 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDicyanobenzene and dicyanopyrazine derived X-shaped charge-transfer chromophores: comparative and structure–property relationship study†
L.
Dokládalová‡
a,
F.
Bureš
*a,
W.
Kuznik
bc,
I. V.
Kityk
c,
A.
Wojciechowski
c,
T.
Mikysek
d,
N.
Almonasy
a,
M.
Ramaiyan
a,
Z.
Padělková
e,
J.
Kulhánek
a and
M.
Ludwig
a
aInstitute of Organic Chemistry and Technology, University of Pardubice, Faculty of Chemical Technology, Studentská 573, Pardubice, 53210, Czech Republic. E-mail: filip.bures@upce.cz
bDepartment of Experimental Physics, University of Debrecen, Bem sq. 18/a, 4026, Debrecen, Hungary
cElectrical Engineering Department, Czestochowa University of Technology, Armii Krajowej 17, Czestochowa, 42201, Poland
dDepartment of Analytical Chemistry, University of Pardubice, Faculty of Chemical Technology, Studentská 573, Pardubice, 53210, Czech Republic
eDepartment of General and Inorganic Chemistry, University of Pardubice, Faculty of Chemical Technology, Studentská 573, Pardubice, 53210, Czech Republic
First published on 3rd June 2014
Abstract
A series of novel X-shaped push–pull compounds based on benzene-1,2-dicarbonitrile has been designed, synthesized and further investigated by X-ray analysis, electrochemistry, absorption and emission spectra, SHG experiment and quantum-chemical calculations. The obtained data were compared with those for isolobal 5,6-disubstituted pyrazine-2,3-dicarbonitriles. Structure–property relationships were elucidated. The extension, composition and planarization of the π-linker used as well as the electron-withdrawing ability of both dicyano-substituted acceptor units affect the linear and nonlinear properties of the target charge-transfer chromophores most significantly.
Introduction
Organic molecules based on the sp2/sp hybridized carbon/hetero atom scaffold end-capped with electron donors and acceptors represent an intensively investigated area of organic chemistry. In these molecules, intramolecular charge-transfer (ICT) from the donor (D) to the acceptor (A) via a π-conjugated system takes place and the D–π-A systems become polarized and gain dipolar character. Due to the ICT, push–pull D–π-A molecules possess distinct linear as well as nonlinear optical (NLO) properties. Charge-transfer chromophores have found widespread applications in modern branches of materials chemistry such as optoelectronics, data processing and storage devices, NLO bioimaging, organic light-emitting diodes (OLEDs), dye-sensitizing solar cells (DSSCs), semiconductors, switches, etc.1–3 Spatial arrangement of a push–pull system involves prevailing linear (D–π-A), quadrupolar (D–π-A–π-D or A–π-D–π-A) and octupolar/tripodal ((D-π)3-A or (A-π)3-D) systems or less common V- (ref. 4), Y- (ref. 5), H- (ref. 6) and X-shaped molecules. X-shaped chromophores based on tetrasubstituted ethenes (tetraethynylethenes, TEEs, and cyanoethynylethenes, CEEs) were introduced and extensively studied mainly by Diederich and co-workers.7,8 Besides the ethene central π-linker, the 1,2,4,5-tetrasubstituted benzene pattern represents another class of X-shaped chromophores. Benzene-derived X-shaped chromophores can conveniently be prepared from catechol, o-phenylenediamine or tetrahalogenated benzenes via functionalization with suitable electron donors (e.g. NR2 and OR groups) and acceptors (e.g. NO2 group and π-deficient heterocycles). Such molecules were mainly investigated for their unique absorption/emission properties and chelating abilities towards metal ions.9Recently, we have designed and synthesized X-shaped chromophores based on the pyrazine skeleton.10 Herein we would extend the concept of X-shaped chromophores with two electron withdrawing cyano groups saturated with two electron donors attached opposite to a six-membered (hetero)aromate. Thus, the previous series of pyrazines P1–P10 was completed with analogous push–pull chromophores B1–B10 featuring the benzene central π-system.
Both chromophore types possess a N,N-dimethylamino donor connected via systematically extended π-linkers that comprise a combination of up to three acetylene and 1,4-phenylene subunits. In contrast to polyenes and polyynes, (oligo)phenylene ethynylene charge-transfer chromophores possess generally higher thermal and (photo)chemical stability. 4,5-Disubstituted benzene-1,2-dicarbonitrile derivatives are a currently intensively investigated class of compounds due to their easy transformation to (sub)phthalocyanines. Peripherally substituted phthalocyanines found widespread applications in NLO,11 DSSCs,12 supramolecular assemblies,13 discotic liquid crystals14 and photodynamic therapy.15 Recently, Adachi et al. showed highly efficient OLEDs from delayed fluorescence that utilize similar push–pull dicyanobenzene derivatives functionalized with carbazole moieties.16

Results and discussion
Synthesis
The synthesis of target chromophores B1–B10 has been carried out in a modular manner starting from 4,5-diiodobenzene-1,2-dicarbonitrile 11, which can be prepared from commercially available phthalimide. In contrast to Terekhov et al., iodination of phthalimide with I2/oleum (30%) and one recrystallization from acetone afforded directly 4,5-diiodophthalimide in 61% yield without the need for Soxlet extraction and column chromatography.17 However, it should be noted that regioselective outcome and yield of the iodination step depend strongly on the concentration of the oleum used. Subsequent ammonolysis to 4,5-diiodophthaldiamide and dehydration with (CF3CO)2O/pyridine afforded compound 11 in 42% overall yield (see the ESI† for more details).17 Similarly to pyrazines P1–P10, benzene derivatives B1–B10 were obtained by Suzuki–Miyaura and Sonogashira cross-coupling reactions. N,N-Dimethylamino-substituted π-linkers with boronic acid (ester) function and terminal acetylene moiety were prepared from commercially available or known short π-linkers18 and their extension/alternation by cross-coupling reactions with [(4-iodophenyl)ethynyl]trimethylsilane.10,19 With the extended π-linkers and 4,5-diiodobenzene-1,2-dicarbonitrile 11 in hand, we have carried out twofold Suzuki–Miyaura cross-coupling reactions leading to target molecules B1, B3 and B4 in yields of 83, 87 and 79% (Scheme 1). These chromophores possess the π-linker connected directly to the dicyanobenzene moiety via a 1,4-phenylene unit. This twisted arrangement usually represents a barrier of efficient D–A interaction.10 Therefore, partial planarization of the π-linker has been achieved via introduction of an additional acetylene unit by Sonogashira reaction. The reaction of 11 with terminal acetylenes afforded target chromophores B2 and B5–B10 with the yields ranging from 62 to 92% (Scheme 2). All target compounds B1–B10 were isolated in similar yields as pyrazines P1–P10 and were purified by column chromatography and recrystallization from 1,2-dichloroethane/hexane. In contrast to compounds B7 and B8, pyrazine analogues P7 and P8 could not be prepared. In these cases, the final Sonogashira reactions were dominated by homocoupling of the starting acetylenes and pyrazine derivatives P7 and P8 were only detected in the crude reaction mixture as minor products. Due to the dipolar character and extended π-system of B1–B10, some target compounds are only sparingly soluble in halogenated solvents. However, their chemical structures have been confirmed by NMR, HR-MALDI-MS and IR spectra as well as elemental analysis and X-ray analysis.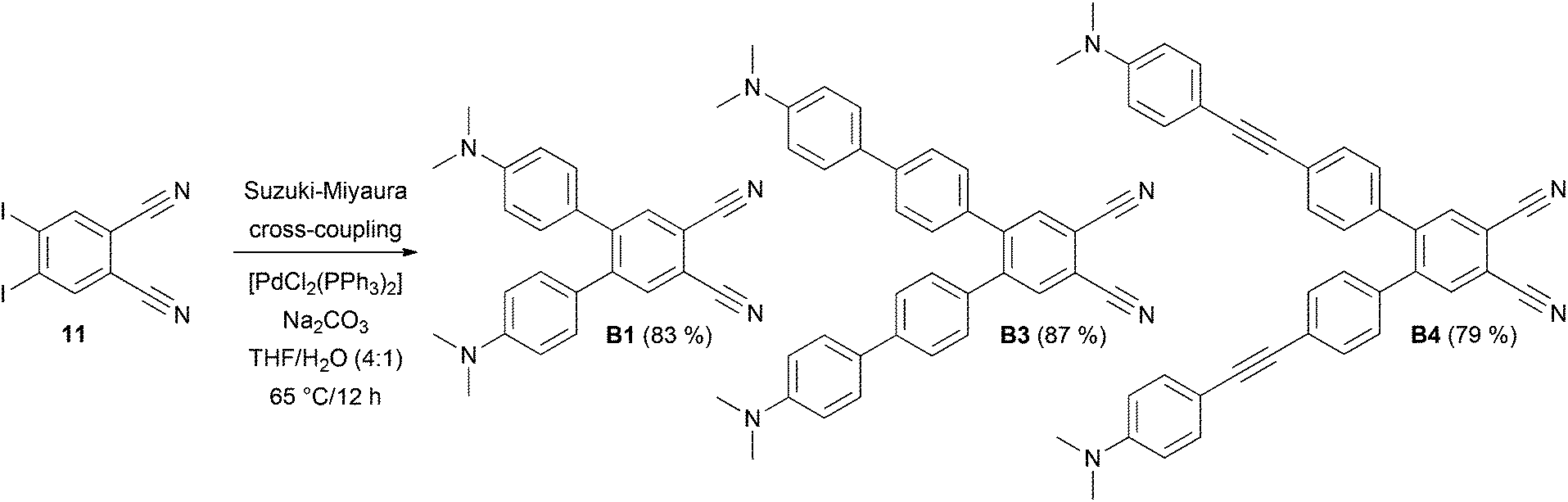 | ||
| Scheme 1 Suzuki–Miyaura reaction leading to chromophores B1, B3 and B4. | ||
 | ||
| Scheme 2 Twofold Sonogashira reaction leading to chromophores B2 and B5–B10. | ||
Crystallography
Slow diffusion of hexane into solution of chromophores B1 and B4 in CH2Cl2/CDCl3 afforded crystals suitable for X-ray analysis. Fig. 1(a/b) shows the ORTEP plots and side views of both molecules and confirms the proposed molecular structure. The ORTEP plot of previously crystallized pyrazine P1 has also been included as a reference (Fig. 1c).10 The spatial arrangement of the central dicyano acceptor moieties and the adjacent 1,4-phenylene units in B1, B4 and P1 can be evaluated by the dihedral angles ϕ1–ϕ6 as shown in Fig. 1 and Table 1. | ||
| Fig. 1 ORTEP representations and spatial arrangements of chromophores B1 (a), B4 (b) and P1 (c) measured at 150 K and the overlap wire model for isolobal chromophores B1/P1 (d). The thermal ellipsoids are shown at 50% probability with arbitrary spheres for H atoms. | ||
| Compd |
ϕ
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
ϕ
2a |
ϕ
3a |
ϕ
4a |
ϕ
5/ϕ6a |
δr |
I
6c |
|---|---|---|---|---|---|---|---|
| a The torsion angles ϕ1–6 are in deg. b The average quinoid characters δr of 1,4-phenylene and DMA units are given in Å. c The Bird index of the central benzene/pyrazine rings. | |||||||
| B1 | 7 | 2 | 2 | 3 | 51/45 | 0.011 | 89.9 |
| B4 | 10 | 5 | 4 | 4 | 48/42 | 0.010/0.013 | 88.9 |
| P1 | 25 | 18 | 13 | 7 | 30/29 | 0.027 | 82.2 |
As can be seen, both donor-substituted π-linkers attached on the benzene-1,2-dicarbonitrile acceptor moiety via a 1,4-phenylene unit are significantly twisted out of the chromophore molecular plane. Pyrazine P1 adopts a similar angular arrangement which is most likely caused by the steric repulsion of the ortho-hydrogens. Whereas in benzene derivatives B1/B4 are both 1,4-phenylene moieties twisted perpendicularly to the adjacent benzene-1,2-dicarbonitrile moiety and adopt mutually almost parallel arrangement with minimal ϕ1 = 7/10° and high torsion angles ϕ5/ϕ6 = 42–51°, the derivative P1 possesses these substituents twisted mainly above/below the pyrazine plane with relatively high both torsion angles ϕ1 = 25° and ϕ5/ϕ6 = 30/29°. This arrangement results in substantial deformation of the pyrazine ring compared to almost planar benzene rings (see ϕ2–4 in Table 1). The differences in spatial arrangements of the C4/C5 and C5/C6 substituents in benzene B1/B4 and pyrazine P1 derivatives are visualized in the side views (Fig. 1). The overlap wire model for isolobal compounds B1/P1 is also provided in Fig. 1d.
Bond length alternation (BLA) in the N,N-dimethylanilino (DMA) and 1,4-phenylene rings can easily be evaluated by the quinoid character (δr). Whereas in benzene, δr is equal to 0, in a fully quinoid ring, δr would be on the order of 0.100–0.120 Å.20 A comparison with known CT chromophores bearing DMA moieties such as CEEs (δr up to 0.070 Å)8 and TEEs (δr up to 0.025 Å)7 places the nonplanar benzene and pyrazine chromophores B1/B4 and P1 below/at the level of donor-substituted tetraethynylethenes. Further comparison of the δr values for B1/P1 shown in Table 1 clearly indicates higher BLA in the pyrazine chromophore P1. Estimation of the aromaticity of both central benzene and pyrazine rings by the calculation of the Bird index (I6)21 provides similar insight. Whereas the I6 values of unsubstituted benzene/pyrazine are 100/88.8, the calculated values for B1, B4 and P1 decrease in the same order of 89.9→88.9→82.2 as a consequence of the π-system extension and replacement of the central benzene by the pyrazine ring.
1H NMR and IR spectroscopy
We analysed the 13C NMR spectra of all B and P chromophores measured in CDCl3 (100 MHz) as well as IR spectra measured neat using an HATR adapter. Electron saturation of the CN group indicated by 13C NMR chemical shift and frequency of vibrational stretching would serve as good indicators of the ICT extent (Table 2).![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group
N group
| Compd |
δ(CN)a |
ν(CN)b |
|---|---|---|
| a 13C NMR chemical shift of the CN group given in ppm – measured in CDCl3 at 100 MHz. b Frequency of the vibrational stretching of the CN group given in cm−1 – measured by IR (neat, HATR). c Not observed due to sparing solubility in CDCl3. | ||
| B1/P1 | 116.27/114.33 | 2223/2230 |
| B2/P2 | 112.59/112.17 | 2177/2165 |
| B3/P3 | 114.19/113.68 | 2224/2340 |
| B4/P4 | 114.62/113.43 | 2202/2194 |
| B5/P5 | 113.83/c | 2200/2182 |
| B6/P6 | 114.29/c | 2206/2189 |
| B7/— | /— | 2203/— |
| B8/— | 114.41/— | 2202/— |
| B9/P9 | 114.21/c | 2202/2188 |
| B10/P10 | 114.47/c | 2202/2190 |
The chemical shift of C atoms strongly depends on hybridization and the electron density at the nucleus. The higher the electron density, the more shielding occurs and an upfield shift will be observed.8c The data in Table 2 clearly demonstrate that 13C NMR shifts of the CN group in pyrazine series of compounds (P1–P4) are generally upfielded as a consequence of higher saturation of the CN group compared to compounds B1–B4. Introduction of more electronegative acetylene units as in chromophores B6–B10 reduces the electron saturation of the CN group, which is then engaged similarly in the ICT, and the chemical shifts became steady at around 114 ppm. However, when comparing the particular compounds B1/B2 or P1/P2 that differ in the type of connection of the π-linker to the acceptor moiety (without/with an additional acetylene unit), the chemical shifts of the latter were upfielded by 3.68 and 2.16 ppm. This observation can be attributed to planarization of the entire π-system and thus resulting better D–A interaction in B2/P2. The chemical shifts δ(CN) = 112–116 ppm measured for the dicyanobenzene and dicyanopyrazine acceptor moieties are slightly downfielded compared to donor substituted CEEs (δ(CN) = 108–115 ppm)8c which further confirms the aforementioned trends seen by the BLA.
The frequency of the stretching vibration of the CN group showed very similar trends, as seen by the NMR. Namely, the CN stretching appeared generally at lower frequencies for pyrazine series. Extension and (partial) planarization of the π-linker (e.g.1vs.2 or 3vs.4) shifted the CN wavenumber to higher energy and became steady at around 2200/2190 cm−1.
Electrochemistry
Electrochemical measurements of all chromophores B1–B10 and P1–P10 (ref. 10) were carried out under standard conditions (N,N-dimethylformamide containing 0.1 M Bu4NPF6) in a three electrode cell by cyclic voltammetry (CV) and rotating disk voltammetry (RDV). The working electrode was a platinum disc (2 mm in diameter) for CV and RDV experiments. As the reference and auxiliary electrodes were used a saturated calomel electrode (SCE) separated by a bridge filled with a supporting electrolyte and a Pt wire, respectively. The acquired data are summarized in Table 3, and representative CV diagrams of chromophores B2 and B10 are shown in the ESI (Fig. S1–S2†).| Compd |
EoF(ox1)a [V] |
EoF(red1)a [V] |
ΔEb [V] |
|---|---|---|---|
| a E oF = (Ep,c + Ep,a)/2 where Ep,c and Ep,a correspond to the cathodic and anodic peak potentials, respectively. b ΔE = EoF(ox1) − EoF(red1). | |||
| B1/P1 | +0.92/+1.02 | −1.62/−1.24 | 2.54/2.26 |
| B2/P2 | +0.93/+1.04 | −1.28/−0.87 | 2.21/1.91 |
| B3/P3 | +0.80/+0.81 | −1.48/−1.05 | 2.28/1.86 |
| B4/P4 | +0.84/+0.83 | −1.41/−0.97 | 2.25/1.80 |
| B5/P5 | +0.81/+0.84 | −1.14/−0.74 | 1.95/1.58 |
| B6/P6 | +0.89/+0.85 | −1.07/−0.69 | 1.96/1.54 |
| B7/— | +0.78/— | −1.32/— | 2.10/— |
| B8/— | +0.78/— | −1.09/— | 1.87/— |
| B9/P9 | +0.84/+0.84 | −1.10/−0.70 | 1.94/1.54 |
| B10/P10 | +0.84/+0.83 | −1.03/−0.67 | 1.87/1.50 |
All chromophores 1–10 in both series B and P showed two close, mostly reversible, one-electron oxidation processes (differences of about 100 mV measured by CV) that were, in most cases, merged into one two-electron wave, as indicated by RDV. This electrochemical behavior reflected the presence of two equivalent N,N-dimethylamino donor groups appended to the dicyanobenzene/pyrazine acceptor. The first oxidation potentials of chromophores in both series B and P range from +0.93 to +0.78 V and +1.02 to 0.81 V, respectively. As can be seen, extension of the π-conjugated path affects the first reduction potentials only negligibly. However, the EoF(ox1) values slightly decrease with the extension of the π-system as a result of the gradual N,N-dimethylamino donor isolation. From chromophores B3/P3 onwards, the first oxidation potentials were concentrated in the range of about +0.80 V, and therefore, the N,N-dimethylamino donor in chromophores 4–10 can be considered as similarly involved in the ICT.
In contrast to the first oxidations, the first reductions of chromophores in the series B and P were observed in a wide range from −1.62 to −1.03 and −1.24 to −0.67 V, respectively. The difference between the first reduction potentials of the particular chromophores in both B and P series ranges from 0.36 to 0.44 V. This reflects a higher withdrawing ability of the dicyanopyrazine moiety over the dicyanobenzene caused by the presence of two electronegative nitrogen atoms. However, when considering the dicyanobenzene/pyrazine acceptor as the main reduction center and the same electron nature and number of appended donors, the observed differences within the particular series B or P must be elucidated as a consequence of the π-linker. The first reduction potentials of the chromophores in the series B or P gradually decrease and become steady at about −1.10 and −0.70 V. When going from the chromophore B1/P1 (one 1,4-phenylene moiety as a π-linker) to B3/P3 (biphenyl π-linker) and B4/P4 (phenylethynylphenyl π-linker), the first reduction potentials decrease from −1.62/−1.24 to −1.48/−1.05 and −1.41/−0.97 V as a result of the π-linker gradual extension. However, chromophores B2/P2 bearing a shorter π-linker showed more positively shifted EoF(red1) up to −1.28/−0.87 V than extended chromophores B3/P3 and B4/P4. This can be attributed to partial extension of the π-linker by an acetylene unit connected directly to the acceptor and, in particular, to planarization of the entire π-systems. A similar effect can be observed when comparing chromophores B3/P3 and B5/P5 or B4/P4 and B6/P6. Hence, a separation of the π-linker from the dicyanobenzene/pyrazine acceptor by the linear ethynyl spacer plays a very crucial role and affects the electrochemical behavior of X-shaped chromophores considerably. In contrast to this, the first reduction of the chromophore B7 (EoF(red1) = −1.32 V) is significantly impeded by the propeller-shape of the terphenyl linker. Except B7, chromophores B6/P6–B10/P10 showed almost constant first reduction potentials which implies that a combination of two/three 1,4-phenylene moieties in conjunction with acetylene units caused complete isolation of the N,N-dimethylamino donor and the dicyanobenzene/pyrazine acceptor.
The corresponding electrochemical gaps ΔE (Table 3) are mainly affected by the variation in the first reduction potentials and, therefore, mimic the same trends as seen for EoF(red1). The lowest electrochemical gaps were recorded for chromophores B10/P10 with the largest and fully planar π-linkers (see also the energy level diagram below).
UV/Vis spectroscopy
Optical properties of all chromophores 1–10 were studied by absorption and emission spectra. The positions of the longest-wavelength absorption and fluorescence maxima λAmax and λFmax as well as the molar absorption coefficients ε and the fluorescence quantum yields qF are given in Table 4. Representative UV/Vis spectra of selected chromophores are shown in Fig. 2; for complete spectra listing see the ESI (Fig. S3–S4†). All spectra were measured in CH2Cl2.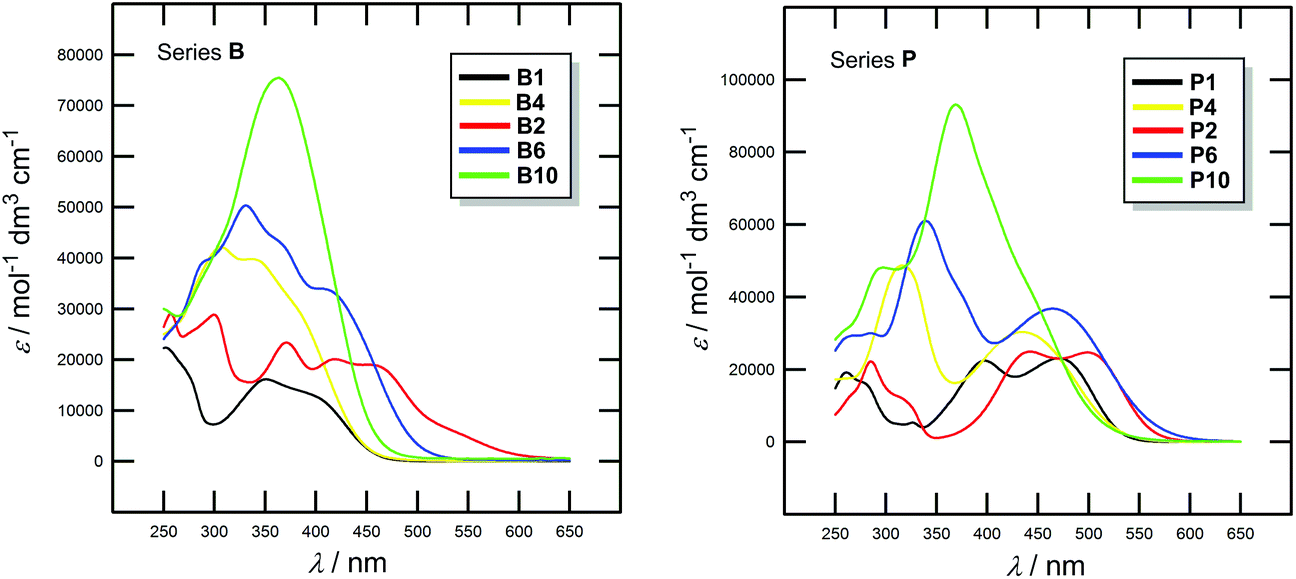 | ||
| Fig. 2 Representative UV/Vis absorption spectra of chromophores B1/P1, B4/P4 and B2/P2, B6/P6, B10/P10 measured in CH2Cl2 (10−5 M). | ||
| Compd | λ Amax [nm(eV)] | ε [103 mol−1 dm3 cm−1] | λ Fmax [nm (eV)] | q F |
|---|---|---|---|---|
| a Observed as a shoulder. b Emission maxima/fluorescence quantum yields of the chromophores in series B. | ||||
| B1/P1 | 410(3.02)a/471(2.63) | 12.82/23.18 | 548b | 0.79b |
| B2/P2 | 458(2.71)a/499(2.48) | 18.98/24.73 | 589b | 0.76b |
| B3/P3 | 380(3.26)a/425(2.92) | 19.05/25.83 | 640b | 0.52b |
| B4/P4 | 378(3.28)a/433(2.86) | 30.29/30.38 | 658b | 0.10b |
| B5/P5 | 422(2.94)a/465(2.67) | 26.66/35.52 | — | — |
| B6/P6 | 424(2.92)/466(2.66) | 33.99/36.81 | — | — |
| B7/— | 335(3.70)/— | 53.73/— | — | — |
| B8/— | 345(3.59)/— | 75.96/— | — | — |
| B9/P9 | 342(3.63)/354(3.50) | 84.51/87.26 | — | — |
| B10/P10 | 362(3.43)/371(3.34) | 75.55/93.31 | — | — |
The absorption spectra of chromophores in series B and P showed longest-wavelength absorption maxima (CT-bands) appearing within the range of 335–458 and 354–499 nm, respectively. The absorption spectra of representative B and P chromophores shown in Fig. 2 are very similar. However, the absorption maxima of P chromophores are generally bathochromically shifted, which again confirms the previous conclusion on higher electron withdrawing character of the dicyanopyrazine moiety. Chromophores B1/P1 and B2/P2 showed multiple CT-bands which reflects complex conjugated pathways between the particular N,N-dimethylamino donor and cyano acceptor. The spectra of chromophores B feature more vibronic structure than those of chromophores P. When going from chromophore B1/P1 to B2/P2, thus extending and planarizing the π-linker by one acetylene unit, the absorption maxima were shifted by 48 and 28 nm (Table 4), respectively. However, moving the acetylene unit between two 1,4-phenylene units as in B4/P4 or further π-linker extension as in B6/P6 and B10/P10 led to disappearance of the CT-band and a pronounced hypsochromic shift. This is in accordance with the observation made by Nielsen et al.22 that an extension of the π-system by more nonplanarly arranged 1,4-phenylene units led to a blue-shift of the CT-band measured in a solvent. However, this is also in contrast to gas phase measurement excluding solvation and H-bonding in which a systematic red-shift was observed. Hence, from B7 onwards, the CT-bands were observed as hardly distinguishable shoulders and were merged with the bands appearing at around 350 nm with rising intensity. These bands probably correspond to the transition between the N,N-dimethylamino donor and the extended electronegative π-linker. Thus, insertion of three 1,4-phenylene units in combination with one or more acetylene units can completely quench D–A interaction and none or very weak CT-band is observed. These observations are in accordance with the aforementioned electrochemical conclusions.
In contrast to all N,N-dimethylamino-substituted π-linkers, which showed emission properties,19 only less extended chromophores B1–B4 showed fluorescent behaviour (Table 4, the ESI Fig. S5†). Moreover, the pyrazine chromophores did not show any fluorescence at all. Thus, extension of the π-system or attachment of a stronger acceptor moiety caused emission disappearance and a gradual reduction of the fluorescence quantum yield.
Calculations and NLO properties
Quantum-chemical calculations and simulation of NLO properties of all target chromophores were performed at the semi-empirical level of theory with the AM1 method and with the time-dependent Hartree–Fock (TDHF) calculations, respectively. All the calculations were performed using the PCGAMESS package. The calculated data such as the ground stated dipole moment μ, energies of the HOMO and the LUMO and their differences ΔE as well as β coefficients are presented in Table 5.| Compd | μ [D] | E HOMO [eV] | E LUMO [eV] | ΔEHOMO–LUMO [eV] | β (AM1) [esu × 10−30] | β (exp.) [pm V−1] |
|---|---|---|---|---|---|---|
| a Below noise level (see the ESI). | ||||||
| B1/P1 | 8.56/9.91 | −8.28/−8.40 | −0.92/−1.43 | 7.36/6.97 | 245/349 | —a/0.53 |
| B2/P2 | 9.74/11.89 | −8.22/−8.33 | −1.07/−1.75 | 7.15/6.58 | 649/942 | 0.9/1.1 |
| B3/P3 | 9.67/9.07 | −8.15/−8.28 | −1.18/−1.78 | 6.97/6.50 | 375/408 | —a/0.6 |
| B4/P4 | 10.43/10.69 | −8.20/−8.23 | −1.31/−1.69 | 6.89/6.54 | 681/787 | —a/1.1 |
| B5/P5 | 10.77/11.49 | −8.21/−8.32 | −1.47/−1.92 | 6.74/6.40 | 809/956 | 1.2/1.2 |
| B6/P6 | 11.48/11.3 | −8.16/−8.38 | −1.55/−2.01 | 6.61/6.37 | 1263/1444 | 3.6/1.4 |
| B7/— | 10.9/— | −8.18/— | −1.53/— | 6.65/— | 867/— | 2.1/— |
| B8/— | 11.3/— | −8.16/— | −1.61/— | 6.55/— | 1 268/— | 2.8/— |
| B9/P9 | 10.89/11.38 | −8.11/−8.15 | −1.56/−2.00 | 6.55/6.15 | 1218/1484 | 3.2/3.0 |
| B10/P10 | 11.78/11.27 | −8.11/−8.29 | −1.64/−2.07 | 6.47/6.22 | 1701/1912 | 4.9/3.1 |
The first oxidation/reduction potentials measured in DMF were recalculated23 to the corresponding HOMO/LUMO energies and gaps (see also the ESI†) and were further compared with the AM1 calculated values. Although both HOMO−LUMO gaps differ in their absolute values (differences of 4.6–4.9 eV), they tightly correlate in both series of compounds (see the ESI Fig. S6–S7†). Hence, even though they are very time-efficient, the used semi-empirical calculations are obviously capable of properly describing the trends seen by the electrochemical measurements and can be considered as a reasonable tool for the electronic property description of chromophores 1–10.
Fig. 3a shows the energy level diagram derived from the electrochemical measurements. This diagram nicely visualizes the aforementioned trends, namely: (i) the principal changes are observed in the LUMO, (ii) the LUMO is significantly lowered when going from series B (in black) to P (in red), (iii) the HOMO remains practically unchanged throughout the whole series of compounds, (iv) from chromophores B6/P6 onwards, the LUMOs as well as the HOMO−LUMO gaps are steady, (v) although it does not contain a large π-linker, the chromophore B2 showed a significantly lowered LUMO level as a result of its planar arrangement, (vi) in contrast, the chromophore B7 showed significantly raised LUMO due to its twisted π-linker, and (vii) the lowest HOMO−LUMO gaps were measured/calculated for chromophores 9 and 10 that feature the longest π-linkers. As expected, the visualizations of frontier molecular orbitals for representative pairs of chromophores B2/B10 and P2/P10 showed acceptor-centered LUMO and donor-centered HOMO (Fig. 3b; for complete listing see the ESI Fig. S20–29†). A significant charge-separation is being observed when extending the π-system.
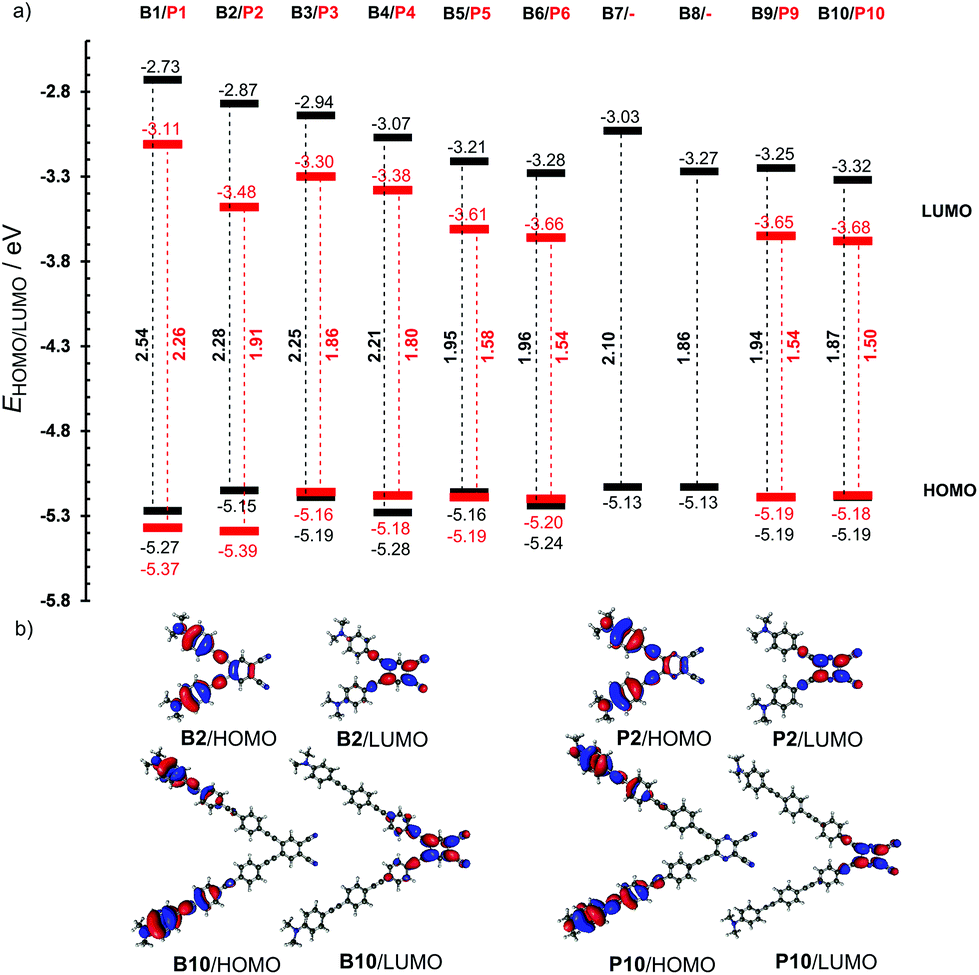 | ||
| Fig. 3 Energy level diagram (a) and representative HOMO and LUMO localizations in B2/B10 and P2/P10 (b). | ||
The second-order optical nonlinearities of the newly synthesized chromophores B1–B10 were also determined experimentally by investigation of the Second Harmonic Generation (SHG). The SHG experiment was performed with powdered samples that were oriented by dc-electric field. The studies were performed for the angles that gave the maximal SHG output. A Nd-YAG laser (pulse duration 15 ns) was used as a fundamental laser beam. Powdered BiB3O6 microcrystallites with known parameters of the second order susceptibilities were used as the reference samples. The experimental β coefficients along with the theoretical values are summarized in Table 5. Both hyperpolarizabilities showed sufficiently good correlation (see the ESI Fig. S8–S9†). Chromophores bearing short and nonplanar arrangement of the π-linkers as in 1, 3 and 4 showed weak (P series) or no (B series) SHG behavior (see also the SHG readouts in the ESI Fig. S10–S19†). From the series of compounds 1–5, bearing up to two 1,4-phenylene and one acetylene units, chromophores B2/P2 are exceptional (β(exp) = 0.9 and 1.1 pm V−1). Although B2/P2 contains only one 1,4-phenylene and one acetylene unit, they showed almost the same nonlinear optical response as B5/P5 having one additional 1,4-phenylene moiety (β(exp) = 1.2 pm V−1). This must be elucidated as an effect of its planar arrangement. The effect of the planar π-system arrangement can be further demonstrated by chromophores B6 and B10 that showed strong SHG behavior (β(exp) = 3.6 and 4.9 pm V−1), which can be attributed to their extended and fully planarized π-linkers (two/three 1,4-phenylene units separated by two/three triple bonds). Chromophores P1–P5, bearing the short π-linker and the stronger dicyanopyrazine acceptor, showed generally higher nonlinearities than isolobal B1–B5. However, from B6/P6 onwards, the impact of the stronger acceptor diminished or even opposite and higher nonlinearities were measured for chromophores in the B series. Hence, as the donor and the acceptor of a given push–pull molecule become completely isolated by a large π-linker, the NLO response is dictated not solely by the ICT but also by the size of the molecule (the number of polarizable electrons).24
Conclusions
A new series of X-shaped push–pull molecules based on 4,5-disubstituted benzene-1,2-dicarbonitrile has been synthesized. Three compounds B1, B3 and B4 were obtained by twofold Suzuki–Miyaura reactions, and seven target chromophores B2 and B5–B10 with the π-linker separated from the acceptor by an additional triple bond were obtained by Sonogashira cross-coupling. All compounds were compared with isolobal chromophores P1–P10 based on 5,6-disubstituted pyrazine-2,3-dicarbonitrile and subsequent structure–property relationships were further evaluated. X-ray analysis revealed different spatial arrangements of both acceptor moieties and the appended N,N-dimethylamino-substituted π-linkers. The dicyanopyrazine core showed significant out of plane deformation while the dicyanobenzene remained almost fully planar. The extent of the ICT has been studied by BLA (δr) and the Bird index (I6). Both quantities indicated higher ICT in the P series of compounds. This has been further confirmed by the 13C NMR and IR spectra that showed higher electron saturation of the CN group in dicyanopyrazine compounds. 13C NMR chemical shift and the frequency of the stretching vibration of the CN group have also been affected by the length, composition and spatial arrangement of the π-linker. Electrochemical measurements carried out by the CV and RDV confirm the observations made above. Principal changes were recorded in the first reduction potential. Significantly positively shifted first reduction potentials have been observed for compounds with fully planar π-linkers connected to the acceptor moiety via triple bonds (e.g.B2/P2, B6/P6 and B10/P10). All chromophores from B6/P6 onwards showed almost constant first reduction potentials which implies that a combination of two/three 1,4-phenylene moieties in conjunction with acetylene units caused complete isolation of the N,N-dimethylamino donor and the dicyanobenzene/pyrazine acceptor. Optical properties of the studied chromophores are dominated by one or more CT-bands, which reflects multiple conjugated pathways between each N,N-dimethylamino and cyano groups. In general, chromophores in the series P showed bathochromically shifted longest-wavelength absorption maxima which reflect higher electron withdrawing character of the dicyanopyrazine moiety. The aforementioned structural changes in particular chromophores affect also their absorption spectra. Extension of the π-system and isolation of the D and A parts of the X-shaped push–pull molecules led to a hypsochromic shift of the CT-bands that are, from B7/P6 onwards, observed as a joint peak at about 350 nm with large molar absorption coefficients. In contrast to P1–P4, chromophores B1–B4 showed also fluorescent properties that gradually decrease with the extension of the π-system.Quantum-chemical calculations further confirmed the experimental data. In general, from the experiments and calculations carried out for X-shaped molecules 1–10 we can conclude the following:
• replacement of the dicyanobenzene with the dicyanopyrazine acceptor affects the optoelectronic properties most significantly (tuning by the LUMO)
• the HOMO of 1–10 remained almost unchanged independently of the size and structure of the π-linker
• the HOMO is localized on the peripheral donors while the LUMO is placed on the acceptor units
• the LUMO can be further lowered by planarization of the entire π-system (e.g.B2vs.B7)
• a position of the acetylene unit within the π-linker plays also an important role (e.g.B4/P4vs.B5/P5)
• π-linker bearing two acetylene units in combination with two 1,4-phenylene moieties completely isolates A and D parts of X-shaped push–pull molecules
• the lowest HOMO−LUMO gap and the highest nonlinearities have been measured/calculated for molecules B9/P9 and B10/P10 with the largest π-system
• planar molecules (e.g.B2, B6 and B10) delivered a strong SHG response
• first-order hyperpolarizability β depends not only on the extent of the ICT but also on the number of polarizable electrons (length of the π-system).
In view of the current interest in push–pull systems and their wide applications mainly in NLO and DSSC (both cyano groups can be hydrolysed to well-anchoring carboxylic acid functions),25 we believe that this structure–property study on X-shaped molecules would serve as a useful guideline.
Experimental section
For general information and synthesis of 11 see the ESI.† Full spectral characterization of pyrazine chromophores P1–P10 is given in our previous publication.10General procedure for Suzuki–Miyaura cross-coupling
4,5-Diiodobenzene-1,2-dicarbonitrile (152 mg, 0.4 mmol) and an appropriate boronic acid or its ester (0.84 mmol, 2.1 eq.) were dissolved in a mixture of THF–H2O (20 ml, 4:1). Argon was bubbled through the solution for 15 min whereupon [PdCl2(PPh3)2] (28 mg, 0.04 mmol) and Na2CO3 (89 mg, 0.84 mmol) were added and the reaction mixture was stirred at 65 °C for 12 h. The reaction was diluted with water (50 ml) and extracted with CH2Cl2 (2 × 50 ml). The combined organic extracts were dried (Na2SO4), the solvents were evaporated in vacuo and the crude product was purified by column chromatography (SiO2; indicated solvent system).
General procedure for Sonogashira cross-coupling
4,5-Diiodobenzene-1,2-dicarbonitrile (152 mg, 0.4 mmol) and an appropriate terminal acetylene (0.84 mmol, 2.1 eq.) were dissolved in THF (20 ml) and Et3N (5 ml). Argon was bubbled through the solution for 15 min whereupon [PdCl2(PPh3)2] (28 mg, 0.04 mmol) and CuI (8 mg, 0.04 mmol) were added and the reaction mixture was stirred at 60 °C for 24 h. The solvents were evaporated in vacuo and the crude product was purified by column chromatography (SiO2; indicated solvent system).:1). 1H-NMR (400 MHz, 25 °C, CDCl3): δH = 7.80 (s, 2H, 2 × ArH), 7.46 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 6.66 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 3.03 (s, 12H, 2 × N(CH3)2). 13C-NMR (100 MHz, 25 °C, CDCl3): δC = 151.19, 135.71, 133.68, 131.05, 115.49, 112.59, 111.92, 108.23, 102.91, 85.51, 40.32. IR (HATR): νmax/cm−1 = 2920, 2177 (CN), 1605, 1360, 1222, 1037, 808. HR-FT-MALDI-MS (DHB) m/z: 414.1826 (M+), C28H22N4+ requires 414.1839. Anal. Calcd for C28H22N4 (414.51): C 81.13, H 5.35, N 13.52; Found C 81.04, H 5.54, N 13.31.
:1). 1H–NMR (400 MHz, 25 °C, CDCl3): δH = 7.91 (s, 2H, 2 × ArH), 7.60 (s, 8H, 4 × Ph), 7.54 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 6.80 (d, 3J(H,H) = 8.4 Hz, 4H; 2 × Ph), 3.01 (s, 12H, 2 × N(CH3)2). 13C-NMR (100 MHz, 25 °C, CDCl3): δC = 150.61, 142.93, 136.22, 132.7, 131.13, 127.91, 127.53, 126.31, 118.86, 115.10, 113.83, 112.82, 101.43, 86.33, 40.64. IR (HATR): νmax/cm−1 = 2880, 2200 (CN), 1595, 1441, 1360, 1222, 1092, 811. HR-FT-MALDI-MS (DHB) m/z: 566.2450 (M+), C40H30N4+ requires 566.2465. Anal. Calcd for C40H30N4 (566.71): C 84.78, H 5.34, N 9.89; Found C 84.38, H 5.19, N 9.66.
:1). 1H-NMR (400 MHz, 25 °C, CDCl3): δH = 7.91 (s, 2H, 2 × ArH), 7.53–7.48 (m, 8H, 4 × Ph), 7.43–7.40 (m, 4H; 2 × Ph), 6.66 (d, 3J(H,H) = 8.4 Hz, 4H; 2 × Ph), 3.00 (s, 12H, 2 × N(CH3)2). 13C-NMR (100 MHz, 25 °C, CDCl3): δC = 150.64, 143.40, 136.35, 133.15, 132.11, 131.62, 131.00, 126.46, 120.22, 114.98, 114.29, 112.00, 100.86, 94.56, 87.41, 87.16, 40.39. IR (HATR): νmax/cm−1 = 3004, 2206 (CN), 1608, 1525, 1356, 1222, 1130, 846, 814. HR-FT-MALDI-MS (DHB) m/z: 614.2470 (M+), C44H30N4+ requires 614.2465. Anal. Calcd for C44H30N4 (614.75): C 85.97, H 4.92, N 9.11; Found C 85.50, H 5.01, N 9.21.
:1). 1H-NMR (400 MHz, 25 °C, CDCl3): δH = 7.95 (s, 2H, 2 × ArH), 7.70–7.66 (m, 16H, 8 × Ph), 7.55 (d, 3J(H,H) = (9.2 Hz, 4H; 2 × Ph), 6.81 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 3.01 (s, 12H, 2 × N(CH3)2). 13C-NMR (100 MHz, 25 °C, CDCl3): δC = 136.20, 135.73, 135.48, 133.73, 132.76, 127.62, 127.30, 40.08 (11 signals are missing). IR (HATR): νmax/cm−1 = 3003, 2203 (CN), 1652, 1418, 1361, 1222, 1092, 809. HR-FT-MALDI-MS (DHB) m/z: 718.3088 (M+), C52H38N4+ requires 718.3091. Anal. Calcd for C52H38N4 (718.90): C 86.88, H 5.33, N 7.79; Found C 86.51, H 5.22, N 7.62.
:1). 1H-NMR (400 MHz, 25 °C, CDCl3): δH = 7.93 (s, 2H, 2 × ArH), 7.56–7.51 (m, 20H, 10 × Ph), 6.80 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 3.00 (s, 12H, 2 × N(CH3)2). 13C-NMR (100 MHz, 25 °C, CDCl3): δC = 150.45, 136.34, 132.31, 132.13, 131.99, 130.93, 127.84, 126.21, 125.65, 120.98, 120.14, 114.90, 114.41, 112.89, 100.56, 93.27, 89.16, 87.30, 40.71 (2 signals are missing). IR (HATR): νmax/cm−1 = 3004, 2202 (CN), 1590, 1418, 1361, 1221, 1093, 838, 812. HR-FT-MALDI-MS (DHB) m/z: 766.3062 (M+), C56H38N4+ requires 766.3091. Anal. Calcd for C56H38N4 (766.95): C 87.70, H 4.99, N 7.31; Found C 86.95, H 5.10, N 7.38.
:1). 1H-NMR (400 MHz, 25 °C, CDCl3): δH = 7.93 (s, 2H, 2 × ArH), 7.65 (br s, 8H, 4 × Ph), 7.57 (br s, 8H, 4 × Ph), 7.41 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 6.66 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 2.99 (s, 12H, 2 × N(CH3)2). 13C-NMR (100 MHz, 25 °C, CDCl3): δC = 150.40, 142.24, 138.70, 136.33, 133.69, 132.99, 132.73, 131.03, 127.34, 127.04, 124.32, 120.62, 114.93, 114.21, 112.01, 109.96, 100.76, 92.39, 87.33, 86.65, 40.41. IR (HATR): νmax/cm−1 = 2962, 2202 (CN), 1592, 1516, 1357, 1222, 1093, 821. HR-FT-MALDI-MS (DHB) m/z: 766.3121 (M+), C56H38N4+ requires 766.3091. Anal. Calcd for C56H38N4 (766.95): C 87.70, H 4.99, N 7.31; Found C 87.51, H 5.28, N 7.08.
:1). 1H-NMR (400 MHz, 25 °C, CDCl3): δH = 7.93 (s, 2H, 2 × ArH), 7.55 (br s, 8H, 4 × Ph), 7.48 (br s, 8H, 4 × Ph), 7.40 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 6.65 (d, 3J(H,H) = 8.8 Hz, 4H; 2 × Ph), 2.99 (s, 12H, 2 × N(CH3)2). 13C-NMR (100 MHz, 25 °C, CDCl3): δ = 150.48, 136.36, 133.04, 132.16, 132.04, 131.78, 131.43, 130.89, 125.29, 124.93, 121.61, 121.26, 114.88, 114.47, 112.00, 109.74, 100.43, 93.45, 92.79, 87.43, 87.38, 40.41 (1 signal is missing). IR (HATR): νmax/cm−1 = 2920, 2202 (CN), 1609, 1591, 1523, 1360, 1122, 836, 818. HR-FT-MALDI-MS (DHB) m/z: 814.3066 (M+), C60H38N4+ requires 814.3091. Anal. Calcd for C60H38N4 (814.99): C 88.43, H 4.70, N 6.87; Found C 88.68, H 4.48, N 7.02.
Computational procedure
All calculations were performed with a semi-empirical AM1 method implemented in the PCGAMESS package.27 Computational files were prepared and interpreted with the aid of GABEDIT 2.4.6 (ref. 28) including the preparation of graphics.Acknowledgements
This research was supported by the Czech Science Foundation (P106/12/0392). L. D. and F. B. are indebted to the Technology Agency of the Czech Republic (TE01020022, Flexprint). M.R. and F. B./M. L. are indebted to the Ministry of Education, Youth and Sports of the Czech Republic (CZ.1.07/2.3.00/30.0021 and LG13053). W. K. is grateful to the Balassi Institute for financial support.Notes and references
- P. N. Prasad and D. J. Williams, Introduction to Nonlinear Optical Effects in Organic Molecules and Polymers, Wiley, New York, 1991 Search PubMed.
- For general reviews on NLO active compounds see: (a) S. R. Marder, ed., Special issue on “Nonlinear optics”, J. Mater. Chem, 2009, 19, 7392–7566 Search PubMed; (b) D. R. Kanis, M. A. Rathner and T. J. Marks, Chem. Rev., 1994, 19, 195–242 CrossRef; (c) J. L. Brédas, C. Adant, P. Tackx and A. Persoons, Chem. Rev., 1994, 94, 243–278 CrossRef; (d) L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS PubMed; (e) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- For general reviews on organic π-conjugated materials and their applications, see: (a) S. R. Forrest and M. E. Thompson, ed., Special issue on “Organic electronics and optoelectronics”, Chem. Rev., 2007, 107, 923–1386 Search PubMed; (b) R. D. Miller and E. A. Chandross, ed., Special issue on “Materials for electronics”, Chem. Rev., 2010, 110, 1–574 Search PubMed; (c) C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed; (d) Y. Ohmori, Laser Photonic Rev., 2009, 4, 300–310 Search PubMed; (e) B. J. Coe, Chem. – Eur. J., 1999, 5, 2464–2471 CrossRef CAS.
- (a) M. A. Ramírez, A. M. Cuadro, J. Alvarez-Builla, O. Castaño, J. L. Andrés, F. Mendicuti, K. Clays, I. Asselberghs and J. J. Vaquero, Org. Biomol. Chem., 2012, 10, 1659–1669 RSC; (b) M. A. Ramirez, T. Cañeque, A. M. Cuadro, F. Mendicuti, K. Clays, I. Asselbergh and J. J. Vaquero, ARKIVOC, 2011, iii, 140–155 Search PubMed.
- (a) J. Kulhánek and F. Bureš, Beilstein J. Org. Chem., 2012, 8, 25–49 CrossRef PubMed; (b) J. Kulhánek, F. Bureš, T. Mikysek, J. Ludvík and O. Pytela, Dyes Pigm., 2011, 90, 48–55 CrossRef PubMed.
- (a) W. Wu, C. Wang, C. Zhong, C. Ye, G. Qui, J. Qin and Z. Li, Polym. Chem., 2013, 4, 378–386 RSC; (b) J. Zhu, C. Lu, Y. Cui, C. Zhang and G. Lu, J. Chem. Phys., 2010, 133, 244503 CrossRef PubMed.
- TEEs: (a) Y. Rubin, C. B. Knobler and F. Diederich, Angew. Chem., Int. Ed. Engl., 1991, 30, 698–700 CrossRef; (b) N. K. Pahadi, D. H. Camacho, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2006, 71, 1152–1155 CrossRef CAS PubMed; (c) M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed.
- CEEs: (a) N. N. P. Moonen, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross and F. Diederich, Angew. Chem., Int. Ed., 2002, 41, 3044–3047 CrossRef CAS; (b) N. N. P. Moonen, R. Gist, C. Boudon, J.-P. Gisselbrecht, P. Seiler, T. Kawai, A. Kishioka, M. Gross, M. Irie and F. Diederich, Org. Biomol. Chem., 2003, 1, 2032–2034 RSC; (c) N. N. P. Moonen, W. C. Pomerantz, R. Gist, C. Boudon, J.-P. Gisselbrecht, T. Kawai, A. Kishioka, M. Gross, M. Irie and F. Diederich, Chem. – Eur. J., 2005, 11, 3325–3341 CrossRef CAS PubMed; (d) F. Bureš, W. B. Schweizer, J. C. May, C. Boudon, J.-P. Gisselbrecht, M. Gross, I. Biaggio and F. Diederich, Chem. – Eur. J., 2007, 13, 5378–5387 CrossRef PubMed.
- (a) J. A. Marsden, J. J. Miller, L. D. Shirtcliff and M. M. Haley, J. Am. Chem. Soc., 2005, 127, 2464–2476 CrossRef CAS PubMed; (b) E. L. Spitler, L. D. Shirtcliff and M. M. Haley, J. Org. Chem., 2007, 72, 86–96 CrossRef CAS PubMed; (c) D. T. Chase, B. S. Young and M. M. Haley, J. Org. Chem., 2011, 76, 4043–4051 CrossRef CAS PubMed; (d) H. Kang, G. Evmenenko, P. Dutta, K. Clays, K. Song and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 6194–6205 CrossRef CAS PubMed; (e) S. Shotwell, P. M. Windscheif, M. D. Smith and U. H. F. Buntz, Org. Lett., 2004, 6, 4151–4154 CrossRef CAS PubMed; (f) C. Aronica, A. Venancio-Marques, J. Chauvin, V. Robert and G. Lemercier, Chem. – Eur. J., 2009, 15, 5047–5055 CrossRef CAS PubMed; (g) V. Kriegish and C. Lambert, Eur. J. Inorg. Chem., 2005, 4509–4515 CrossRef.
- F. Bureš, H. Čermáková, J. Kulhánek, M. Ludwig, W. Kuznik, I. V. Kityk, T. Mikysek and A. Růžička, Eur. J. Org. Chem., 2012, 529–538 CrossRef.
- (a) B. del Rey, U. Keller, T. Torres, G. Rojo, F. Agulló-López, S. Nonell, C. Martí, S. Brasselet, I. Ledoux and J. Zyss, J. Am. Chem. Soc., 1998, 120, 12808–12817 CrossRef CAS; (b) S. H. Kang, Y.-S. Kang, W.-C. Zin, G. Olbrechts, K. Wostyn, K. Clays, A. Persoons and K. Kim, Chem. Commun., 1999, 1661–1662 RSC.
- (a) M. Kimura, H. Nomoto, N. Masaki and S. Mori, Angew. Chem., Int. Ed., 2012, 51, 4371–4374 CrossRef CAS PubMed; (b) S. Eu, T. Katoh, T. Umeyama, Y. Matano and H. Imahori, Dalton Trans., 2008, 5476–5483 RSC.
- (a) F. D'Souza, E. Maligaspe, A. S. D. Sandanayaka, N. K. Subbaiyan, P. A. Karr, T. Hasobe and O. Ito, J. Phys. Chem. A, 2010, 114, 10951–10959 CrossRef PubMed; (b) X. Chen, J. Thomas, P. Gangopadhyay, R. A. Norwood, N. Peyghambarian and D. V. McGrath, J. Am. Chem. Soc., 2009, 131, 13840–13843 CrossRef CAS PubMed.
- (a) D. Miyajima, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2010, 132, 8530–8531 CrossRef CAS PubMed; (b) K. Ban, K. Nishizawa, K. Ohta and H. Shirai, J. Mater. Chem., 2000, 10, 1083–1090 RSC.
- (a) J.-W. Hofman, F. van Zeeland, S. Turker, H. Talsma, S. A. G. Lambrechts, D. V. Sakharov, W. E. Hennink and C. F. van Nostrum, J. Med. Chem., 2007, 50, 1485–1494 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shitzu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- D. S. Terekhov, K. J. M. Nolan, C. R. McArthur and C. C. Leznoff, J. Org. Chem., 1996, 61, 3034–3040 CrossRef CAS PubMed.
- J. Kulhánek, F. Bureš and M. Ludwig, Beilstein J. Org. Chem., 2009, 5, 11 Search PubMed.
- N. Almonasy, F. Bureš, M. Nepraš, H. Přichystalová and G. Grampp, Dyes Pigm., 2014, 108, 50–56 CrossRef CAS PubMed.
- C. Dehu, F. Meyers and J. L. Brédas, J. Am. Chem. Soc., 1993, 115, 6198–6206 CrossRef CAS.
- (a) C. W. Bird, Tetrahedron, 1986, 42, 89–92 CrossRef CAS; (b) C. W. Bird, Tetrahedron, 1985, 41, 1409–1414 CrossRef CAS; (c) S. I. Kotelevskii and O. V. Prezhdo, Tetrahedron, 2001, 57, 5715–5729 CrossRef CAS.
- M.-B. S. Kirketerp, M. Å. Petersen, M. Wanko, L. A. E. Leal, H. Zettergren, F. M. Raymo, A. Rubio, M. B. Nielsen and S. B. Nielsen, ChemPhysChem, 2009, 10, 1207–1209 CrossRef PubMed.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894–7899 CrossRef CAS PubMed.
- (a) M. Kuzyk, J. Mater. Chem., 2009, 19, 7444–7465 RSC; (b) J. C. May, I. Biaggio, F. Bureš and F. Diederich, Appl. Phys. Lett., 2007, 90, 251106 CrossRef PubMed.
- (a) K. E. Fairfull-Smith, F. Brackmann and S. E. Bottle, Eur. J. Org. Chem., 2009, 1902–1915 CrossRef CAS; (b) T. Tsuda, K. Fujishima and H. Ueda, Agric. Biol. Chem., 1981, 45, 2129–2130 CrossRef CAS; (c) L. E. Hinkel, G. O. Richards and O. Thomas, J. Chem. Soc., 1937, 1432–1437 RSC.
- T. Sugimori, J. Nojima, T. Ozawa, M. Handa and K. Kasuga, Chem. Lett., 2004, 33, 1014–1015 CrossRef CAS.
- (a) Alex A. Granovsky, PC GAMESS version 7.1.E (Firefly), Copyright(c) 1994, Moscow State University, Moscow, Russia, 2008 Search PubMed; (b) PC GAMESS is partially based on GAMESS (US) ver. 6, June 1999, Iowa State Univ. M. W. Schmidt, K. K. Balridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- A. R. Allouche, Gabedit is a free Graphical User Interface for computational chemistry packages. It is available from http://gabedit.sourceforge.net/.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, synthesis and characterization of 11, further crystallographic, electrochemical, spectroscopic data, SHG results and HOMO/LUMO localizations in B1–B10, 1H and 13C NMR spectra of B1–B10. CCDC 984735 and 984736. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob00901k |
| ‡ Present address: COC Ltd, Rybitví 296, 53354, Pardubice, Czech Republic. |
| This journal is © The Royal Society of Chemistry 2014 |