 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of tripeptide derivatized cyclopentadienyl complexes of technetium and rhenium as radiopharmaceutical probes†
Qaisar
Nadeem
a,
Daniel
Can
b,
Yunjun
Shen
b,
Michael
Felber
b,
Zaid
Mahmood
a and
Roger
Alberto
*b
aInstitute of Chemistry, University of the Punjab, Quaid-e-Azam Campus 54590, Lahore, Pakistan
bInstitute of Inorganic Chemistry, University of Zurich, Winterthurerstr. 190, 8057, Zurich, Switzerland. E-mail: ariel@aci.uzh.ch
First published on 23rd January 2014
Abstract
We describe the syntheses of half-sandwich complexes of the type [(η5-Cp(CONH-R))M(CO)3] with M = Re or 99mTc. The R group represents different tri-peptides (tpe) which display high binding affinities for oligopeptide transporters PEPT2. The 99mTc complexes were prepared directly from [99mTc(OH2)3(CO)3]+ and Diels–Alder dimerized, cyclopentadienyl derivatized peptides in water. This approach corroborates the feasibility of metal-mediated retro Diels–Alder reactions for the preparation of not only small molecules but also peptides carrying a [(η5-Cp)99mTc(CO)3] tag. We synthesized the Diels–Alder product [(HCpCONH-tpe)2] from Thiele's acid [(η5-HCpCOOH)2] via double peptide coupling. The Re-complexes [(η5-CpCONH-tpe)Re(CO)3] were obtained by attaching [(Cp-COOH)Re(CO)3] directly to the N-terminus of peptides as received from SPPS. The authenticity of the 99mTc-complexes is confirmed by chromatographic comparison with the corresponding rhenium complexes, fully characterized by spectroscopic techniques.
Introduction
Labeling of peptides with radionuclides is a major strategy to obtain compounds for SPECT or PET imaging. One particular example is 111In radiolabeled Octreotide (Octreoscan®) which was introduced into clinical routine oncology many years ago. Increasing knowledge of targeted receptor-binding peptides has established successful procedures for molecular peptide receptor imaging.1–4 Peptide labeling with a multitude of radionuclides for diagnosis and therapy has been reviewed.5–7 Besides routine applications in the clinic of e.g. somatostatin derivatives for neuroendocrine tumours, other promising candidates such as neuropeptide Y (NPY), c(RCDyK), c(RCDfK), Lys3-BBN, monocyte chemotactic peptide-1 and MG (minigastrin) are amongst the subjects of current preclinical evaluation.8,9 Small, synthetic, receptor-binding peptides are excellent vectors for specific receptor-imaging, primarily due to the availability of numerous peptides with a broad range of biological activities against many chronic diseases and their favourable pharmacokinetic characteristics, especially combined with their flexibility in chemical modification and radiolabeling. The high binding affinity of a peptide for its receptor induces its retention in receptor over-expressing tissues.10–13Di- and tri-peptides as well as peptidomimetics such as β-lactam antibiotics, valganciclovir, angiotensin-converting enzyme inhibitors, the antineoplastic agent bestatin and other drugs are transported across biological membranes through PEPT1 and PEPT2 transporters, membrane proteins14–20 which are expressed in a variety of organs including kidney, lung, enteric nervous system, mammary gland, pituitary gland, testis, prostate, ovary, uterus, eye and intestine.21–29 An example is the di-peptide, Ala-Lys-AMCA, which is specifically accumulated in both ganglionic layers of the enteric nervous system via PEPT2, as revealed by fluorescence microscopy in all regions and species studied such as rat, mouse, and guinea pig.30 The distribution of carnosine in HT-1080 tumours via oligopeptide transporters (PEPT1 & PEPT2) was examined among various human cancer cell lines to distinguish cancer from normal cells.31
There are only very few reports on the affinity of tripeptides to PEPT2. The first evidence for intact tripeptide transport via PEPT2 has been provided by Tiruppathi and co-workers.32–35 According to structure–activity relationship studies by Thondorf and co-workers, the requirements for tripeptides and small pharmacophores to bind firmly to PEPT2 are a terminal amino and a carboxylic acid group, amide bonds and lipophilicity. Ki values for tripeptides cover a broad range and the highest affinities are in the low μM range.36 Small structural changes have a dramatic influence on the affinity. For some cases, amidation of the carboxylato terminus in tripeptides abolishes recognition for PEPT2.
Labeling of peptides with radionuclides is a major imaging strategy. Since peptides are relatively small molecules, the selection of chelators and complexes (in the case of metallic radionuclides) is decisive since their molecular weights and structural requirements often compare with the ones of the carriers. Di- and tripeptides for PEPT1 and PEPT2 targeting with metallic radionuclides are rare and, to the best of our knowledge, have not been studied with 99mTc. 99mTc is still one of the most favourable radionuclides due to its convenient availability from generators, low cost and ideal decay characteristics.37,38 These advantages are somewhat counter-balanced by its non-routine chemistry, but it has to be kept in mind that the availability of different oxidation states allows adapting the physico-chemical properties of the complex label to those of the vector.
The introduction of the organometallic complex [99mTc(CNR)6]+ for imaging purposes by Davison and co-workers39 opened the way for low-valent organometallic complexes. In this tradition, [99mTc(OH2)3(CO)3]+ represents a versatile precursor for the labeling of biomolecules under aqueous conditions.40 Due to the small size of peptides, small complexes are desirable41–43 and the cyclopentadienyl (η5-Cp) ligand is therefore a consequent choice. Notably, it mimics phenyl rings in pharmaceuticals or biomolecules such as amino acids.44–49 Starting from [99mTcO4]−, Wenzel and co-workers reported the preparation of half-sandwich complexes [(η5-Cp)99mTc(CO)3] by a double ligand transfer approach.50 That methodology was applied to the synthesis of 99mTc radiolabeled peptides comprising a Cp core structure, utilizing octreotide in organic solvent and under autoclave conditions.51,52 Several peptides, including octreotide, were labelled with the ground state 99Tc along the same double ligand transfer strategy in a decent 70% yield by Katzenellenbogen and co-workers.52
In extending our ongoing research towards the labeling of biomolecules with [(η5-Cp-R)M(CO)3] (R = targeting group, M = 99mTc), we report herein the syntheses of PEPT2 targeting, radiolabeled tripeptides. These potential molecular imaging agents have been prepared by a metal mediated retro Diels–Alder reaction as reported for other small molecules.53,54 Here, we show that the concept can be extended to peptides as well. We selected tripeptides since different sequences exhibit low micromolar binding affinities for the PEPT2 receptor (Leu-Gly-Gly, Ki = 18 μM; Trp-Gly-Tyr, 1.7 μM; Met-Met-Met, 2.0 μM; Leu-Arg-Pro, 1.2 μM and Val-Ala-Leu, 9.0 μM) and to provide the proof of principle for peptide labeling with cyclopentadienyl based complexes.55
Results and discussion
Synthesis of peptidic, di-cyclopentadiene derivatives
Thiele's acid, (HCpCOOH)2 the Diels–Alder dimer of C5H5(COOH), was reported by Top et al. for the preparation of [(η5-CpCOOH)Re(CO)3] from [ReO4]−.56 We used the amide form of Thiele's acid [(HCpCONH-R)2] for the preparation of complexes [(η5-CpCONH-R)99mTc(CO)3] with R being natural biomolecules such as glycine or amino acids,53,54 pharmacophores like 2-benzylpiperidin-4-yl57 or carbonic anhydrase inhibitors of the aryl-sulfonamide type.58 Pursuing this approach, different tripeptide sequences were conjugated to Thiele's acid by peptide bond formation. The dimer 2 has been synthesized in a two-step reaction. The carboxylic group of Leu-Gly-Gly was activated/protected with acetylchloride–methanol to obtain 1 which was then coupled with Thiele's acid (Scheme 1). After silica gel chromatography, 2 was obtained as a white solid in 22% yield and characterized by ESI-MS and elemental analysis. To obtain analogous compounds but with other tripeptide sequences, the Thiele's acid linked bis-tripeptides 5–8 were prepared by standard solid phase peptide synthesis (SPPS) based on 2-chlorotrityl chloride resin (Scheme 2). First, Fmoc amino acids with side chain protecting groups were coupled to the resin. Then, Fmoc protecting groups were removed and the sequence extended by standard SPPS procedures (HOBt, HBTU in the presence of DIEA) to obtain resin bound, side chain protected tripeptides. Finally, dimerization was achieved directly on the solid support by cross linking the N-terminal amino groups of the peptides with the two carboxylic acid groups of Thiele's acid, activated by HATU in the presence of DIEA. It should be emphasized at this point that this coupling reaction was done with the pure endo–endo isomer of Thiele's acid. Subsequently, the product was cleaved from the resin and deprotected with TFA, H2O and TIPS. After purification by RP-HPLC, dimers 5–8 were obtained as white solids in 25–30% yield and characterized by RP-HPLC and mass spectra (see ESI†). | ||
| Scheme 1 Synthesis of labelling precursor 2 and Re-complex 4 (a) CH3COCl–MeOH; (b) [(CpCOOH)Re(CO)3], Et3N, DCC HOBt; (c) [(HCpCOOH)2], Et3N, DCC HOBt; (d) LiOH·H2O, THF. | ||
 | ||
| Scheme 2 Solid phase synthesis of ligands and their corresponding Re-complexes: (a) the first Fmoc-amino acid, DIEA; (b) DIEA, MeOH, DCM; (c) 20% piperidine in DMF; (d) the second Fmoc-amino acid, HOBt, HBTU, DIEA; (e) repeat steps c, d and c for the coupling of the next amino acid; (f) [(HCpCOOH)2], HATU, DIEA; (g) TFA, TIPS, H2O; (h) [(CpCOOH)Re(CO)3], HATU, DIEA. | ||
Synthesis of rhenium complexes
Rhenium complexes [(η5-CpCONH-R)Re(CO)3] (R = peptide) are rare. Katzenellenbogen and co-workers reported one example in which [η5-CpCOOCH3M(CO)3] was synthesized by double ligand transfer reaction from pertechnetate/perrhenate, ferrocene derivative (as a Cp source) and [Cr(CO)6] (as a CO source and a reducing agent) in the presence of CrCl3. Ester hydrolysis and coupling to a peptide gave the corresponding imaging agent.52 The key intermediate [(η5-CpCOOH)M(CO)3] was also synthesized from perrhenate via reduction to [Re2(CO)10] with CO, followed by thermal reaction of C5H5COOH or Thiele's acid.56To assess the authenticity of the later prepared, homologues 99mTc complexes [(η5-CpCONH-tpe)99mTc(CO)3] by comparison of HPLC retention times, we have synthesized [(η5-CpCONH-tpe)Re(CO)3] with selected tripeptides directly from [(η5-CpCOOH)Re(CO)3]. The rhenium complex 4 (tpe = Leu-Gly-Gly) was synthesized in two steps. Compound 3 was obtained after amide coupling of the protected tripeptide 1 to [(η5-CpCOOH)Re(CO)3]. Alkaline deprotection with LiOH and column purification on silica gel gave 4 as a white solid in 42% yield (Scheme 1). The other rhenium complexes [(η5-CpCONH-tpe)Re(CO)3] (tpe = Trp-Gly-Tyr, 9, Leu-Arg-Pro 10, Val-Ala-Leu, 11 and Met-Met-Met 12) were directly accessible on a solid support by amide coupling of [(η5-CpCOOH)Re(CO)3] to the corresponding resin bound tripeptides, received as described for 5–8 (Scheme 2). After RP-HPLC purification, complexes 9–12 were obtained as white solids in 40–45% yield. The 1H NMR spectra (see ESI†) of all Re complexes 4, 9–12, in CD3OD supported the proposed structures. The η5-Cp region displayed three peaks as doublets, triplets or multiplets, depending on the conjugated tripeptide to the Cp ring, between 5.5 and 6.3 ppm.
Stereochemical activity due to the vicinity of CpCoNHCH-carbon is observed in all [(η5-Cp(CONH-tpe))Re(CO)3] complexes, as confirmed by 2D NMR spectra (see ESI†). As Cp is planar, the two protons next to the α-carbon of the first amino acid appear separately at about 6.3 and 6.1 ppm, while the other two, more remote Cp protons are represented by one signal around 5.5 ppm. The aromatic protons of the conjugate 9 are multiplets between 6.6 and 7.5 ppm. 13C NMR spectra of 4 and 9–12 in CD3OD indicate five well resolved carbon signals between 94 and 86 ppm.
The infrared spectra in KBr show the two characteristic strong vibration bands at 2028 and 1932 cm−1, associated with the fac-[Re(CO)3]+ fragment. Furthermore, ESI-MS measurements in methanol assessed the correct m/z peaks at 809.1 ([9 + Na]+), 685.9 ([11 + Na]+), 747.0 ([10 + H]+) and 773.9 [12 + H]+ respectively. Complex 4 in MeOH showed a main peak at 605.9 in negative mode [M − H]− (see ESI†).
Synthesis of 99mTc complexes, labelling
Wenzel et al.50,59 reported the first approach to half-sandwich complexes of the type [(η5-Cp-R)99mTc(CO)3] by the so-called double ligand transfer reaction directly from [99mTcO4]−, ferrocene, Mn(CO)5Br and SnCl2 as a reducing agent in organic solvents at 150 °C. Although performed in one step, these conditions are not versatile for radiopharmaceutical purposes due to the concerted formation of manganese homologues. Improvements by Katzenellenbogen have been mentioned above but temperature was still as high as 160 °C.52 Direct reaction of (HCpCOOH)2 with [99mTc(OH2)3(CO)3]+ or [99mTcO4]− in aqueous solution is more convenient and leads directly to a multitude of [(η5-CpCONH-R)99mTc(CO)3] in very good yields with different bioactive groups “R”.53,54,57,58 Peptides, in contrast to small pharmacophores, possess often competing coordinating groups in their side chains and it is not immediately obvious if Cp can compete for these binding sites. On the basis of the reported aqueous synthesis of [(η5-Cp-R)99mTc(CO)3] type complexes, we extend the approach to [(η5-Cp-tpe)99mTc(CO)3] compounds (Scheme 3). We used two different procedures to synthesize the 99mTc complexes 13–17 starting from their corresponding Diels–Alder precursors 2 and 5–8. In particular, at alkaline pH, the reaction time and temperature were varied in order to find optimal conditions for dimer cleavage and labelling. Method A involved the classical “one pot” reaction of the dimerized tripeptides with [99mTcO4]− in the presence of boranocarbonate [H3BCOOH]− without changing the pH of the vial. Method B is a two-step procedure in which [99mTc(OH2)3(CO)3]+ was synthesized first under strongly alkaline conditions, the solution neutralized and only then reacted with the corresponding ligands. Method B was not suitable for the preparations of 13–14 and 16–17 since reactions were slow and yielded less product after heating at 90 °C for 30 to 200 min than with method A. Side reactions were observed especially for 14 and 16. Complex 15 was not formed at all by this method. It is likely that the guanidine group in arginine (NH2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH) of 15 coordinates faster with [99mTc(OH2)3(CO)3]+, thereby preventing coordination of the Cp ring. However, 15 was synthesized by method A in 26% yield (see ESI†). The direct, one pot method A was faster and gave much better yields for all complexes. Only small amounts of side products were observed. In labelling reactions at high dilution (99mTc), concentrations are important. Labelling yields with less than 1 mg of peptide strongly decreased for kinetic reasons. The labelling proceeded more slowly with smaller amounts (thermal heating), leading to less products and more side products. At amounts higher than 2 to 5 mg, labelling yields for all peptides 2, 5–8 were comparable. It was generally observed that yields started to decrease after a certain time of heating, indicating decomposition of the product after prolonged thermal treatment. For example, the 99mTc-complex 13 was formed after 45 min in 8%, after 120 min in 30% and finally in 54% yield after 210 min. The complex 14 reached only 25% after 60 min and yields then continuously decreased due to decomposition. The yield of complex 15 remained almost constant at 26% during 60–210 min and 16 achieved about 26% after 160 min. The reaction with ligand 8 (complex 17) gave the best yields, 65% after 60 min, but then began to decrease slowly to reach 50% after 190 min of heating The time dependence of the radiochemical yield by the “one pot” method at 90 °C is given in ESI.†
NH) of 15 coordinates faster with [99mTc(OH2)3(CO)3]+, thereby preventing coordination of the Cp ring. However, 15 was synthesized by method A in 26% yield (see ESI†). The direct, one pot method A was faster and gave much better yields for all complexes. Only small amounts of side products were observed. In labelling reactions at high dilution (99mTc), concentrations are important. Labelling yields with less than 1 mg of peptide strongly decreased for kinetic reasons. The labelling proceeded more slowly with smaller amounts (thermal heating), leading to less products and more side products. At amounts higher than 2 to 5 mg, labelling yields for all peptides 2, 5–8 were comparable. It was generally observed that yields started to decrease after a certain time of heating, indicating decomposition of the product after prolonged thermal treatment. For example, the 99mTc-complex 13 was formed after 45 min in 8%, after 120 min in 30% and finally in 54% yield after 210 min. The complex 14 reached only 25% after 60 min and yields then continuously decreased due to decomposition. The yield of complex 15 remained almost constant at 26% during 60–210 min and 16 achieved about 26% after 160 min. The reaction with ligand 8 (complex 17) gave the best yields, 65% after 60 min, but then began to decrease slowly to reach 50% after 190 min of heating The time dependence of the radiochemical yield by the “one pot” method at 90 °C is given in ESI.†
 | ||
| Scheme 3 Reaction of [99mTcO4]− with dimers 2, 5–8 in the presence of Na[H3BCO2H], Na2B4O7·10H2O and Na2tartrate·2H2O. | ||
From a practical point of view, the one step procedure directly from [99mTcO4]− is favoured and more efficient indeed. It allows to simply merge the [99mTcO4]− generator eluate with a vial containing all the compounds for the preparation of the labelled peptides. The two step procedure would demand two vials and two reactions.
In the recent literature, the application of microwave heating has been described as a viable and often superior method as compared to thermal heating. Reaction times are shorter and yields often higher.60–63 We labelled compounds 2 and 5–8 with the microwave technique in order to compare conditions with the purely thermal process.
Under microwave irradiation, complexes 13–17 formed after 50 min at 110 °C in much better yields as compared to the thermal process, 56% of 13, 66% of 14, 24% of 15, 56% of 16 and 90% of 17 respectively, even with relatively small amounts of the corresponding ligands (Fig. 1). Although yields strongly depend on the nature of the tripeptides, microwave heating is clearly superior. It also supports the result of Cp directed labelling even in the presence of competing side chains in peptides such as the three thioether groups in Met-Met-Met of compound 12. Furthermore, these reaction conditions also hydrolyzed the terminal ester group in 2 leading directly to 13. To make high specific activity radiolabelled peptides available, the 99mTc-complexes 13–17 can be purified by semi-preparative RP-HPLC. The differences in retention times of complexes 13–17 from their corresponding precursors 2 and 5–8 are sufficiently large to enable good separation. Purities >98% can be achieved and coinjection with the rhenium homologues 4, 9–12 and HPLC retention times comparison confirmed the authenticities of the 99mTc compounds. An example is given in Fig. 2 and in ESI,† respectively. It should be noted that the labelling process will lead to the formation of one molecule of unlabelled peptide, resulting from the cleavage of the dimers 2, 5–8. Without separation, they could compete for receptor binding with the labelled peptides; however, their quantities are minute (in the sub-picomol range) and, in principle, equal to the concentration of the 99mTc labelled peptides.
 | ||
| Fig. 1 Time dependent radiochemical yields of 99mTc complexes 13–17 from ligands 2, 5–7 (0.6 mg), and 8 (1.5 mg) under microwave conditions at 110 °C. | ||
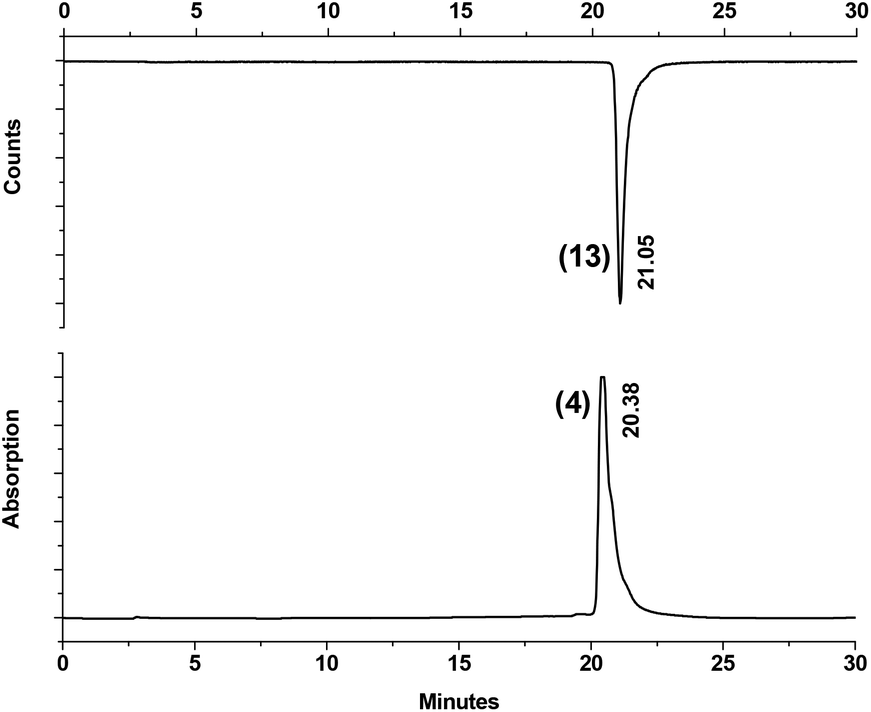 | ||
| Fig. 2 HPLC traces of Re-complex 4 and the corresponding 99mTc-complex 13 as received by coinjection of the two compounds. Time difference due to detector separation. | ||
Conclusion
The preparation of piano-stool type complexes [(η5-C5H4CONH-R)99mTc(CO)3)] conjugated to small, bioactive molecules “R” in a one pot synthesis from [99mTcO4]− was established. Since small, pharmaceutically active compounds resist labelling conditions, yields are generally quantitative. Due to the important role of peptides in molecular imaging, we extended the concept to those, more complex biomolecules. Peptides, conjugated via Thiele's acid to form dimers, as prepared in this study, represent the direct precursors for labelled peptides of the form [(η5-C5H4CONH-pep)99mTc(CO)3]. Direct labelling in one step from [99mTcO4]− by thermal heating or in a microwave oven resulted in the desired products and in yields between 25 and 90%, depending on the peptide sequence and the presence of coordinating groups in the peptide side chains. HPLC purification gave radiochemically pure and stable 99mTc labelled peptides. Thus, we exemplified with potentially PEPT2 binding tripeptides that the concept of retro-Diels–Alder reactions can be extended to more complex biomolecules such as peptides.Experimental procedures
General
Leu-Gly-Gly-OH was obtained from NovaBiochem. The other tripeptides were synthesized manually using standard Fmoc synthesis procedures. Fmoc-amino acids with side chain protecting groups, 2-chlorotrityl chloride resin, were obtained from Iris Biotech GmbH, DIEA from Fluka, HOBt from Acros Organics, HBTU from Fluorochem and HATU from Sigma Aldrich. Thiele's acid and [(Cp-COOH)Re(CO)3] were prepared according to literature procedures.56,64,65 ESI-MS, Bruker Esquire HCT (ESI) instrument, 1H NMR and 13C NMR spectra were recorded on Bruker Advance 500 MHz or 400 MHz spectrometers. RP-HPLC was performed on Merck Hitachi LaChrom L7200 with a tuneable UV detector, a radiodetector, separated by a Teflon tube causing about 0.4–0.7 min delay compared to UV/vis detection. Analytical separations were performed on a Nucleosil C-18 column (100 Å, 5 μm, 250 × 4 mm) which was eluted with a flow rate of 0.5 ml min−1, using 0.1% TFA in H2O (solvent A) and methanol (solvent B) as eluents with variable gradients (0–3 min, 100% A; 3–3.1 min, 0 to 25% B; 3.1–9 min, 25% B; 9–9.1 min, 25% B to 34% B; 9.1–20 min, 34% B to 100% B; 20–25 min, 100% B; 25–25.1 min, 100% B to 100% A; 25.1–30 min, 100% A). Preparative HPLC was performed on a Varian Pro Star system using a Macherey-Nagel VP 250/30 Nucleosil 100-7 C18 column with flow rates of 30 ml min−1. The solvents were 0.1% TFA in H2O (solvent A) and methanol (solvent B).The microwave oven was a power controlled Biotage Initiator EXP microwave system instrument. Labelling was performed in a septum closed vial at constant temperature.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:1) as an eluent, which afforded 40 mg of 2 (20%). Elemental analysis calcd (%) for C34H50N6O10: C 58.11, H 7.17, N 11.96; found: C 58.48, H 7.31, N 11.56; MS-ESI (m/z): [M + Na]+ calcd for C34H50N6O10Na, 725.3; found, 725.3.
:20:1 as an eluent to give 70 mg of 3 (38%). 1H NMR (400 MHz, d4-MeOD) δ ppm: 6.35–6.34 (m, 1H, Cp), 6.19–6.18 (m, 1H, Cp), 5.59–5.56 (m, 2H, Cp), 4.51–4.48 (m, 1H, CH), 4.01–3.81 (m, 4H, CH2), 3.71 (s, 3H, OCH3), 1.75–1.65 (m, 3H, CH, CH2), 0.98–0.93 (m, 6H, CH3); MS-ESI (m/z): [M + Na]+ calcd for C20H24N3O8ReNa, 644.1; found, 644.0.
:3:1:0.2 as an eluent which afforded 22 mg of 4 (42%). IR (cm−1, KBr): 2028, 1932, 1638, 1543, 1417, 1369, 1263, 1186, 1033. 1H NMR (500 MHz, d4-MeOD) δ ppm: 6.35 (t, J = 1.3 Hz, 1H, Cp), 6.19 (d, J = 1.5 Hz, 1H, Cp), 5.59–5.57 (m, 2H, Cp), 4.54–4.50 (m, 1H, CH), 3.98–3.83 (m, 4H, CH2), 1.75–1.65 (m, 3H, CH2, CH), 0.98–0.94 (m, 6H, CH3); 13C NMR (125 MHz, d4-MeOD) δ ppm: 194.1, 175.3, 173.0, 172.1, 165.5, 94.6, 89.5, 88.1, 86.8, 86.4, 53.8, 43.5, 41.9, 41.4, 26.2, 23.7, 21.8; elemental analysis calcd (%) for C19H22N3O8Re: C 37.62, H 3.66, N 6.93; found: C 37.25, H 3.91, N 6.69; MS-ESI (m/z): [M − H]− calcd for C19H21N3O8Re, 606.1; found, 605.9.
:15:1) as an eluent, which afforded 40 mg of 2 (20%). Elemental analysis calcd (%) for C34H50N6O10: C 58.11, H 7.17, N 11.96; found: C 58.48, H 7.31, N 11.56; MS-ESI (m/z): [M + Na]+ calcd for C34H50N6O10Na, 725.3; found, 725.3.
:20:1 as an eluent to give 70 mg of 3 (38%). 1H NMR (400 MHz, d4-MeOD) δ ppm: 6.35–6.34 (m, 1H, Cp), 6.19–6.18 (m, 1H, Cp), 5.59–5.56 (m, 2H, Cp), 4.51–4.48 (m, 1H, CH), 4.01–3.81 (m, 4H, CH2), 3.71 (s, 3H, OCH3), 1.75–1.65 (m, 3H, CH, CH2), 0.98–0.93 (m, 6H, CH3); MS-ESI (m/z): [M + Na]+ calcd for C20H24N3O8ReNa, 644.1; found, 644.0.
:3:1:0.2 as an eluent which afforded 22 mg of 4 (42%). IR (cm−1, KBr): 2028, 1932, 1638, 1543, 1417, 1369, 1263, 1186, 1033. 1H NMR (500 MHz, d4-MeOD) δ ppm: 6.35 (t, J = 1.3 Hz, 1H, Cp), 6.19 (d, J = 1.5 Hz, 1H, Cp), 5.59–5.57 (m, 2H, Cp), 4.54–4.50 (m, 1H, CH), 3.98–3.83 (m, 4H, CH2), 1.75–1.65 (m, 3H, CH2, CH), 0.98–0.94 (m, 6H, CH3); 13C NMR (125 MHz, d4-MeOD) δ ppm: 194.1, 175.3, 173.0, 172.1, 165.5, 94.6, 89.5, 88.1, 86.8, 86.4, 53.8, 43.5, 41.9, 41.4, 26.2, 23.7, 21.8; elemental analysis calcd (%) for C19H22N3O8Re: C 37.62, H 3.66, N 6.93; found: C 37.25, H 3.91, N 6.69; MS-ESI (m/z): [M − H]− calcd for C19H21N3O8Re, 606.1; found, 605.9.
Procedure for synthesis of resin bound tripeptides
The resin bound tripeptides were synthesized manually by standard Fmoc solid-phase peptide synthesis; the 2-chlorotrityl chloride resin (1 g, 1.56 mmol g−1) was loaded into a 20 ml fritted glass column under N2 and washed with DMF (3 × 8 ml) and DCM (3 × 8 ml). The first amino acid (0.78 mmol, 1 eq.) was dissolved in 10 ml of dry DCM and then DIEA (3.12 mmol, 4 eq.) was added. The reaction mixture was transferred into a column and then shaken for 2 h. The solvent was filtered, the resin was washed with DCM, DMF and then with DCM, 3 × 7 ml each. For capping, the resin was treated with DCM–MeOH–DIEA 30:4:2 (3 × 12 ml) for 2 min each, followed by washing with DMF and DCM 3 × 7 ml. The Fmoc protecting group was removed by repetitive treatment with 20% piperidine in DMF (10 × 7 ml, 2 min) followed by washing with DMF, DCM and then with DMF 3 × 7 ml each. The deprotected resin and the progress of amino acid coupling were monitored by the colour change with ninhydrin. The second Fmoc protected amino acid (3.12 mmol, 4 eq.), HOBt (3.12 mmol, 4 eq.), and HBTU (3.12 mmol, 4 eq.) were dissolved in 7 ml of DMF, and then DIEA (4.68 mmol, 6 eq.) was added. This coupling mixture was transferred into the fritted glass vessel with resin and was reacted for 2 h. After Fmoc deprotection and washing, the third amino acid was coupled analogously to obtain resin bound tripeptide with a free N-terminus.
General procedure for synthesis of 5–8
Thiele's acid (HCpCOOH)2 (0.5 eq., based on the initial amount of the first amino acid) and HATU (2 eq.) were dissolved in 10 ml DMF. DIEA (5 eq.) was added and the reaction mixture transferred into the column containing the resin bound peptide and reacted for 48 h. This resulted in cross linking of adjacent peptides on the resin to the two carboxylic acid groups from Thiele's acid. The resulting peptide-dimers were cleaved from the resin and their side chains deprotected after treatment with 95% TFA, 2.5% H2O and 2.5% TIPS under N2 for 2 h. For peptide-dimers without side chain protecting groups, milder conditions 1% TFA in DCM (30 ml) were applied for 30 min. The solutions were filtered, the resins rinsed with TFA and the combined solutions concentrated. Precipitation with ice-cold MTBE and centrifugation gave the crude dimers [(HCpCONH-tpe-OH)2]. The precipitate was dissolved in MeOH–H2O, purified by preparative RP-HPLC and lyophilized to afford the products as white solids in 25–30% yield. The purified peptides 5–8 were characterized by RP-HPLC and mass spectrometry.General procedure for the syntheses of [(η5-CpCONH-tpe-OH)Re(CO)3] 9–12
[(η5-Cp-COOH)Re(CO)3] was coupled to resin bound tripeptides by a standard coupling procedure with HATU (2 eq.) and DIEA (5 eq.) for 22 h at r.t. After cleavage from the resin, deprotection and concentration, the crude complexes were precipitated with cold MTBE. All solvents were removed in vacuo and the residues dissolved in a small amount of DCM and precipitated with hexane. Finally, the crude Re-complexes [(η5-CpCONH-tpe-OH)Re(CO)3] were purified by preparative RP-HPLC and lyophilized to obtain 9–12 as white solids in 40–45% yield. The purified Re-complexes were characterized by RP-HPLC, mass spectrometry, and 1H and 13C NMR (see ESI†). Analytical data are as follows.Tripeptide labelling. The tripeptide dimers 5–8 (2–5 mg) were loaded into a vial which was sealed and flushed with N2. Freshly prepared [99mTc(H2O)3(CO)3]+ (1 ml) was added and the vial heated at 85–90 °C for 30–300 min. Coinjection with the corresponding Re-complex allowed identification of the 99mTc product. Before HPLC purification of the labelled peptide, specific activity was estimated to be about 2 mCi μmol−1. Since unlabelled and labelled peptides can be well separated on HPLC, a maximum specific activity of up to 500 Ci μmol−1 is feasible.
Acknowledgements
This work was financially supported by the Higher Education Commission, Pakistan (PIN no. IRSIP 18 Ps 06) and the University of Zürich, Switzerland.Notes and references
- W. A. P. Breeman, D. J. Kwekkeboom, E. de Blois, M. de Jong, T. J. Visser and E. P. Krenning, Anticancer Agents Med. Chem., 2007, 7 Search PubMed
.
- J. D. G. Correia, A. Paulo, P. D. Raposinho and I. Santos, Dalton Trans., 2011, 40 Search PubMed
- J. C. Reubi and H. R. Maecke, J. Nucl. Med., 2008, 49, 1735 CrossRef CAS PubMed
- M. Schottelius and H. J. Wester, Methods, 2009, 48, 161 CrossRef CAS PubMed
- C. Bolzati, F. Refosco, A. Marchiani and P. Ruzza, Curr. Med. Chem., 2010, 17, 2656 CrossRef CAS
- J. Fichna and A. Janecka, Bioconjugate Chem., 2003, 14, 3 CrossRef CAS
- S. Liu and S. Chakraborty, Dalton Trans., 2011, 40, 6077 RSC
- C. Bolzati, D. Carta, N. Salvarese and F. Refosco, Anticancer Agents Med. Chem., 2012, 12, 428 CrossRef CAS
- G. R. Morais, A. Paulo and I. Santos, Organometallics, 2012, 31, 5693 CrossRef CAS
- D. Blok, R. I. J. Feitsma, P. Vermeij and E. J. K. Pauwels, Eur. J. Nucl. Med., 1999, 26, 1511 CrossRef CAS
- O. C. Boerman, W. J. G. Oyen and F. H. M. Corstens, Semin. Nucl. Med., 2000, 30, 195 CrossRef CAS PubMed
- S. M. Okarvi, Nucl. Med. Commun., 1999, 20, 1093 CrossRef CAS PubMed
- R. E. Weiner and M. L. Thakur, Semin. Nucl. Med., 2001, 31, 296 CrossRef CAS PubMed
- M. E. Ganapathy, M. Brandsch, P. D. Prasad, V. Ganapathy and F. H. Leibach, J. Biol. Chem., 1995, 270, 25672 CrossRef CAS PubMed
- M. E. Ganapathy, P. D. Prasad, B. Mackenzie, V. Ganapathy and F. H. Leibach, BBA-Biomembranes, 1997, 1324, 296 CrossRef CAS
- J. Neumann, M. Bruch, S. Gebauer and M. Brandsch, Eur. J. Biochem., 2004, 271, 2012 CrossRef CAS PubMed
- H. Shen, R. F. Keep, Y. J. Hu and D. E. Smith, J. Pharmacol. Exp. Ther., 2005, 315, 1101 CrossRef CAS PubMed
- H. Shen, S. M. Ocheltree, Y. J. Hu, R. F. Keep and D. E. Smith, Drug Metab. Dispos., 2007, 35, 1209 CrossRef CAS PubMed
- C. Shu, H. Shen, U. Hopfer and D. E. Smith, Drug Metab. Dispos., 2001, 29, 1307 CAS
- M. Sugawara, W. Huang, Y. L. Fei, F. H. Leibach, V. Ganapathy and M. E. Ganapathy, J. Pharm. Sci., 2000, 89, 781 CrossRef CAS
- U. V. Berger and M. A. Hediger, Anat. Embryol., 1999, 199, 439 CrossRef CAS
- S. T. Dieck, H. Heuer, J. Ehrchen, C. Otto and K. Bauer, Glia, 1999, 25, 10 CrossRef CAS
- T. Fujita, T. Kishida, M. Wada, N. Okada, A. Yamamoto, F. H. Leibach and V. Ganapathy, Brain Res., 2004, 997, 52 CrossRef CAS PubMed
- D. A. Groneberg, F. Doring, M. Nickolaus, H. Daniel and A. Fischer, Pharm. Res., 2002, 19, 1209 CrossRef CAS
- D. A. Groneberg, F. Doring, S. Theis, M. Nickolaus, A. Fischer and H. Daniel, Am. J. Physiol. Endocrinal. Metab., 2002, 282, E1172 CAS
- D. A. Groneberg, M. Nickolaus, J. Springer, F. Doring, H. Daniel and A. Fischer, Am. J. Pathol., 2001, 158, 707 CrossRef CAS
- S. M. Ocheltree, R. F. Keep, H. Shen, D. L. Yang, B. A. Hughes and D. E. Smith, Pharm. Res., 2003, 20, 1364 CrossRef CAS
- S. Ramamoorthy, W. Liu, Y. Y. Ma, T. L. Yangfeng, V. Ganapathy and F. H. Leibach, BBA-Biomembranes, 1995, 1240, 1 CrossRef
- H. Shen, D. E. Smith, T. X. Yang, Y. N. G. Huang, J. B. Schnermann and F. C. Brosius, Am. J. Physiol. Endocrinal. Metab., 1999, 276, F658 CAS
- A. Ruhl, S. Hoppe, I. Frey, H. Daniel and M. Schemann, J. Comp. Neurol., 2005, 490, 1 CrossRef PubMed
- T. Nakanishi, I. Tamai, A. Takaki and A. Tsuji, Int. J. Cancer, 2000, 88, 274 CrossRef CAS
- M. Brandsch, C. Brandsch, P. D. Prasad, V. Ganapathy, U. Hopfer and F. H. Leibach, FASEB J., 1995, 9, 1489 CAS
- H. Daniel, E. L. Morse and S. A. Adibi, J. Biol. Chem., 1992, 267, 9565 CAS
- T. Terada, K. Sawada, M. Irie, H. Saito, Y. Hashimoto and K. Inui, Pflug. Arch. Eur. J. Phys., 2000, 440, 679 CrossRef CAS
- C. Tiruppathi, V. Ganapathy and F. H. Leibach, J. Biol. Chem., 1990, 265, 14870 CAS
- A. Biegel, I. Knutter, B. Hartrodt, S. Gebauer, S. Theis, P. Luckner, G. Kottra, M. Rastetter, K. Zebisch, I. Thondorf, H. Daniel, K. Neubert and M. Brandsch, Amino Acids, 2006, 31, 137 CrossRef CAS PubMed
- J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43 RSC
- S. S. Jurisson and J. D. Lydon, Chem. Rev., 1999, 99, 2205 CrossRef CAS PubMed
- M. J. Abrams, A. Davison, A. G. Jones, C. E. Costello and H. Pang, Inorg. Chem., 1983, 22, 2798 CrossRef CAS
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 123, 3135 CrossRef CAS
- M. Langer and A. G. Beck-Sickinger, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 71 CrossRef CAS
- S. Liu and D. S. Edwards, Bioconjugate Chem., 2001, 12, 653 CrossRef CAS
- S. M. Okarvi, Med. Res. Rev., 2004, 24, 685 CrossRef CAS
- G. Jaouen, Chem. Brit., 2001, 37, 36 CAS
- G. Jaouen, S. Top, A. Vessieres and R. Alberto, J. Organomet. Chem., 2000, 600, 23 CrossRef CAS
- G. Jaouen, S. Top, A. Vessieres, P. Pigeon, G. Leclercq and I. Laios, Chem. Commun., 2001, 383 RSC
- S. Top, B. Dauer, J. Vaissermann and G. Jaouen, J. Organomet. Chem., 1997, 541, 355 CrossRef CAS
- S. Top, H. Elhafa, A. Vessieres, J. Quivy, J. Vaissermann, D. W. Hughes, M. J. Mcglinchey, J. P. Mornon, E. Thoreau and G. Jaouen, J. Am. Chem. Soc., 1995, 117, 8372 CrossRef CAS
- S. Top, A. Vessieres, P. Pigeon, M. N. Rager, M. Huche, E. Salomon, C. Cabestaing, J. Vaissermann and G. Jaouen, ChemBioChem, 2004, 5, 1104 CrossRef CAS PubMed
- M. Wenzel and C. Klinge, J. Labelled Compd. Radiopharm., 1994, 34, 981 CrossRef CAS
- T. W. Spradau, W. B. Edwards, C. J. Anderson, M. J. Welch and J. A. Katzenellenbogen, Nucl. Med. Biol., 1999, 26, 1 CrossRef CAS
- T. W. Spradau and J. A. Katzenellenbogen, Bioconjugate Chem., 1998, 9, 765 CrossRef CAS PubMed
- Y. Liu, B. Spingler, P. Schmultz and R. Alberto, J. Am. Chem. Soc., 2008, 130, 1554 CrossRef CAS PubMed
- H. W. P. N'Dongo, Y. Liu, D. Can, P. Schmutz, B. Spingler and R. Alberto, J. Organomet. Chem., 2009, 694, 981 CrossRef PubMed
- A. Biegel, S. Gebauer, M. Brandsch, K. Neubert and I. Thondorf, J. Med. Chem., 2006, 49, 4286 CrossRef CAS PubMed
- S. Top, J. S. Lehn, P. Morel and G. Jaouen, J. Organomet. Chem., 1999, 583, 63 CrossRef CAS
- H. W. P. N'Dongo, P. D. Raposinho, C. Fernandes, I. Santos, D. Can, P. Schmutz, B. Spingler and R. Alberto, Nucl. Med. Biol., 2010, 37, 255 CrossRef PubMed
- D. Can, B. Spingler, P. Schmutz, F. Mendes, P. Raposinho, C. Fernandes, F. Carta, A. Innocenti, I. Santos, C. T. Supuran and R. Alberto, Angew. Chem., Int. Ed., 2012, 51, 3354 CrossRef CAS PubMed
- M. Wenzel, J. Labelled Compd. Radiopharm., 1992, 31, 641 CrossRef CAS
- P. W. Causey, T. R. Besanger, P. Schaffer and J. F. Valliant, Inorg. Chem., 2008, 47, 8213 CrossRef CAS PubMed
- P. W. Causey, T. R. Besanger and J. F. Valliant, J. Med. Chem., 2008, 51, 2833 CrossRef CAS PubMed
- A. E. C. Green, P. W. Causey, A. S. Louie, A. F. Armstrong, L. E. Harrington and J. F. Valliant, Inorg. Chem., 2006, 45, 5727 CrossRef CAS PubMed
- S. H. Park, H. J. Gwon, S. H. Jang and H. Lee, Chem. Lett., 2006, 35, 1304 CrossRef CAS
- A. P. Marchand, I. N. N. Namboothiri, S. B. Lewis, W. H. Watson and M. Krawiec, Tetrahedron, 1998, 54, 12691 CrossRef CAS
- J. Thiele, Chem. Ber., 1990, 33, 666 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, HPLC chromatograms and mass spectra of new compounds. See DOI: 10.1039/c3ob41866a |
| This journal is © The Royal Society of Chemistry 2014 |