 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSteric vs. electronic effects in the Lactobacillus brevis ADH-catalyzed bioreduction of ketones†
Cristina
Rodríguez‡
a,
Wioleta
Borzęcka‡
a,
Johann H.
Sattler
b,
Wolfgang
Kroutil
b,
Iván
Lavandera
*a and
Vicente
Gotor
*a
aDepartamento de Química Orgánica e Inorgánica, Instituto Universitario de Biotecnología de Asturias, Universidad de Oviedo, C/Julián Clavería 8, 33006 Oviedo, Spain. E-mail: lavanderaivan@uniovi.es; vgs@uniovi.es; Fax: (+) 34 985 103448; Tel: (+) 34 985 103452 Tel: (+) 34 985 103448
bDepartment of Chemistry, Organic and Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
First published on 15th November 2013
Abstract
Lactobacillus brevis ADH (LBADH) is an alcohol dehydrogenase that is commonly employed to reduce alkyl or aryl ketones usually bearing a methyl, an ethyl or a chloromethyl as a small ketone substituent to the corresponding (R)-alcohols. Herein we have tested a series of 24 acetophenone derivatives differing in their size and electronic properties for their reduction employing LBADH. After plotting the relative activity against the measured substrate volumes we observed that apart from the substrate size other effects must be responsible for the activity obtained. Compared to acetophenone (100% relative activity), other small substrates such as propiophenone, α,α,α-trifluoroacetophenone, α-hydroxyacetophenone, and benzoylacetonitrile had relative activities lower than 30%, while medium-sized ketones such as α-bromo-, α,α-dichloro-, and α,α-dibromoacetophenone presented relative activities between 70% and 550%. Moreover, the comparison between the enzymatic activity and the obtained final conversions using an excess or just 2.5 equiv. of the hydrogen donor 2-propanol, denoted again deviations between them. These data supported that these hydrogen transfer (HT) transformations are mainly thermodynamically controlled. For instance, bulky α-halogenated derivatives could be quantitatively reduced by LBADH even employing 2.5 equiv. of 2-propanol independently of their kinetic values. Finally, we found good correlations between the IR absorption band of the carbonyl groups and the degrees of conversion obtained in these HT processes, making this simple method a convenient tool to predict the success of these transformations.
Introduction
In the last few years, the use of alcohol dehydrogenases [ADHs, EC 1.1.1.x., also called ketoreductases (KREDs) or carbonyl reductases (CRs)] applied for synthetic purposes has largely increased, mainly due to the usually high catalytic efficiency and selectivity displayed to achieve carbonyl reductions or enantioselective alcohol oxidations under very mild reaction conditions.1–7 These enzymes are probably the most employed oxidoreductases to date, and several applications to obtain fine chemicals have already been found due to the use of very efficient systems to recycle the expensive nicotinamide cofactors [NAD(P)H or NAD(P)+], needed to transfer electrons into and from the target substrate.8–11Lactobacillus brevis ADH (LBADH)12 is a versatile short-chain alcohol dehydrogenase discovered by Hummel's group in 1997,13 which catalyzes the stereoselective reduction of ketones into the corresponding (R)-configured alcohols,14 or the oxidative kinetic resolution of sec-alcohols to obtain the remaining enantioenriched (S)-hydroxy derivatives,14 therefore displaying the so-called ‘anti-Prelog’ selectivity.15 Furthermore, it requires NADP(H) as a cofactor and usually works under the ‘coupled-substrate’ approach,11 thus using 2-propanol (2-PrOH) or acetone to recycle the cofactor. This enzyme appears as a highly valuable tool since only a limited number of available ADHs show such good stereopreference and can be used with 2-PrOH.16–19
Its amino acid sequence and three-dimensional structure have been determined,20,21 and several docking and modeling studies have been performed to explain the stereospecificity of this biocatalyst.20–23 The stereoselectivity emerges from an active site that possess two hydrophobic pockets, a small flexible one and a large open area, which host the small and large substituents of the substrate, respectively. Concerning the substrate spectra, this ADH is able to accept a more variable (often aromatic) bulky moiety such as a large ketone substituent;12,24 however, it has demonstrated a narrower acceptance for the small ketone substituent, accommodating preferentially methyl, ethyl, or chloromethyl groups.12,25
The reduction of ketones coupled with the oxidation of alcohols in a hydrogen transfer (HT) fashion is a typical example where the redox potential between the hydrogen donor and the acceptor is not usually large enough to achieve quantitative conversions (conv), unless a huge excess of a sacrificial alcohol/ketone such as 2-propanol/acetone is used,11 or activated ketones with an electron-withdrawing group at α-position such as chlorine,26,27 bromine,28 or a carbonyl moiety at α-23 or β-position23,29 are employed. In a recent contribution,22 we observed that for some substrates LBADH activity did not correlate with the conversions obtained, appearing some remarkable exceptions, and suggesting that mainly thermodynamic effects were involved in these processes. Furthermore, a method based on the infrared (IR) absorption band of the carbonyl group to predict the degree of conversion achieved in a HT-reduction of a ketone, was also proposed.
Herein, we have extended this study to a series of 24 α-substituted acetophenone derivatives (Fig. 1), differing in their steric and electronic properties, in order to provide a rationale for the final conversion of their HT reductions, as well as the influence of kinetics and thermodynamics using a huge excess of 2-propanol as a hydrogen donor (standard conditions), or just 2.5 equivalents (minimal conditions). Consequently the relative activities of LBADH with each substrate were measured compared with the calculated volume of the substrate. We have also measured the IR absorption band of the carbonyl groups to check this predictive method with the enzymatic conversions. As a result, it will be shown that sterically demanding substrates can also be reduced by this enzyme.
 | ||
| Fig. 1 Substrates employed to study the LBADH activity. | ||
Results and discussion
Enzyme activity of LBADH in the bioreduction of ketones 1–24a
A series of α- or β-substituted acetophenone derivatives (1–24a, Fig. 1) and the corresponding alcohols (1–24b) were selected based on their different steric and electronic properties in order to see the possible influence of the small substituent in their LBADH-catalyzed bioreductions. Thus, alkylated (1–3a), halogenated (4–13a), oxygenated (14–16a), nitrogenated (17–20a), and esterified (21–24a) derivatives were used as substrates in this study (see the ESI† for synthetic protocols).Accordingly, NADPH depletion was used to determine the catalytic efficiency (kcat/KM) of the LBADH-catalyzed reduction of ketones 1–24a to subsequently calculate the relative activity with regard to acetophenone (1a, 100%). Ketone 1a has been used in other studies as a model substrate for this enzyme due to its high reactivity.20–22 As shown in Table 1, compounds bearing one halogen atom at α-position (4–6a), α,α-difluoro- (7a), α,α-dichloro- (8a), and α-bromo-α-chloroacetophenone (9a) were the best substrates, being reduced approx. between 1.6 and 32-times more effectively than 1a. Another halogenated compound such as α,α-dibromoacetophenone (10a) was also a good substrate while α,α,α-trifluoro (11a) and α,α,α-tricholoro (12a) derivatives could be reduced at levels around 0.2 times compared to 1a. Ketone 2a and α-keto esters 21a and 22a showed relative activities around 30%, while ketones 3a, 14a, 15a, 17a, 18a, 20a, 23a, and 24a can be considered as poor substrates from a kinetic point of view.
| Entry | Compound | Relative activitya (%) |
|---|---|---|
a 100% value corresponds to kcat/KM = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 M−1 s−1 (see ESI).
b Solubility problems.
c Absorbance interference. n.d. not determined. 100 M−1 s−1 (see ESI).
b Solubility problems.
c Absorbance interference. n.d. not determined.
|
||
| 1 | 1a | 100 |
| 2 | 2a | 29 |
| 3 | 3a | <1 |
| 4 | 4a | 3187 |
| 5 | 5a | 2358 |
| 6 | 6a | 547 |
| 7 | 7a | 780 |
| 8 | 8a | 472 |
| 9 | 9a | 159 |
| 10 | 10a | 73 |
| 11 | 11a | 21 |
| 12 | 12a | 26 |
| 13 | 13a | n.d.b |
| 14 | 14a | 5 |
| 15 | 15a | <1 |
| 16 | 16a | n.d.c |
| 17 | 17a | 3 |
| 18 | 18a | 4 |
| 19 | 19a | n.d.c |
| 20 | 20a | 1 |
| 21 | 21a | 27 |
| 22 | 22a | 27 |
| 23 | 23a | <1 |
| 24 | 24a | <1 |
It was noticeable that going from acetophenone to propiophenone (2a) and butyrophenone (3a), the activity considerably dropped down (entries 1–3), supporting previous results obtained with this enzyme showing that a bulkier group than ethyl can hardly be accepted.25 As mentioned before, when a family of halogenated derivatives was employed, it was observed that, on the one hand, monohalogenated compounds (entries 4–6) showed higher activities than dihalogenated ketones (entries 7–10), and these, in turn, exhibited higher velocities than the trihalogenated ones (entries 11 and 12). On the other hand, fluorinated compounds demonstrated to be better substrates than chlorinated and these ones better than brominated. These results can be explained based on steric reasons (see below) and also due to the higher electrophilicity of the carbonyl group when fluorine is present at α-position with regard to chlorine or bromine atoms.30 For the oxygenated compounds (entries 14 and 15), it seemed that a polar hydroxyl group at α-location (14a) did not avoid completely the enzymatic activity, while a neutral methoxy moiety (15a) negatively affected it. The presence of a nitrile (17a), an azide (18a) or an acetamide (20a), caused a dramatic drop in LBADH reactivities (entries 17–20). The catalytic efficiencies of these nitrogenated derivatives mainly decreased due to the high KM values (see ESI†), leading to very low relative activities. Finally, we observed differences between keto esters (entries 21–24) depending on the carbonylic position, since α-substituted derivatives 21a and 22a showed a moderate enzymatic activity, while the β-substituted ones (23a and 24a) seemed not to be appropriate substrates for LBADH.
Comparison of enzymatic activities and molecular volumes
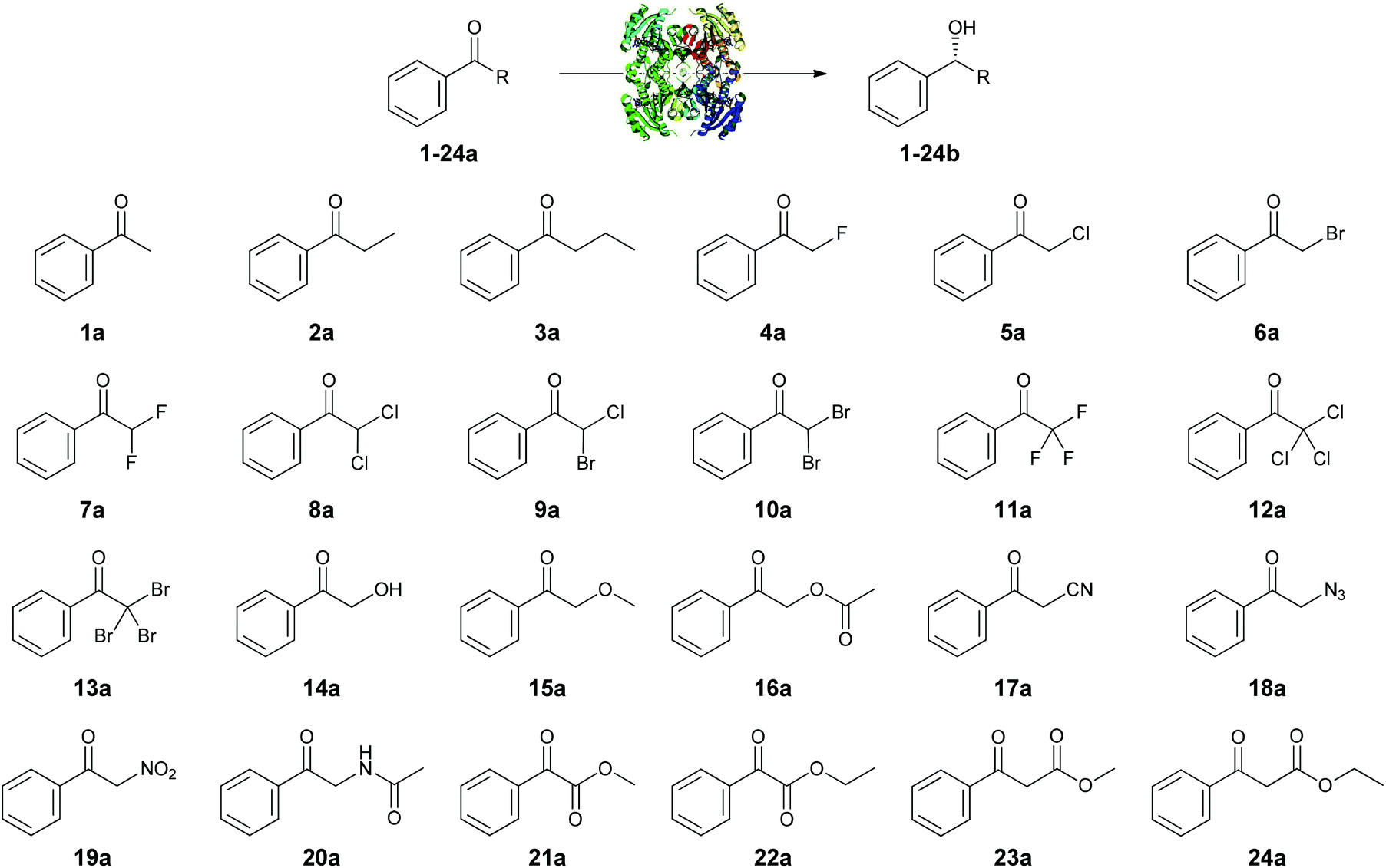
Steric hindrance is usually taken as the most prominent reason to explain the low reactivity of a substrate with a (bio)catalyst. For enzymes it is well known that (de)stabilizing interactions between the substrate and the enzyme can determine the activity and selectivity of the biocatalysts. For instance, based on these molecular interactions successful empiric rules have appeared for lipases (Kazlauskas’ rule)31 or alcohol dehydrogenases (Prelog's rule).15 In an ADH, a ternary complex between an enzyme, ketone or alcohol substrate, and a cofactor in the reduced or oxidized form, must be produced to correctly transfer the electrons. Taking into account a model where two hydrophobic pockets differing in size would accommodate the acetophenone substrates 1–24a, we decided to explore the correlation between the enzymatic activities with LBADH and the calculated substrate sizes. Moreover, the large group would be maintained (a phenyl moiety), while the small one would change in terms of steric demand.Subsequently, the volume of all acetophenone compounds were estimated using the generalized AMBER force field (GAFF).32 For each derivative, a conformational search was performed using the Multiconf–Dock tool generating conformers by rotating all single, non-terminal, acyclic bonds.33 The MSMS program34 was used to carry out the numerical computation of the molecular volume. Both the solvent-excluded molecular volume (VSES) for a probe sphere of 1.4 Å radius and the van der Waals molecular volume (Vmol) were computed for each compound and the results for different conformers were averaged (see the Experimental section and ESI† for more details). Thus, the molecular volume ranged from 117.49 Å3 (1a, R = CH3) to 177.87 Å3 (24a, R = CH2CO2Et), meaning that the biggest substrate had a volume difference of approximately 1.5 times with regard to the smallest one. With these data in hand, a graphic correlation between the molecular volumes and the enzymatic activities for all ketones tested could be done as shown in Fig. 2.
 | ||
| Fig. 2 Comparison of ketone volumes (black line) with relative activities (bars, logarithm scale). Ketone substrates are divided as small-sized (1 < V/V1a < 1.15), medium-sized (1.15 < V/V1a < 1.3), and large-sized (1.3 < V/V1a). Substrates are ordered from left to right by an increasing value of calculated molecular volume. | ||
Although there was a general trend correlating higher velocities with the smaller substrates, some noticeable deviations were observed. For instance, small ketones such as 2a (R = CH2CH3), 11a (R = CF3), 14a (R = CH2OH), and 17a (R = CH2CN) had relative activities lower than 30%, while medium-sized ketones such as 6a (R = CH2Br), 8a (R = CHCl2), or 9a (R = CHBr2) presented relative activities between 150% and 550%. Even one of the biggest substrates, 22a (R = CO2CH2CH3, V = 162.50 Å3) was moderately well accepted by this enzyme. This study shows that although undoubtedly the steric demand is a very important factor, other effects, e.g. electronic, must also have an influence and therefore should also be taken into account.35
Comparison of enzymatic activities and conversions: electronic effects and thermodynamic implications
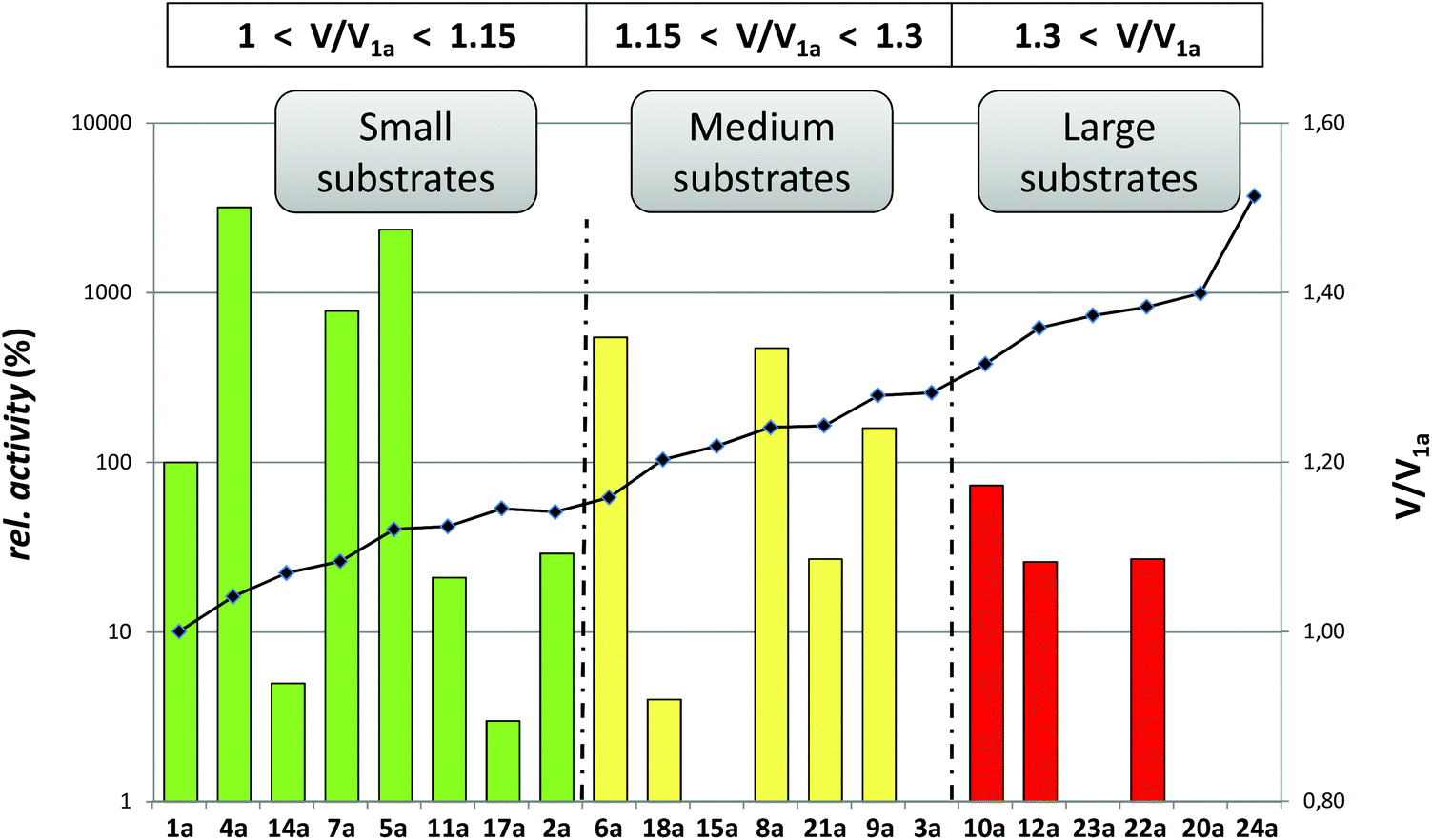
Trying to answer whether a good substrate leads to complete conversion while a poor substrate affords low conversion, we analyzed the correlation between the kinetic results with the conversions obtained in the LBADH-catalyzed bioreductions. In a previous study22 it was demonstrated with a small set of ketones that high conversions were obtained with substrates bearing a chlorine atom at α-position. In fact, there was no clear correlation between the enzymatic conversions and the initial rates of the reactions. Although a trend was observed in some cases, there were some remarkable exceptions such as methoxyacetone, suggesting that the bioreduction of ketones was not under kinetic, but mainly under thermodynamic control.Herein we have reduced the ketones listed in Fig. 1 with LBADH adding a catalytic amount of NADPH (1 mM) and employing a huge excess of 2-PrOH (5% v/v, standard conditions) or just using 2.5 equiv. of 2-PrOH (minimal conditions).27 The variety of substituents enabled the drawing of a structure–activity relationship, so in a subsequent step, conversions obtained were plotted against the relative activities shown in Table 1 to easily detect possible deviations (Fig. 3).
 | ||
| Fig. 3 Comparison of the initial rates (white line, logarithm scale) with LBADH-catalyzed bioreduction conversions (t = 48 h) under standard (5% v/v of 2-PrOH, black bars) and minimal conditions (2.5 equiv. of 2-PrOH, grey bars). Substrates (30 mM) are ordered from left to right by a decreasing value of enzymatic activity. In all cases, ee measured for the alcohol obtained was >99%. | ||
Although acetophenone (1a) was reduced 4 times faster than propiophenone (2a), LBADH produced both alcohols with a conversion higher than 90% after 48 h using an excess of 2-propanol and approximately 50% when using just 2.5 equiv. Since in these cases the alcohol formation is not highly favored with regard to the 2-PrOH–acetone counterpart (<0.5 kcal mol−1),22 the thermodynamic equilibrium does not allow a very high conversion unless a huge excess of the hydrogen donor is applied. To ensure that the equilibrium was reached using 2.5 equiv. of 2-PrOH, a time study of the bioreduction with 1a was performed (Fig. 4), observing always a conversion lower than 50% within the time. Butyrophenone (3a) afforded very low formation of the corresponding alcohol (conv <5%), showing that a propyl group was not suitable for this enzyme. The introduction of electronegative groups at the small substituent generally led to excellent conversions. Thus, α-halogenated derivatives 4–11a could be reduced nicely (conv >90%) under these mild conditions even just employing 2.5 equiv. of 2-PrOH (Fig. 3 and 4), independently of their relative activity (i.e., the catalytic efficiency of 4a was around 40 times higher than that of 10a). This is due to the fact that the thermodynamic equilibrium in these HT reactions favors the halohydrin formation (between 4 and 6 kcal mol−1),22 being probably involved in an intramolecular H-bond between the hydroxyl group and the halogen atom(s).36 Just as in the case of very bulky substrates such as 12a (R = CCl3) and 13a (R = CBr3), the conversions were not high.
 | ||
| Fig. 4 Conversions of selected substrates (30 mM) within the time using 2.5 equiv. of 2-PrOH. | ||
Concerning the oxygenated derivatives, only α-hydroxyacetophenone 14a appeared as a suitable substrate for LBADH achieving quantitative conversion to 14b under standard conditions, while 90% for the minimal setup, even at 8 h (Fig. 4). This suggested that in this case the intramolecular H-bond was not as effective as in the case of the halogenated compounds, although the equilibrium was clearly more shifted towards the alcohol product than for alkylated derivatives 1a and 2a.
Although nitrogenated ketones 19a and 20a were not appropriate substrates for this biocatalyst affording low conversions (<15%), keto nitrile 17a led to a formation of 17b of approx. 70% with an excess of 2-PrOH and 55% employing 2.5 equiv. of the hydrogen donor. Remarkably, α-azidoacetophenone 18a, despite its low relative activity (4%), could be reduced by LBADH in a quantitative manner under the standard HT conditions. While a conversion around 60% was achieved after 24 h with a small excess of 2-PrOH, when the reaction was left for 48 h (Fig. 4), LBADH already led to 85% of alcohol 18b.
For keto esters, while α-substituted 21a and 22a led to high conversions using both reaction settings (>70%), β-keto esters 23a and 24a were not accepted by the ADH. In these cases, the existence of a carbonyl group at α-position seems to highly favor the bioreduction, possibly due to an internal H-bond interaction between the alcohol and the carbonyl ester moiety.23,29 Again, since these ketones were moderately good substrates for LBADH, we performed the experiments with 22a at different reaction times to confirm that when using an excess of the hydrogen donor we obtained a final conversion of around 75% at 24 and 48 h, while employing 2.5 equiv. (Fig. 4), the conversion still increased from 60% to 74%.
Substrate solubility is another important issue that can significantly affect these bioreductions.24 Due to the fact that enzymatic reactions catalyzed by ADHs are generally carried out in an aqueous buffer solution to keep the enzyme stability, and since the majority of ketones of interest are highly hydrophobic, the use of organic cosolvents in biphasic37–39 and monophasic systems40–42 has been proposed. In the standard HT conditions, 5% v/v of 2-PrOH was used as a hydrogen donor and also helped to solubilize the substrates, but under the minimal setup 2% v/v of dimethylsulfoxide (DMSO) was added for solid ketone substrates that did not afford high conversions, to check whether their water-solubility could be an issue that would hamper higher degrees of alcohol formation. Thus, several bioreductions were repeated, observing a remarkable effect in some cases. Ketone 8a was reduced quantitatively in the presence of DMSO while only affording 63% of 8b with just 2.5 equiv. of 2-PrOH. Conversion with ketone 9a was enhanced from 75% to >99% employing DMSO, and a similar effect (from 11% to 57%) was observed for keto nitrile 17a. It must also be mentioned that in all cases stereoselectivities were higher than 99% for the formation of the anti-Prelog alcohols, thus demonstrating that the binding of these substrates was not altered (phenyl linked to the big pocket, and the smaller ketone derivative to the small pocket).
Enzyme stability should also be taken into account when measuring enzymatic conversions. Therefore, LBADH was incubated for 24 h at 30 °C in a buffer in the presence or in the absence of 2-PrOH before addition of 1a, in order to measure the enzymatic conversion after 24 h. In both cases, transformations around 90% were achieved, confirming the excellent enzyme stability during the experimental time-frames studied.
IR carbonyl stretching bands to predict biocatalyzed HT conversions
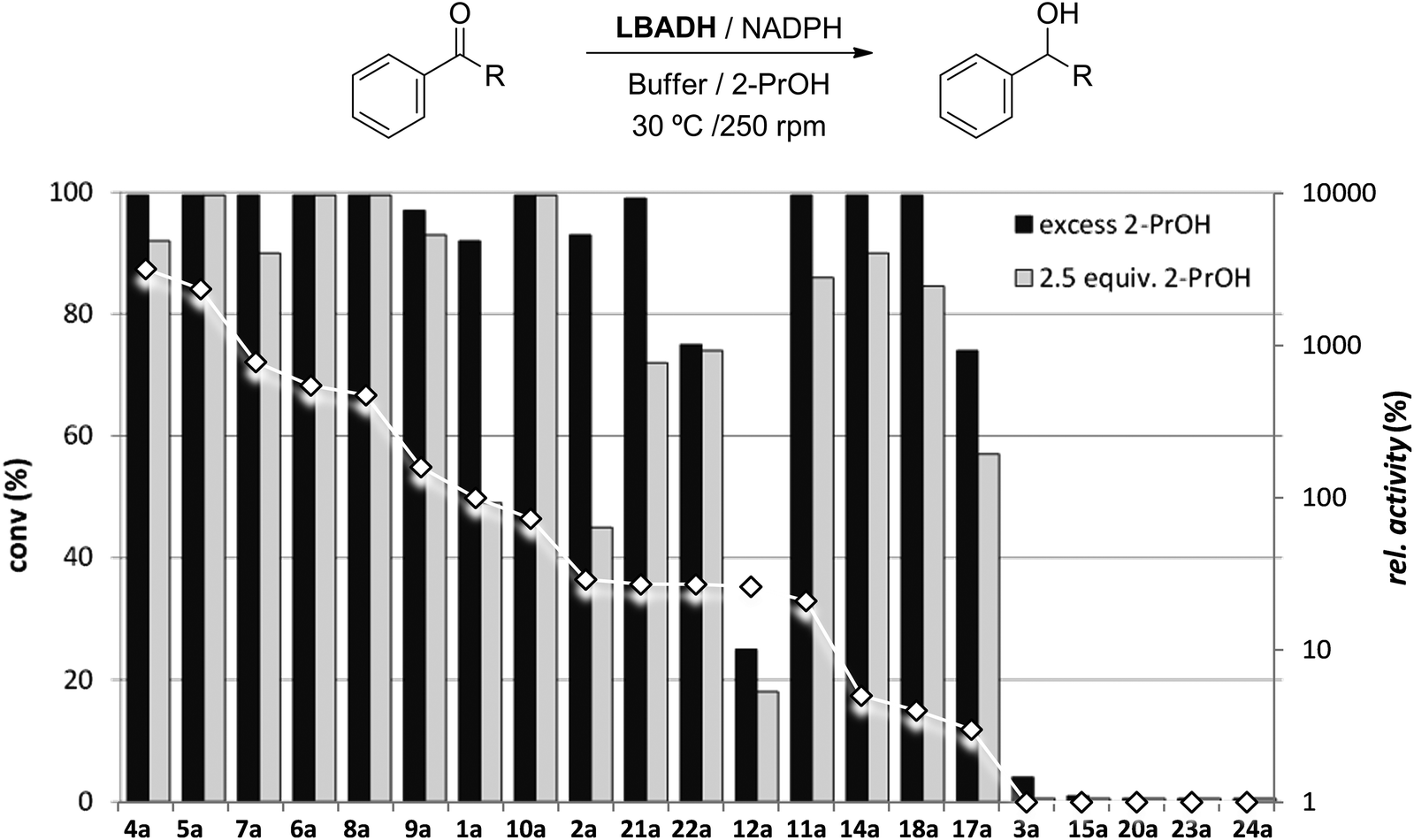
We previously measured the IR carbonyl absorption bands of a small set of ketones and plotted them against the degrees of conversion at equilibrium using a slight excess of the hydrogen donor, obtaining a good correlation between them.22 The presence of electron-withdrawing groups led to higher IR frequencies, since the dipolar form of these ketones (Fig. 5) was disfavored, therefore destabilizing their ground resonance form and making them more reactive.43 It was concluded that IR could be employed as a reliable methodology to qualitatively predict if a ketone could be a good hydrogen acceptor under HT conditions. | ||
| Fig. 5 Ketone resonance structures and plot of IR carbonyl stretching bands of acetophenone derivatives against the normalized degrees of conversion for LBADH-catalyzed HT reactions. The exception found is circled in red. | ||
Due to the fact that we had a broad set of structurally related ketones, we decided to further explore the reliability of this methodology. IR carbonyl stretching bands were measured for all reactive acetophenones and then these values were plotted against the normalized enzymatic conversions, that is, the LBADH-catalyzed conversion obtained with 2.5 equiv. of 2-PrOH divided by the one achieved using an excess of the hydrogen donor (Fig. 5).44
From our previous study,22 ketones having a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O value lower than 1690 cm−1 did not show quantitative conversions under the minimal conditions while those presenting values higher than 1690 cm−1 were highly reduced even with a small excess of the hydrogen donor. As can be seen, from the 15 ketones studied, just 6a did not fit with this criterion. α-Bromoacetophenone showed a lower value (1683.9 cm−1) compared to other halogenated derivatives. This is because α-brominated and α-iodinated carbonylic derivatives present lower CO shifts in comparison with fluorinated or chlorinated ketones due to the dihedral angle (φ) between CO and C–Br or C–I bonds.45
O value lower than 1690 cm−1 did not show quantitative conversions under the minimal conditions while those presenting values higher than 1690 cm−1 were highly reduced even with a small excess of the hydrogen donor. As can be seen, from the 15 ketones studied, just 6a did not fit with this criterion. α-Bromoacetophenone showed a lower value (1683.9 cm−1) compared to other halogenated derivatives. This is because α-brominated and α-iodinated carbonylic derivatives present lower CO shifts in comparison with fluorinated or chlorinated ketones due to the dihedral angle (φ) between CO and C–Br or C–I bonds.45
In contrast, a correlation to the 13C-NMR signal of the carbonyl moiety did not lead to any conclusive data.
Conclusions
Lactobacillus brevis ADH is a very robust and versatile oxidoreductase which is often employed since it can work under the highly convenient ‘coupled-substrate’ approach displaying ‘anti-Prelog’ selectivity. Until now it has been described as a very active biocatalyst for alkyl or aryl ketones possessing methyl, ethyl, or chloromethyl as a small substituent in the ketone failing for other bulkier substrates. Herein we have shown that not only these moieties, but also other groups such as α,α-dibromomethyl, α,α,α-trichloromethyl or α-ethoxycarbonyl can be accepted by this enzyme achieving good to excellent conversions and stereoselectivities to the corresponding alcohols. When the molecular volumes of the substrates were measured, a trend was observed regarding the enzymatic activity, although there was a clear deviation for some ketones. Hence, steric effects are important in these biocatalyzed reductions, but other influences, i.e. electronic, cannot be ignored.These hydrogen transfer reactions are a good example where ΔE0 between the hydrogen donor and the acceptor is usually not large enough to achieve quantitative conversions necessitating the use of a large excess of a co-substrate to drive the equilibrium in the desired direction. In this sense, the use of α-halo- and α- or β-carbonyl ketones as hydrogen acceptors in nearly stoichiometric amounts has been utilized for synthetic purposes.26–30 This behavior has been studied for LBADH obtaining excellent conversions with substrates bearing electron-withdrawing groups such halogen(s), azido, cyano, hydroxyl, and carbonyl. Although some of these derivatives appeared as poor substrates for this enzyme from a kinetic point of view, they could be converted into the alcohol products at high conversion, since these hydrogen transfer processes are mainly driven by thermodynamics. While for some of the formed alcohols a stabilizing intramolecular H-bond is assumed, for others an inductive effect can be ascribed for the low alcohol oxidation rates.
Finally, IR frequencies of the carbonyl band correlated well with the normalized conversions, and except for the special case of α-brominated ketone 6a, this method appears as an easy and straightforward predictive methodology for the conversion obtained by a ketone under HT conditions.
Our data demonstrate that the frequently employed statement “due to steric reasons” to explain the low reactivity or conversion of a substrate with an (enzyme) catalyst must be careful assessed since other important effects contribute as well. Furthermore, a low (enzyme) activity does not necessarily imply low conversion, especially in the case of HT transformations catalyzed by ADHs.
Experimental
General methods
NADPH was acquired from Codexis. Chemical reactions were monitored by analytical TLC, performed on Merck silica gel 60 F254 plates and visualized by UV irradiation. Flash chromatography was performed using silica gel 60 (230–400 mesh). IR spectra were recorded on a Perkin-Elmer 1720-X infrared Fourier transform spectrophotometer on NaCl pellets dissolving the compounds with a drop of CH2Cl2. 1H-, 13C-NMR, and DEPT were obtained using a Bruker DPX-300 and NAV-400 (1H, 300.13 MHz and 13C, 75.5 MHz) spectrometer for routine experiments. The chemical shifts (δ) are given in ppm and the coupling constants (J) in hertz (Hz). ESI+ mode was used to record mass spectra (MS) and ESI-TOF for HRMS. Gas chromatography (GC) analyses were performed on a Hewlett Packard 6890 Series II chromatograph. Optical rotations were measured using a Perkin-Elmer 241 polarimeter and are quoted in units of 10−1 deg cm2 g−1. Steady-state kinetic parameters were determined using a Varian Cary50Bio UV/Vis spectrophotometer.Protein expression and purification
E. coli/LBADH was grown overnight at 37 °C in 250 mL of Luria–Bertani medium supplemented with 100 μg mL−1 ampicillin. The protein expression was induced by the addition of anhydrotetracycline hydrochloride (0.2 mg L−1, 0.4 μM final concentration). The cultivation was performed overnight at 20 °C and 130 rpm to avoid the formation of inclusion bodies. The cells were harvested by centrifugation (5600 rpm, 20 min, r.t., Heraeus SEPATECH, Megafuge 1.0) and washed with phosphate buffer 50 mM, pH 7.5. Cells were disrupted by ultrasonication (Branson, S250D CE, 200 W, 5 mm spike, 50 mL tubes, 1 s impulse, 4 s pause, amplitude 40%, 2 min and 30 s, 4 °C) and subsequently subjected to centrifugation (5600 rpm, 20 min, r.t.). The supernatant was used for protein purification.The sample was filtered off and LBADH-Strep was purified via Strep-Tag chromatography following the procedure described by the company (GE Healthcare, StrepTrap HP 5 mL column) and LBADH-Strep was obtained at a concentration of 145 μM. The purity of protein samples was assessed by SDS polyacrylamide gel electrophoresis.
Steady-state kinetic analysis of LBADH
The enzymatic activities of LBADH in the reduction of ketones 1a–24a were determined spectrophotometrically by monitoring the NADPH consumption at 340 nm (εNADPH,340 = 6.22 mM−1 cm−1). In some cases, high concentrations of the substrate or the proper compound resulted in absorption values exceeding 1 unit. In these experiments, the coenzyme consumption was measured at 370 nm (εNADPH,370 = 2.7 mM−1 cm−1).Stock solutions of substrates (1.0 M) were made in DMSO since the presence of 1% DMSO resulted in a slight decrease in LBADH activity, while higher solubility of certain compounds could be achieved. A reaction mixture (1.0 mL) usually contained the corresponding ketone in 50 mM Tris-HCl, pH 7.5, 100 μM NADPH, 1% v/v DMSO, and 0.02–0.15 μM of LBADH.
Molecular volume calculations
The generalized AMBER force field (GAFF) was employed for carrying out the molecular modeling of the selected PhCOR compounds.32 The GAFF parameters including AM1-BCC atomic charges were generated automatically using the antechamber module included in the Amber11 package.46 For each compound, a conformational search using the Multiconf–Dock tool33 was performed generating conformers by rotating all single, non-terminal, acyclic bonds. Subsequently, the SANDER module in Amber11 was employed to minimize and score all the non-redundant conformers in terms of their relative GAFF energies.The MSMS program34 was used to carry out the numerical computation of the molecular volume. To improve the accuracy of the results, a high value (10 vertex points per Å2) for the triangulation density was employed. We computed both the solvent-excluded molecular volume for a probe sphere of 1.4 Å radius and the van der Waals molecular volume. The Bondi atomic radii were used in all the volume calculations and the results for different conformers were averaged (see ESI†).
LBADH-catalyzed reduction of ketones
000 rpm, 2 min) and dried over Na2SO4. Conversions and ee of the corresponding alcohols were determined by GC (see ESI†). For α-brominated ketones, 50 mM Tris-H2SO4 buffer, pH 7.5 (600 μL, 1 mM NADPH, 1 mM MgBr2) was used to avoid undesired SN2 reactions.28
000 rpm, 2 min) and dried over Na2SO4. Conversions and ee of the corresponding alcohols were determined by GC (see ESI†). For α-brominated ketones 50 mM Tris-H2SO4 buffer, pH 7.5 (600 μL, 1 mM NADPH, 1 mM MgBr2) was used.28 In some cases, 12 μL of DMSO (2% v/v) were added to improve substrate solubilization.
Acknowledgements
W. B. thanks the Ministerio de Educación, Cultura y Deporte for her predoctoral fellowship (FPU Program). I. L. thanks the Spanish Ministerio de Ciencia e Innovación (MICINN) for personal funding (Ramón y Cajal Program). Financial support of this work by the Spanish MICINN (Project MICINN-12-CTQ2011-24237) is gratefully acknowledged. We thank Prof. Dimas Suárez (University of Oviedo) for ab initio molecular volume calculations and Dr Eduardo García-Urdiales for helpful discussions.Notes and references
- H. Gröger, W. Hummel, S. Borchert and M. Krauβer, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012, vol. 2, pp. 1037–1110 Search PubMed.
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2313 RSC.
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC.
- E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, PR110–PR180 CrossRef PubMed.
- M. M. Musa and R. S. Phillips, Catal. Sci. Technol., 2011, 1, 1311–1323 CAS.
- K. Faber, Biotransformations in Organic Chemistry, Springer-Verlag, Heidelberg, 6th edn, 2011 Search PubMed.
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed.
- C. Rodríguez, I. Lavandera and V. Gotor, Curr. Org. Chem., 2012, 16, 2525–2541 CrossRef.
- M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088–4110 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- S. Leuchs and L. Greiner, Chem. Biochem. Eng. Q., 2011, 25, 267–281 CAS , and references cited therein.
- W. Hummel, Adv. Biochem. Eng./Biotechnol., 1997, 58, 145–184 CrossRef CAS.
- This is true when the formal Cahn–Ingold–Prelog priorities (CIP) match with the steric demand of the substituents at the chiral alcohol center.
- V. Prelog, Pure Appl. Chem., 1964, 9, 119–130 CrossRef CAS.
- M. M. Musa, N. Lott, M. Laivenieks, L. Watanabe, C. Vieille and R. S. Phillips, ChemCatChem, 2009, 1, 89–93 CrossRef CAS.
- K. Inoue, Y. Makino and N. Itoh, Tetrahedron: Asymmetry, 2005, 16, 2539–2549 CrossRef CAS PubMed.
- P. Hildebrandt, T. Riermeier, J. Altenbuchner and U. T. Bornscheuer, Tetrahedron: Asymmetry, 2001, 12, 1207–1210 CrossRef CAS.
- C. W. Bradshaw, W. Hummel and C.-H. Wong, J. Org. Chem., 1992, 57, 1532–1536 CrossRef CAS.
- N. H. Schlieben, K. Niefind, J. Muller, B. Riebel, W. Hummel and D. Schomburg, J. Mol. Biol., 2005, 349, 801–813 CrossRef CAS PubMed.
- K. Niefind, J. Muller, B. Riebel, W. Hummel and D. Schomburg, J. Mol. Biol., 2003, 327, 317–328 CrossRef CAS.
- F. R. Bisogno, E. García-Urdiales, H. Valdés, I. Lavandera, W. Kroutil, D. Suárez and V. Gotor, Chem.–Eur. J., 2010, 16, 11012–11019 CrossRef CAS PubMed.
- M. Kurina-Sanz, F. R. Bisogno, I. Lavandera, A. A. Orden and V. Gotor, Adv. Synth. Catal., 2009, 351, 1842–1848 CrossRef CAS.
- H. G. Naik, B. Yeniad, C. E. Koning and A. Heise, Org. Biomol. Chem., 2012, 10, 4961–4967 CAS.
- M. Wolberg, W. Hummel and M. Müller, Chem.–Eur. J., 2001, 7, 4562–4571 CrossRef CAS.
- F. R. Bisogno, I. Lavandera, W. Kroutil and V. Gotor, J. Org. Chem., 2009, 74, 1730–1732 CrossRef CAS PubMed.
- I. Lavandera, A. Kern, V. Resch, B. Ferreira-Silva, A. Glieder, W. M. F. Fabian, S. de Wildeman and W. Kroutil, Org. Lett., 2008, 10, 2155–2158 CrossRef CAS PubMed.
- F. R. Bisogno, A. Cuetos, A. A. Orden, M. Kurina-Sanz, I. Lavandera and V. Gotor, Adv. Synth. Catal., 2010, 352, 1657–1661 CrossRef CAS.
- A. Cuetos, A. Rioz-Martínez, F. R. Bisogno, B. Grischek, I. Lavandera, G. de Gonzalo, W. Kroutil and V. Gotor, Adv. Synth. Catal., 2012, 354, 1743–1749 CrossRef CAS.
- Fluorinated ketones have been recently shown to be excellent substrates for this enzyme. See: W. Borzęcka, I. Lavandera and V. Gotor, J. Org. Chem., 2013, 78, 7312–7317 CrossRef PubMed.
- R. J. Kazlaukas, A. N. E. Weissfloch, A. T. Rappaport and L. A. Cuccia, J. Org. Chem., 1991, 56, 2656–2665 CrossRef.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- N. Sauton, D. Lagorce, B. O. Villoutreix and M. A. Miteva, BMC Bioinformatics, 2008, 9, 184 CrossRef PubMed.
- M. F. Sanner, A. J. Olson and J.-C. Spehner, Biopolymers, 1996, 38, 305–320 CrossRef CAS.
- Please note that the shape of the substituents is not taken into account. It is obvious that the volume data obtained do not account for the specific shape of the substituent, that is, globular, tubular, etc., but we think that for some of them, e.g.16a or 20–24a, the shaping is not trivial due to multiple substrate conformations, as observed in the ab initio calculations, and the possible discussion would be incomplete and misleading.
- T. Goldstein, M. S. Snow and B. J. Howard, J. Mol. Spectrosc., 2006, 236, 1–10 CrossRef CAS PubMed.
- M. Singh, S. Singh, S. Deshaboina, H. Krishnen, R. Lloyd, K. Holt-Tiffin, A. Bhattacharya and R. Bandichhor, Catal. Sci. Technol., 2012, 2, 1602–1605 CAS.
- X. Wu, Y. Wang, J. Ju, C. Chen, N. Liu and Y. Chen, Tetrahedron: Asymmetry, 2009, 20, 2504–2509 CrossRef CAS PubMed.
- J. He, X. Mao, Z. Sun, P. Zheng, Y. Ni and Y. Xu, Biotechnol. J., 2007, 2, 260–265 CrossRef CAS PubMed.
- P. Galletti, E. Emer, G. Gucciardo, A. Quintavalla, M. Pori and D. Giacomini, Org. Biomol. Chem., 2010, 8, 4117–4123 CAS.
- I. Lavandera, A. Kern, M. Schaffenberger, J. Gross, A. Glieder, S. de Wildeman and W. Kroutil, ChemSusChem, 2008, 1, 431–436 CrossRef CAS PubMed.
- G. de Gonzalo, I. Lavandera, K. Faber and W. Kroutil, Org. Lett., 2007, 9, 2163–2166 CrossRef CAS PubMed.
- For a similar discussion in the nucleophilic acyl substitution of different esters, see: H. Neuvonen and K. Neuvonen, J. Chem. Soc., Perkin Trans. 2, 1999, 1497–1502 RSC.
- This normalization has been done to compare correctly the conversion obtained employing a small excess of the hydrogen donor with regard to the use of a huge excess of 2-PrOH, independently of the intrinsic reactivity for each substrate with LBADH.
- E. Pretsch, P. Bühlmann and M. Badertscher, Structure Determination of Organic Compounds, Springer, Heidelberg, 4th edn, 2009 Search PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 11, University of California, San Francisco, 2010 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, molecular volume data, analytics and copy of 1H-, 13C-NMR, and DEPT spectra for 9b are described. See DOI: 10.1039/c3ob42057d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |