dichloromethane (150 mL) was set under an argon atmosphere and cooled to −78 °C. After the addition of diisobutylaluminium hydride (11.4 mL, 1 M in dichloromethane) was accomplished using a syringe, the mixture was allowed to warm to −40 °C for a period of 2 h. The reaction was quenched by addition of silica gel (5 g) and water (3 mL) in small portions and the resulting mixture was stirred at 0 °C for 1 h; then, the solution was transferred into an Erlenmeyer flask and a spatula of K2CO3 was added. After drying over MgSO4, the solid was separated off by filtration and rinsed with dichloromethane until no more product was washed out of the silica gel/MgSO4 mixture (control of TLC spots under UV light). Evaporation of the combined organic phases under reduced pressure gave 504 mg (90%) of [3,5-13C2]2,6-dideuteriohydroxybenzaldehyde 25 as a yellow solid. 1H NMR (400 MHz, DMSO-d6): 10.57 (s, 1H, OH), 9.79 (s, 1H, CHO), 7.78–7.73 (m, 0.15H, residual o-CHarom.), 6.92 (dm, J = 160.8 Hz, 1.55 H, 13CHarom.); 13C NMR (100.6 MHz, DMSO-d6): 115.69 (13C); HRMS (EI): calcd for C513C2H4D2O2 [M] 126.0555; found 126.0549.
5-([3,5-13C2]2,6-dideuterio-4-hydroxybenzylidene)hydantoin 26.
The reagents [3,5-13C2]2,6-dideuteriohydroxybenzaldehyde 25 (474 mg), hydantoin (423 mg) and piperidine (575 mg) were stirred in a 10 ml round bottomed flask, equipped with a reflux condenser at 130 °C for 30 min. Addition of warm water (8 mL) was followed by homogenization of the resulting mixture in an ultrasonic bath. Precipitation of a solid was induced by adding HClconc. (0.5 mL). The crude product was separated by filtration and recrystallized from methanol, yielding 5-([3,5-13C2]2,6-dideuterio-4-hydroxybenzylidene)hydantoin 26 (742 mg, 74%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6): 11.09 (s, 1H, NH), 10.30 (s, 1H, NH), 9.83 (s, 1H, OH), 7.50–7.43 (m, 0.12H, residual CHarom.), 6.78 (dm, J = 159.2 Hz, 1.47 H, 13CHarom.), 6.35 (s, 1H, CH); 13C NMR (100.6 Hz, DMSO-d6): 116.07 (13C); HRMS (EI): calcd for C813C2H6D2N2O3 208.0722; found 208.0716.
Sodium 3,3-dideuterio([3,5-13C2]2,6-dideuterio-4-hydroxy phenyl)pyruvate 3.
A 10 mL round bottomed three-necked flask was charged with 5-([3,5-13C2]2,6-dideuterio-4-hydroxybenzylidene)hydantoin 26 (50 mg) and set under an argon atmosphere. A solution of NaOD in D2O (20%, 4 mL) was degassed under argon by ultrasonication and added via a syringe. Throughout the reaction a constant stream of argon was purged through the reaction mixture via a syringe and needle to prevent oxidative degradation of the product. The mixture was stirred at 110 °C for 4 h. After the reaction was allowed to cool to room temperature, the mixture was extracted with diethylether (2 × 20 mL). Subsequent addition of HClconc. (2.5 mL) to the aqueous phase was followed by extraction with diethylether (5 × 30 mL). The organic phases were combined and dried over MgSO4. Evaporation of the solvents under reduced pressure gave a white solid to which D2O was added (10 mL) and the resulting solution was brought to pH 7 by slow addition of NaOD (1 N). Lyophilization yielded sodium 3,3-dideuterio([3,5-13C2]2,6-dideuterio-4-hydroxyphenyl)pyruvate 3 (44 mg, 88%) as a yellow solid. NMR analysis showed residual 1H at C3 (<4%). 1H NMR (400 MHz, D2O): 6.83 (dm, J = 163.9 Hz, 1.5 H, 13CHarom.), 3.99 (s, 0.07 H, residual CHarom.); 13C NMR (100.6 Hz, D2O): 115.69 (13C). HRMS (ESI): calcd for C713C2H3D4O4 [M − Na]− 185.0663; found 185.0663.
3,5-Dideuterio-4-aminophenol 30.
1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 750 μL) was added to a stirred solution of 4-nitrophenol (700 mg) in acetone (25 mL). The reaction mixture was stirred at room temperature for 10 min before iodomethane (934 μL) was added dropwise. After stirring the reaction mixture for 4 h, TLC still showed remaining starting material. Therefore, additional DBU (750 μL) and iodomethane (310 μL) were added and stirring was continued for 1 h. The solvents were then removed under reduced pressure and the residue was dissolved in ethyl acetate (100 mL). This solution was washed with 1 N HCl (10 mL), water (10 mL), a saturated solution of sodium thiosulfate (10 mL) and brine (10 mL). Drying of the organic phase over MgSO4 and evaporation of the solvents yielded 4-nitroanisole 27 (686 mg, 90%). 1H NMR (400 MHz, CDCl3): 8.21 (dm, J = 9.3 Hz, 2H, m-CHarom.), 6.96 (dm, J = 9.3 Hz, 2H, o-CHarom.), 3.91 (s, 3H, CH3); 13C NMR (100.6 Hz, CDCl3): 164.59 (Carom.), 125.92 (CHarom.), 114.02 (CHarom.), 55.95 (CH3). 4-Nitroanisole 27 (546 mg) was dissolved in MeOH (40 mL) and conducted over a Pd/C (10%) catalyst cartridge in a continuous-flow hydrogen reactor (H-cube® – Thalesnano) at a flow-rate of 1 mL min−1 and room temperature. The hydrogen generator was set to full hydrogen mode. Evaporation of methanol under reduced pressure gave p-anisidine 28 (392 mg, 89%). 1H NMR (400 MHz, DMSO-d6): 6.74 (dm, J = 8.9 Hz, 2H, m-CHarom.), 6.65 (dm, J = 8.9 Hz, 2H, o-CHarom.), 3.75 (s, 3H, CH3), 4.41 (bs, 2H, NH2); 13C NMR (100.6 Hz, DMSO-d6): 152.87 (Carom.), 139.94 (Carom.NH2), 116.42 (CHarom.), 114.85 (CHarom.), 55.76 (CH3). A microwave vessel was charged with anisidine 28 (272 mg), D2O (2.5 mL) and HClconc. (50 μL) and heated in the microwave reactor at 180 °C for 40 min. After the solvents had been removed under reduced pressure, the residue was dissolved in methanol (10 mL) and concentrated again to yield 2,6-dideuterio-p-anisidine 29 (245 mg, 90%). 1H NMR (400 MHz, DMSO-d6): 6.73 (s, 2H, CHarom.), 6.37 (bs, 2H, NH2), 3.65 (s, 3H, CH3); 13C NMR (100.6 Hz, DMSO-d6): 153.14 (Carom.), 138.15 (Carom.NH2), 114.95 (CHarom.), 55.81 (CH3). HRMS (EI): C7H2OND2 125.0804; found 125.0803. 2,6-Dideuterio-p-anisidine 29 (96 mg) was treated with HBr (47%, 620 mL) and Aliquat-336® (16 mg) at 105 °C for 6 h. The reaction was quenched by the addition of water (5 mL) and the resulting solution was extracted with ethyl acetate (50 mL). After the aqueous phase was brought to a pH > 12 by addition of 1 M NaOH, the mixture was extracted with ethyl acetate (3 × 50 mL). The organic phases resulting from the second extraction were combined, washed with water (2 × 20 mL) and dried over MgSO4. The crude product was purified over a short silica-gel column using ethyl acetate as an eluent to yield 3,5-dideuterio-4-aminophenol 30 (63 mg, 73%). 1H NMR (400 MHz, DMSO-d6): 8.30 (s, 1H, OH), 6.46 (s, 2H, o-CHarom.), 4.34 (s, 2H, NH2); 13C NMR (100.6 Hz, DMSO-d6): 148.16 (Carom.OH), 140.52 (Carom.NH2), 115.07 (o-CHarom.).
Notes and references
- U. Weininger, C. Diehl and M. Akke, J. Biomol. NMR, 2012, 53, 181–190 CrossRef CAS PubMed.
- L. Lo Conte, C. Chothia and J. Janin, J. Mol. Biol., 1999, 285, 2177–2198 CrossRef CAS PubMed.
-
(a) Z. Lin, Y. Xu, S. Yang and D. Yang, Angew. Chem., Int. Ed., 2006, 45, 1969–1963 (
Angew. Chem.
, 118
, 1994–1997
) Search PubMed;
(b) C. M. Slupsky, L. N. Gentile and L. P. McIntosh, Biochem. Cell Biol., 1998, 76, 379–390 CrossRef CAS PubMed;
(c) G. W. Vuister, S.-J. Kim, C. Wu and A. Bax, J. Am. Chem. Soc., 1994, 116, 9206–9210 CrossRef CAS.
- J. A. Boyer and A. L. Lee, Biochemistry, 2008, 47(17), 4876–4886 CrossRef CAS PubMed.
-
(a) M. Takeda, A. M. Ono, T. Terauchi and M. Kainosho, J. Biomol. NMR, 2010, 46, 45–49 CrossRef CAS PubMed;
(b) P. Lundström, K. Teilum, T. Carstensen, I. Bezsonova, S. Wiesner, D. Flemming Hansen, T. L. Religa, M. Akke and L. E. Kay, J. Biomol. NMR, 2007, 38, 199–212 CrossRef PubMed.
-
(a) V. Kasinath, K. G. Valentine and A. J. Wand, J. Am. Chem. Soc., 2013, 135, 9560–9563 CrossRef CAS PubMed;
(b) K. Teilum, U. Brath, P. Lundström and M. Akke, J. Am. Chem. Soc., 2006, 128, 2506–2507 CrossRef CAS PubMed.
- M. Kainosho, T. Torizawa, Y. Iwashita, T. Terauchi, A. Mei Ono and P. Güntert, Nature, 2006, 440, 52–57 CrossRef CAS PubMed.
- S. Rajesh, D. Nietlispach, H. Nakayama, K. Takio, E. D. Laue, T. Shibata and Y. Ito, J. Biomol. NMR, 2003, 27, 81–86 CrossRef CAS PubMed.
- C. G. Hoogstraten and J. E. Johnson Jr., Concepts. Magn. Reson. A, 2008, 32A, 34–55 CrossRef CAS.
- R. J. Lichtenecker, K. Weinhäupl, W. Schmid and R. Konrat, J. Biomol. NMR, 2013, 57(4), 327–331 CrossRef CAS PubMed.
-
(a) R. Lichtenecker, M. L. Ludwiczek, W. Schmid and R. Konrat, J. Am. Chem. Soc., 2004, 126(17), 5348–5349 CrossRef CAS PubMed;
(b) M. Fischer, K. Kloiber, J. Hauesler, K. Ledolter, R. Konrat and W. Schmid, ChemBioChem, 2007, 8(6), 610–612 CrossRef CAS PubMed;
(c) R. J. Lichtenecker, N. Coudevylle, R. Konrat and W. Schmid, ChemBioChem, 2013, 14(7), 818–821 CrossRef CAS PubMed;
(d) R. J. Lichtenecker, K. Weinhäupl, L. Reuther, J. Schörghuber, W. Schmid and R. Konrat, J. Biomol. NMR, 2013, 57(3), 205–209 CrossRef CAS PubMed.
-
(a) V. Viswanatha and V. J. Hruby, J. Org. Chem., 1979, 44(16), 2892–2896 CrossRef CAS; for a review of synthetic routes to label arenes, see:
(b) T. J. Gregson, J. M. Herbert and E. C. Row, J. Labelled Compd. Radiopharm., 2011, 54, 1–32 CrossRef CAS.
-
(a) A. Martins and M. Lautens, Org. Lett., 2008, 10(19), 4351–4353 CrossRef CAS PubMed; for a review of deuteration methods, see:
(b) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., 2007, 119, 7890–7911 (
Angew. Chem. Int. Ed.
, 2007
, 46
, 7744–7765
) CrossRef; and
(c) T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401–406 RSC.
-
P. E. Fanta, Org. Synth.Collect. Vol. IV, 1963, 844 Search PubMed.
- A. J. Bartlett, J. S. E. Holker, E. O'Brien and T. J. Simpson, J. Chem. Soc., Perkin Trans. 1, 1983, 667–670 RSC.
-
(a) W. J. Musliner and J. W. Gates Jr., J. Am. Chem. Soc., 1966, 88, 4271–4273 CrossRef CAS;
(b) R. A. W. Johnstone and P. J. Price, Tetrahedron, 1985, 41(12), 2493–2501 CrossRef CAS.
- K. Akira, H. Hasegawa and S. Baba, J. Labelled Compd. Radiopharm., 1995, 36(9), 845–853 CrossRef CAS.
-
(a) J. Raap, S. Nieuwenhuis, A. Creemers, S. Hexspoor, U. Kragl and J. Lugtenburg, Eur. J. Org. Chem., 1999, 2609–2621 CrossRef CAS;
(b) G. Billek, Monatsh. Chem., 1961, 92(2), 352–360 CrossRef CAS;
(c) G. Billek, Monatsh. Chem., 1961, 92(2), 343–351 CrossRef CAS;
(d) G. Billek, Monatsh. Chem., 1961, 92(2), 335–342 CrossRef CAS.
-
G. Billek, Org. Synth.Coll. Vol. 5, 1973, 627 Search PubMed.
- D. Mal, A. Jana, S. Ray, S. Bhattacharya, A. Patra and S. R. De, Synth. Commun., 2008, 38(22), 3937–3946 CrossRef CAS.
- T. Pirali, F. Zhang, A. H. Miller, J. L. Head, D. McAusland and M. F. Greaney, Angew. Chem., Int. Ed., 2012, 51(4), 1006–1009 CrossRef CAS PubMed.
- S. B. Waghmode, G. Mahale, V. P. Patil, K. Renalson and D. Singh, Synth. Commun., 2013, 43(24), 3272–3280 CrossRef CAS.
- K. Hanai, S. Kawai and A. Kuwae, J. Mol. Struct., 1991, 245, 21–27 CrossRef CAS.
-
(a) A. J. Wand, Nat. Struct. Biol., 2001, 8(11), 926–931 CrossRef CAS PubMed;
(b) A. G. Palmer, M. J. Grey and C. Wang, Methods Enzymol., 2005, 430–465 CAS;
(c) K. King Frederick, M. S. Marlow, K. G. Valentine and A. J. Wand, Nature, 2007, 448, 325–329 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR and 13C-NMR spectra of the target aromatic α-ketoacid sodium salts 1–3 and the synthetic intermediates. See DOI: 10.1039/c4ob01129e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a