 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of pressure on the post-synthetic modification of a nanoporous metal–organic framework†
Scott C.
McKellar
a,
Alexander J.
Graham
a,
David R.
Allan
b,
M. Infas H.
Mohideen
c,
Russell E.
Morris
c and
Stephen A.
Moggach
*a
aEaStCHEM School of Chemistry, The Centre for Science at Extreme Conditions, University of Edinburgh, Kings Buildings, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: s.moggach@ed.ac.uk; Tel: +44 131 651 7152
bDiamond Light Source Ltd, Harwell Science and Innovation Campus, Diamond House, Oxfordshire, OX11 0DE, UK
cEaStCHEM School of Chemistry, University of St Andrews, Purdie Building, St Andrews, KY16 9ST, UK
First published on 24th February 2014
Abstract
Here we report four post-synthetic modifications, including the first ever example of a high pressure-induced post-synthetic modification, of a porous copper-based metal–organic framework. Ligand exchange with a water ligand at the axial metal site occurs with methanol, acetonitrile, methylamine and ethylamine within a single-crystal and without the need to expose a free metal site prior to modification, resulting in significant changes in the pore size, shape and functionality. Pressure experiments carried out using isopropylalcohol and acetaldehyde, however, results in no ligand exchange. By using these solvents as hydrostatic media for high-pressure single-crystal X-ray diffraction experiments, we have investigated the effect of ligand exchange on the stability and compressibility of the framework and demonstrate that post-synthetic ligand exchange is very sensitive to both the molecular size and functionality of the exchanged ligand. We also demonstrate the ability to force hydrophilic molecules into hydrophobic pores using high pressures which results in a pressure-induced chemical decomposition of the Cu-framework.
Introduction
Nanoporous metal–organic frameworks (MOFs) have an array of potential applications including gas storage,1–3 separation processes4–6 and molecular recognition.7,8 As such, MOF-themed research papers now number in the hundreds per annum,9 with many reporting synthetic or crystal engineering approaches to making more sophisticated, novel frameworks. Recently, this has led to strong interest in the concept of post-synthetic modification (PSM).10Traditionally, structural variation in MOFs is achieved mainly through judicious choice of metal source and chemical modification of the organic linking molecules, with accompanying changes in framework pore size, shape and selectivity giving rise to an increasingly diverse array of sorption and catalytic properties. Though functionalisation of the organic component is typically performed prior to the synthesis of the MOF, it can also be achieved post-synthesis. PSM has proven to be a very elegant technique in which to modify MOFs after they have been formed, since it offers the potential to tune the pore size, shape and functionality of a crystalline framework while conserving the integrity of the structure, and is an attractive route for covalent modification that is unachievable by established synthetic routes.
To date, several approaches have been used for PSM of MOFs. One method involves covalently modifying the organic linker by introducing new functional groups, as exemplified by the post-modification of free pyridyl groups to N-methylpyridinium cations in the 2D homochiral MOF, D-POST-1 ([C72H78N12O31Zn32−]n·2n(H3O+)·7n(H2O)). This was achieved by treating the crystalline material with excess iodomethane in dimethylformamide (DMF).11 Another example is a Zn-based ‘paddle wheel’ MOF, Zn2(TCPB)(DPG) (TCPB = 1,2,4,5-tetrakis(4-carboxyphenyl)-benzene; DPG = meso-1,2-bis(4-pyridyl)-1,2-ethanediol), in which Hupp and co-workers were able to introduce free carboxylic acid groups by reacting the native MOF with succinic anhydride.12 A second method involves exposing a free site on the metal, usually by first removing a ligand. This route has been very successful in modifying the framework HKUST-1 ([Cu3(BTC)2(H2O)3]n, BTC = benzene-1,3,5-tricarboxylate) in which an axial water ligand can be exchanged for pyridines once the water ligand has been removed.13 Hupp et al. also demonstrated in a 3D non-catenated Zn paddle wheel MOF, [Zn2(L)(DMF)2]n(DMF)n (L = (4,4′,4′′,4′′′-benzene-1,2,4,5-tetrayltetrabenzoic acid)), that various pyridine-based ligands could be introduced by removing the coordinated DMF solvent.14 More recently, great success has also been made using a third technique, which involves a post-synthetic ligand exchange reaction. In the mesoposous bio-MOF-100 for example, zinc–adeninate clusters (Zn8Ad4O28+; Ad = adeninate) are periodically linked with 4,4′-biphenyldicarboxylate (BPDC), which can be exchanged with progressively longer dicarboxylate linkers to form progressively larger cavities.15
Here, we have investigated the PSM behaviour of STAM-1 (St. Andrews MOF-1), a Cu-based framework with a remarkably high susceptibility to modification due to highly labile water ligands at the axial metal coordination site. Notably, the PSM occurs within a single crystal without the need to remove the water ligand prior to the ligand exchange. We also present the first ever example of a pressure-induced PSM of a MOF.
STAM-1 ([Cu3O21C30H24]n·5n(H2O); Fig. 1)16 is comprised of monomethyl-esterified BTC ligands linking five-coordinate Cu ‘paddle wheels’; a motif found in other similar Cu–MOFs such as HKUST-113 and MOF-1417 (Cu(II) 4,4′,4′′-benzene-1,3,5-triyl-tris(benzoate)). These paddle wheels consist of dimeric Cu tetracarboxylate units which form four symmetry-equivalent equatorial Cu–O bonds that measure ∼1.96 Å in length. The 5th (axial) coordination site is filled by a Jahn–Teller distorted Cu–O bond to a coordinated water molecule, which measures ∼2.15 Å. Each metal completes a pseudooctahedral coordination sphere with a long Cu–Cu contact (Cu⋯Cu ≈ 2.6 Å). The preparation of both HKUST-1 and STAM-1 involve similar reaction conditions. The synthesis of HKUST-1 involves reacting Cu(NO3)2·3(H2O) with BTC in a Teflon-lined autoclave using ethanol as a solvent. Exchanging the solvent for a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of water–methanol results in the formation of STAM-1, in which the BTC linker is monoesterified during synthesis. The paddle wheels in STAM-1 are connected through the monomethyl BTC ester linkers to form approximately triangular ‘cups’16 (see Fig. 1a) and the resulting framework forms interdigitated layers with two types of channel: one lined by the ester groups (hydrophobic), and others lined by the axial water molecules (hydrophilic), as shown in Fig. 1c. There are one hydrophobic and two hydrophilic channels per unit cell.
:50 mixture of water–methanol results in the formation of STAM-1, in which the BTC linker is monoesterified during synthesis. The paddle wheels in STAM-1 are connected through the monomethyl BTC ester linkers to form approximately triangular ‘cups’16 (see Fig. 1a) and the resulting framework forms interdigitated layers with two types of channel: one lined by the ester groups (hydrophobic), and others lined by the axial water molecules (hydrophilic), as shown in Fig. 1c. There are one hydrophobic and two hydrophilic channels per unit cell.
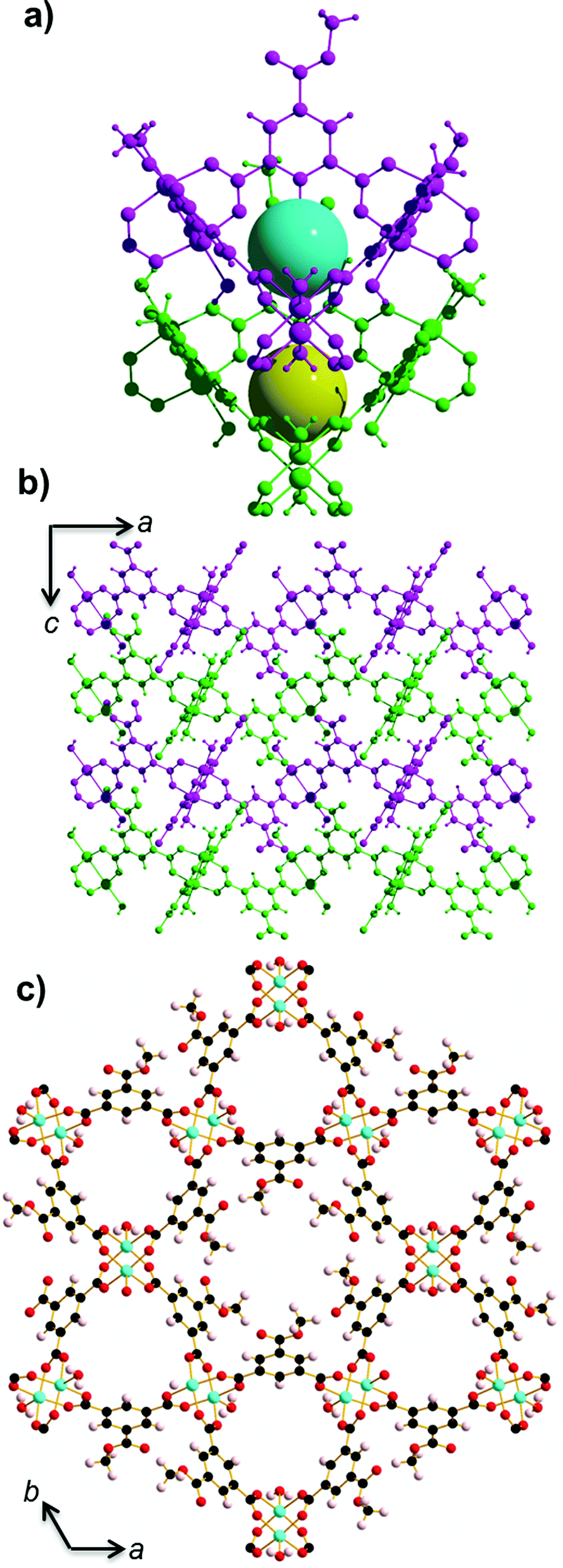 | ||
| Fig. 1 (a) Two cup-like structural units (block coloured purple and green) found in STAM-1 stacked on top of one another. The blue and yellow spheres represent the void space in the centre of each cup. (b) View of STAM-1 parallel to crystallographic b-axis showing how the cups form interdigitated layers. (c) Hydrophobic and hydrophilic pores in STAM-1 which form channels running parallel to the c-axis. The central large hydrophobic channel is surrounded by six hydrophilic channels. The disordered ester groups are shown occupying only one site. Colour scheme: O – red; C – black; Cu – turquoise; H – pale pink. | ||
In this study, single-crystal X-ray diffraction data has been used to show that the water ligands in the axial position of the Cu paddle wheel of STAM-1 can exchange with various organic solvents, both at ambient and above-ambient pressure, causing significant changes in the hydrophilic pore size and functionality. In our high-pressure experiments, the sample is surrounded with a pressure-transmitting (hydrostatic) medium, which is usually a liquid, and ensures that pressure is applied evenly to the sample. In our previous work on MOFs, we have shown that when the molecules encompassing this hydrostatic medium are small enough, they not only apply pressure to the sample, but enter the pores of the framework as well.18 Here, we demonstrate that high pressure can be used as a tool to induce ligand substitution with various solvents used as pressure-transmitting media, and investigate the effect of ligand exchange on the compressibility of the framework. We show that the selection of a hydrostatic medium of appropriate molecular size and functionality can be of paramount importance when investigating the high pressure PSM behaviour of MOFs.
Results and discussion
Post-synthetic ligand exchange of STAM-1 was observed using methanol, acetonitrile, methylamine and ethylamine, yielding four new phases of the material. However, a range of structural responses was observed based on both the pressure and type of solvent used. In particular, pressure was required to drive the acetonitrile ligand exchange. An interesting dependence on size has been observed for solvents with the same functional group (e.g. alcohols). Therefore, results will be discussed sequentially in the manuscript based on the solvent used (methanol, ethanol, isopropylalcohol, acetaldehyde, acetonitrile, methylamine, ethylamine and water).Ambient- and high-pressure ligand exchange with methanol
Single-crystal X-ray diffraction data were collected on a crystal of STAM-1 at room temperature and ambient pressure (see Table S1† for crystallographic data). Under these conditions, STAM-1 contains water in the hydrophilic channels which equates to three molecules of water per channel, and one molecule of water in the hydrophobic channel per unit cell (calculated using the SQUEEZE algorithm within the program PLATON19). The volume of both the hydrophobic and hydrophilic channels measures ∼209 and ∼142 Å3, respectively. The same crystal was then loaded in a diamond anvil cell (DAC), and surrounded with methanol as a hydrostatic medium. Upon loading to 0.2 GPa, a single-crystal to single-crystal phase transition was observed, to a previously unknown form of STAM-1 which we hereafter refer to as STAM-1MeOH (Fig. 2b and d; Table 1). This transition entailed a ligand exchange of the coordinated water at the axial position of the CuII paddle wheel for the methanol that was used as the hydrostatic medium. The ligand exchange reaction was accompanied by an increase in unit cell volume (2.19%), arising not only from the ligand exchange, but also from the filling of structural voids with the hydrostatic liquid. The a/b- and c-axes expanded by 0.34 and 0.30%, respectively. On undergoing the transition, a marked change in the pore volume and content was observed, with the hydrophobic pores increasing by 74 Å3, accompanied by an increase in solvent content equating to inclusion of two methanol molecules per hydrophobic pore (Table 2). This behaviour at pressure could be highly advantageous, as it allows the inclusion of hydrophilic molecules into hydrophobic pore channels that are inaccessible under ambient pressure conditions. Contrary to this, the hydrophilic channels decrease in both size and content due the ligand exchange reaction. The included water molecules observed at ambient pressure in STAM-1 are also forced out of the channels on undergoing the transition to STAM-1MeOH. In order to evaluate whether STAM-1MeOH was stable under ambient conditions, the pressure was decreased and the same crystal was then recovered from the DAC. Single-crystal X-ray diffraction data were then collected under ambient temperature and pressure conditions and the recovered sample was confirmed as the exchanged STAM-1MeOH structure. Ambient-pressure crystallographic data for modified STAM-1, after downloading from the DAC, is shown in Table 1. The exchange reaction had no associated change in crystal symmetry, but there are subtle yet significant changes in unit cell parameters and volume. The volume of the hydrophilic pores remains unchanged on decreasing pressure, but the pore content at ambient pressure is almost negligible. We propose that this evacuation of the pores on decreasing pressure is caused by the greater hydrophobicity of the pores produced by the methyl groups on the exchanged methanol ligand, which could be counteracted at 0.2 GPa, but not at ambient pressure.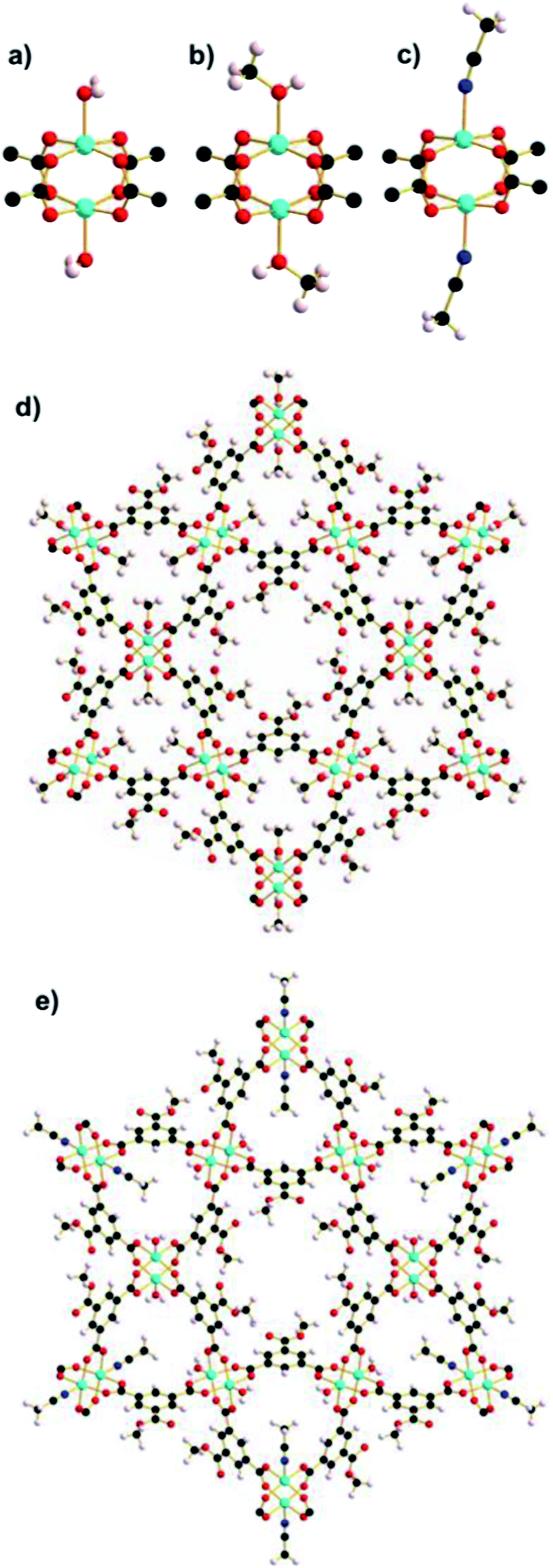 | ||
| Fig. 2 Cu paddle wheel units in (a) STAM-1, (b) STAM-1MeOH and (c) STAM-1MeCN. Reduction in size of the ‘hydrophilic’ channel resulting from ligand exchange in (d) STAM-1MeOH and (e) STAM-1MeCN, viewed parallel to the c-axis. The part-occupied acetonitrile ligands are shown at full occupancy in (c), but the part-exchange is represented in (e) by showing coordination at only one of the potential three coordination sites. | ||
| STAM-1MeOH | STAM-1MeCN | |
|---|---|---|
| Formula | C34.5H30Cu3O22.5 | C35H31Cu3N2.5O21 |
| Temperature (K) | 300 | 150.0(2) |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Crystal system | Trigonal | Trigonal |
| Space group |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 m1 |
Pm1 |
| a (Å) | 18.7034(3) | 18.5877(5) |
| b (Å) | 18.7034(3) | 18.5877(5) |
| c (Å) | 6.8780(1) | 6.8740(2) |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 120 | 120 |
| Volume (Å3) | 2083.69(6) | 2056.80(10) |
| Z | 2 | 2 |
| Density (g cm−3) | 1.545 | 1.602 |
| Mu (mm−1) | 1.600 | 1.613 |
| F(000) | 984.0 | 1004.0 |
| θ range (°) | 1.3–28.4 | 1.3–26.4 |
| Index ranges | −21 ≤ h ≤ 0 | −19 ≤ h ≤ 0 |
| 0 ≤ k ≤ 25 | 0 ≤ k ≤ 23 | |
| 0 ≤ l ≤ 9 | 0 ≤ l ≤ 8 | |
| Reflections collected | 24540 |
20617 |
| Independent reflections | 1894 [R(int) = 0.061] | 1553 [R(int) = 0.118] |
| Restraints | 6 | 86 |
| Parameters | 103 | 100 |
| GooF on F2 | 0.9536 | 0.9830 |
| Final R indices [I > 2σ(I)] | R 1 = 0.032, wR2 = 0.076 | R 1 = 0.057, wR2 = 0.153 |
| R indices (all data) | R 1 = 0.049, wR2 = 0.082 | R 1 = 0.080, wR2 = 0.160 |
| Compound | P (GPa) | Unit cell V (Å3) | Hydrophobic pore | Hydrophilic pore | ||
|---|---|---|---|---|---|---|
| V (Å3) | e − | V (Å3) | e − | |||
| STAM-1 | 0 | 2058.2(4) | 209 | 15 | 142 | 33 |
| STAM-1MeOH | 0.2 | 2103.2(5) | 283 | 51 | 46 | 11 |
| STAM-1MeOH | 0 | 2083.7(1) | 215 | 14 | 48 | 2 |
| STAM-1MeCN | 0.3 | 2067.1(2) | 249 | 68 | 54 | 25 |
| STAM-1MeCN | 0 | 2056.8(1) | 225 | 48 | 38 | 13 |
STAM-1MeOH under ambient conditions has a volume ∼25 Å3 larger than that of native STAM-1, though the density of the frameworks (ignoring included solvent) measures 1.470 and 1.519 g cm−3 for STAM-1 and STAM-1MeOH, respectively. This could indicate that the exchanged sample is more stable, though further investigation is needed to confirm this. On recovering STAM-1MeOH, the hydrophobic pores decrease in size, and are comparable to STAM-1 prior to application of pressure (Table 2). This coincides with a reduction in the pore content as the included methanol molecules observed at 0.2 GPa leave the hydrophobic pores on decreasing pressure, confirming that inclusion of methanol guest molecules is stabilised at high pressure.
At ambient pressure, the coordination environment around the Cu centre is very similar in both STAM-1 and STAM-1MeOH, though the axial Cu–O bond and Cu–Cu distances are shorter in STAM-1MeOH by 0.015(4) and 0.012(1) Å, respectively. On undergoing the ligand exchange reaction, the hydrophilic channels are essentially blocked. The radius of the largest sphere which can fit through these channels (calculated using the Olex2 software suite)20 decreases from 1.79 to 0.49 Å. The ligand exchange reaction therefore turns the hydrophilic channels into discrete hydrophobic pores, measuring ∼50 Å3 (see ESI; Fig. S2, S4–S7 and Table S2†).
In high-pressure crystallographic studies, comparisons of high-pressure crystal structures with the ambient-pressure structure do not usually consider the effect of the hydrostatic medium as an experimental component. X-ray diffraction data at ambient pressure are usually collected on a dry crystal with low temperatures, dry air and mounting oil used where necessary. For high-pressure data collections, the liquid in the DAC is used purely as a means of applying hydrostatic pressure to the sample. The choice of liquid used is based on consideration of the hydrostatic limit of the liquid and the solubility of the sample. However, for porous materials such as STAM-1, where the hydrostatic medium can penetrate the framework voids and react with the framework itself, it seems prudent to understand the behaviour of the sample when in contact with the medium at ambient pressure, thus allowing as accurate a comparison as possible with the high-pressure behaviour. Therefore, a method was developed to obtain the crystal structure of STAM-1 when surrounded by methanol at ambient pressure. Here, a crystal was mounted inside a MiTeGen MicroRT™ polyester capillary which was stuck to a goniometer head, sealed with an epoxy resin and filled with methanol (Fig. 3). This allowed in situ room-temperature diffraction data to be collected on STAM-1 while submerged in methanol. Though there was a high background in the diffraction frames due to scattering by the liquid, the crystal structure could easily be solved, revealing that the methanol ligand exchange occurs at ambient pressure, producing STAM-1MeOH. This is an interesting result, especially considering that methanol is used as a solvent during the original STAM-1 synthesis. This indicates that PSM of STAM-1 is the only route to producing STAM-1MeOH.
 | ||
| Fig. 3 A single crystal of STAM-1 stuck to the top of a MiTeGen Microloop™ and surrounded with methanol in a MicroRT™ polyester capillary, mounted on a goniometer head. | ||
Framework compressibility using alcohols as pressure-transmitting liquids
In order to elucidate the stability of STAM-1MeOH, and discover whether larger alcohols could undergo a similar exchange reaction, further high-pressure experiments were carried out in which another single crystal of STAM-1 was loaded with methanol, and a separate sample loaded with IPA as a pressure transmitting medium. On loading to 0.5 GPa in methanol, the ligand exchange reaction occurred again and high-pressure data were then collected from 0.5 to 5.7 GPa. On loading with IPA to 0.5 GPa, no exchange reaction was observed. High-pressure data were then collected from 0.5 to 2.4 GPa. In methanol, and separately with IPA, increasing pressure above 5.7 and 2.4 GPa, respectively, resulted in the samples becoming polycrystalline, and no further information could be extracted. Structural data could only be reliably determined to 0.9 and 1.3 GPa for compression data in IPA and methanol, respectively – above these pressures, only unit cell dimensions could be determined. Comparing the compressibility of both STAM-1, and STAM-1MeOH, the greater stability of STAM-1MeOH to pressure is apparent from its greater rigidity, and the fact that STAM-1MeOH remains stable to much higher pressures (Fig. 4). This is unsurprising as STAM-1MeOH is a denser framework.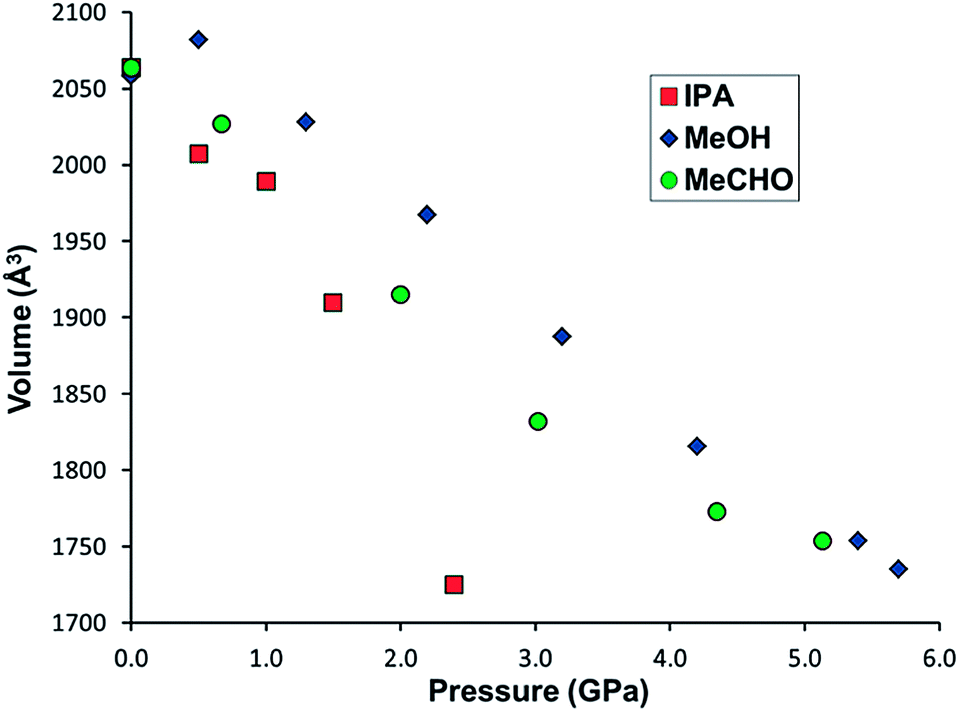 | ||
| Fig. 4 Unit cell volume of STAM-1 in IPA (red squares), acetaldehyde (MeCHO) (green circles) and STAM-1MeOH in methanol (blue diamonds) and as a function of pressure. | ||
On increasing pressure on STAM-1 in IPA to 0.5 GPa, uptake of hydrostatic liquid is similar to that observed with methanol (though no ligand exchange is observed), with an increase in electron density observed in the hydrophobic channels (see Table S3†). The onset of pressure here would therefore seem to override any energy penalty for IPA molecules entering hydrophobic channels. On increasing pressure further, the IPA content of the hydrophobic channels continues to increase, with two molecules of IPA occupying the hydrophobic channels at 0.9 GPa (Table S3†). The content of the hydrophilic channels appears to vary somewhat across this pressure range, both decreasing, and then increasing again on increasing pressure to 0.5, and then further to 0.9 GPa. We suspect, however that this is due to the mobility of water molecules already present in these pores, as the pore size (∼50 Å3) is too small to accommodate IPA, which has a molecular volume of ∼60 Å3 at ambient pressure.20 This argument is also supported by the fact that no exchange reaction takes place which would require the inclusion of IPA in these pores. In STAM-1MeOH, increasing pressure above 0.5 GPa results in more methanol being squeezed into the hydrophobic pores. A clear transition is observed on increasing pressure from 0.5 to 1.3 GPa, with the solvent content increasing from 55 to 159e− (within the hydrophobic pores), equivalent to ∼9 methanol molecules per pore (Table S2†). In a previous compression study of ZIF-8 (Zn(MeIM)2, MeIM = 2-methylimidazolate), a similar jump in pore content was observed on increasing pressure from 0.96 to 1.47 GPa, though the sudden increase in pore content was accompanied by a rotation of the MeIM rings, which increased the available pore volume.18 No significant structural changes were observed in the framework of STAM-1MeOH, though this is unsurprising, as the monoesterified BTC ligands have much fewer degrees of freedom compared to the MeIm ligands in ZIF-8.18
The significant uptake of methanol molecules into all STAM-1MeOH channels explains the greater stability of the framework at higher pressures compared to the native STAM-1 structure in IPA, since inclusion of solvent throughout the whole framework makes it much harder. Similar behaviour has been observed before in the compression behaviour of HKUST-1, where the ‘solvent filling’ regime resulted in a much higher bulk modulus of the sample.21,22 This behaviour appears to be a consistent feature of MOFs put under extreme pressure, with the compressibility changing by several orders of magnitude depending on whether guest inclusion occurs, the nature of the guest species and how the pressure is applied. In MOF-5 [Zn4O(BDC)3], amorphisation can be induced by grinding the sample at 3.5 MPa,23 or by application of hydrostatic pressure above 3.2 GPa.24 More recently, in a high-pressure study of the flexible framework NH2-MIL-53-(In), amorphisation does not take place until >20 GPa.25
Between 2.2 and 4.2 GPa, a clear plateau is reached in the compressibility of the a/b-axes of STAM-1MeOH (Fig. 5), while the c-axis continues to decrease – by 9.96% up to 4.2 GPa, comparative to the ambient-pressure structure. Unfortunately, this could not be correlated to any changes in the structure or pore content reliably, as the resolution of the data deteriorated above this pressure. On increasing pressure above 4.2 GPa, the a/b axes show a rapid decrease in length, and could indicate that the solvent is ‘flushed out’ of the hydrophobic pores above this pressure. Very similar behaviour has been observed in the compressibility of the frameworks MOF-5 (ref. 24) and HKUST-1,21 where a ‘gating’ pressure was reached which resulted in a decrease in pore content at higher pressures, allowing further compression of the crystallographic axes. In MOF-5 and HKUST-1 this was achieved above 0.8 and 3.9 GPa, respectively.
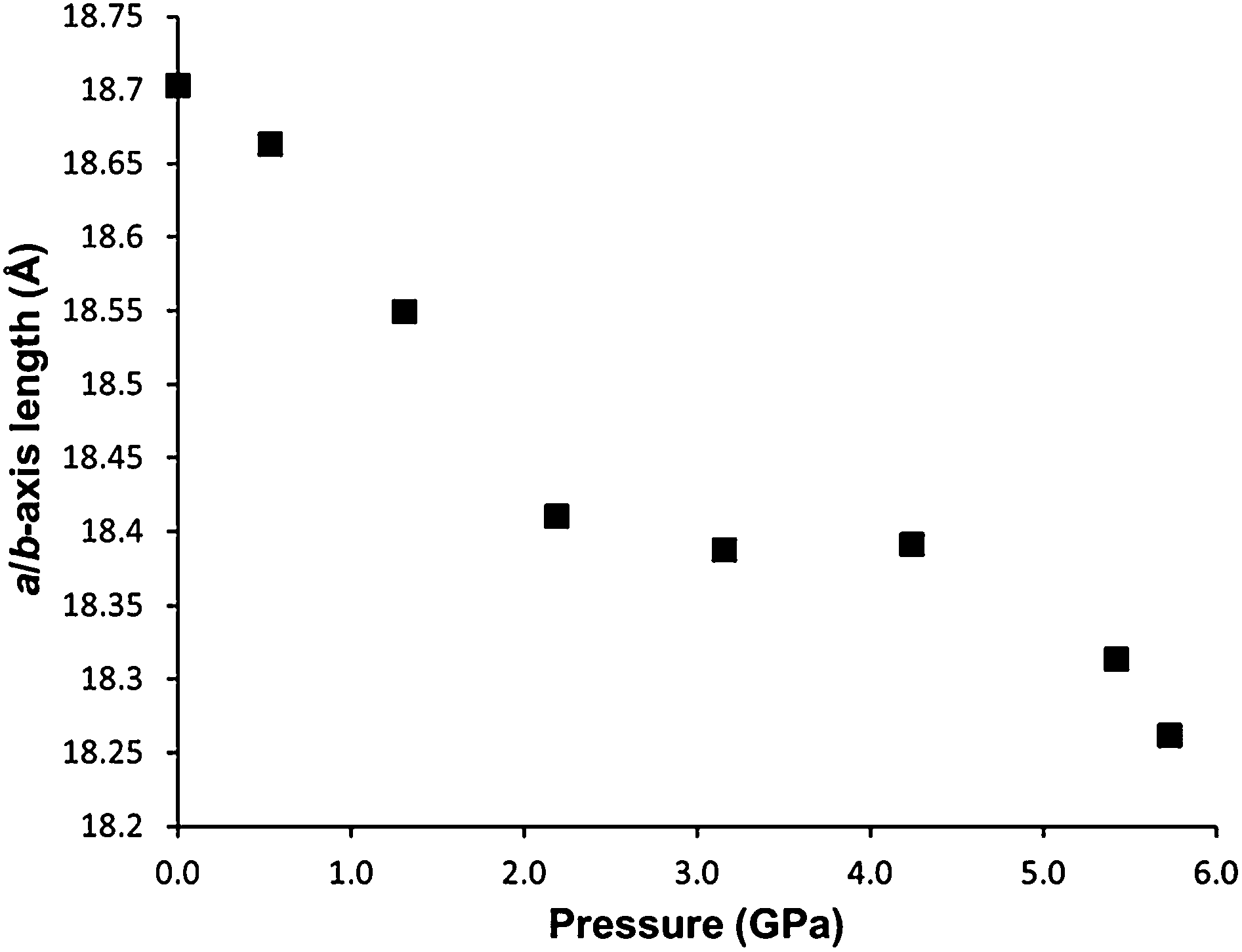 | ||
| Fig. 5 Unit cell a/b-axes length of STAM-1MeOH in methanol as a function of pressure. | ||
To probe the effect of other alcohols on the PSM behaviour of STAM-1, a crystal was initially loaded into a DAC with ethanol. The crystal rapidly fractured and became amorphous, which was unexpected given the previous results with methanol and IPA, and thus structural changes could not be resolved. The crystal also split apart at ambient pressure when surrounded with ethanol in a capillary, as described previously for methanol (Fig. 3). We attribute the breakdown of the crystal to a strain-induced collapse of the framework caused by exchange of water for ethanol at the axial CuII coordination site, as observed with STAM-1MeOH. Sterically, the size of ethanol (kinetic diameter ≈ 4.5 Å; molecular volume ≈ 42 Å3)20 comparative to methanol (∼3.6 Å; ∼32 Å3) likely causes a hindered and therefore energetically unfavourable environment in the smaller hydrophilic pores following PSM. This theory is substantiated by the results obtained when the compression experiments were repeated using acetonitrile (MeCN) and acetaldehyde (MeCHO) as hydrostatic liquids (as outlined below).
The intriguing PSM behaviour of STAM-1 arises from the lability of the coordinated water ligands. STAM-1 ligand exchanges occur in a single experimental step, but most reported ambient-pressure ligand exchange PSMs of MOFs first require the removal of the target ligand. As stated previously, HKUST-1 is a chemically similar compound to STAM-1 which has also been functionalised by PSM after removal of the water ligands via heating.13 HKUST-1 is an extended three-dimensional network with BTC ligands linked by CuII paddle wheels which, like STAM-1, have axially coordinated water molecules along a distorted Jahn–Teller axis (see Fig. S12†).
The cubic HKUST-1 framework contains three interconnected guest-accessible pores linked along the body diagonal. Four axial water molecules protrude into a large pore at (0,0,0) (square aperture) and three into a smaller pore at (1/4,1/4,1/4) (triangular aperture) (see Fig. S13†). Previous compression experiments in methanol–ethanol–water (16:3:1) and IPA have shown no evidence of axial ligand exchange in HKUST-1.21,22
It is clear that the structural and topographical changes in STAM-1, comparative to HKUST-1, have a pronounced effect on the water ligand lability. The reason for this may lie in the extent of the tetragonal Jahn–Teller distortion of the axial ligands in each framework. In the ambient-pressure structures of STAM-1 and HKUST-1, the axial Cu–O bonds are 2.149(3) and 2.167(7) Å, respectively, possibly resulting in a larger energetic stabilisation in HKUST-1. Post-exchange, the Cu–O distance in STAM-1MeOH decreases to 2.129(4) and then continues to decrease until measuring 2.105(3) Å at 1.3 GPa. This is in contrast to the compression behaviour of HKUST-1, in which the axial Cu–O bond length actually increases at 0.5 GPa, decreases slightly thereafter, and then increases again above 3.9 GPa. It is also clear that the shorter axial Cu–O bond in STAM-1 does not actually result in a stronger bond, as may be expected. We propose that the shorter bond and resulting lability of the axial water ligands is caused by the secondary structure, i.e. the framework topology of STAM-1, where the hydrophilic channel is relatively constrained compared to HKUST-1 which is composed of much larger pores.
The large pores at (0,0,0) in HKUST-1 form channels linked in three dimensions directly along all three axis directions. The small pores at (1/4,1/4,1/4) are aligned along the body diagonal of the cell. Like the small hydrophilic pore in STAM-1, in HKUST-1 the small pores contain water molecules and have approximately the same diameter as that of STAM-1 (∼5.85 Å in HKUST-1, ∼5.87 Å in STAM-1). However, the opening to each HKUST-1 pore along [1,1,1] is capped with the aromatic ring of the BTC ligand, and in that respect they are more akin to cages with accessible void space into the large channels at (0,0,0) (see Fig. S14†). This differs to STAM-1, where the esterification of one carboxylate group results in loss of the cubic symmetry and two isolated and chemically distinct channels, running in one dimension along the c-axis. It is also noteworthy that the water ligands which form the square and triangular apertures in HKUST-1 are alternately staggered to each other by ∼45° between adjacent planes, providing a greater spread of the ligand sites throughout the channels. In the layered STAM-1 structure, the water ligands pointing into the small pore are stacked directly on top of each other throughout the channel (Fig. 1). Therefore, the three-dimensional connectivity in HKUST-1 may stabilise the axial water ligands due to a more even partial charge distribution and guest uptake throughout the framework, while the difference in STAM-1 channel functionality results in opposing chemical environments and an uneven charge distribution which may have a destabilising effect on the axial water.
Direct framework compression in acetaldehyde
Though essentially the same molecular size and volume as ethanol (∼42 Å3), the different functional groups of acetaldehyde (∼44 Å3) and acetonitrile (∼43 Å3)20 give rise to marked differences in the PSM behaviour of STAM-1. Loaded in acetaldehyde to 0.7 GPa, the liquid entered both the hydrophobic and hydrophilic channels of the STAM-1 crystal, but no ligand exchange was observed. Compression of the unit cell volume and unit cell axes then occurred steadily as pressure was increased, with an overall compression of 4.68% and 6.23% along the a/b and c-axes, respectively (Fig. 6), with the unit cell volume decreasing by 14.8% (Fig. 4). The framework was stable up to pressures of 5.1 GPa, though the data resolution declined significantly after the first pressure point. Despite this, the crystal structure up to 3.0 GPa was able to be refined satisfactorily using only isotropic displacement parameters. There was uptake of acetaldehyde in both the hydrophobic and hydrophilic pores up to 2.0 GPa, as shown by the pore content in Table 3. Above 3.0 GPa, only unit cell parameters could reliably be extracted from the diffraction data.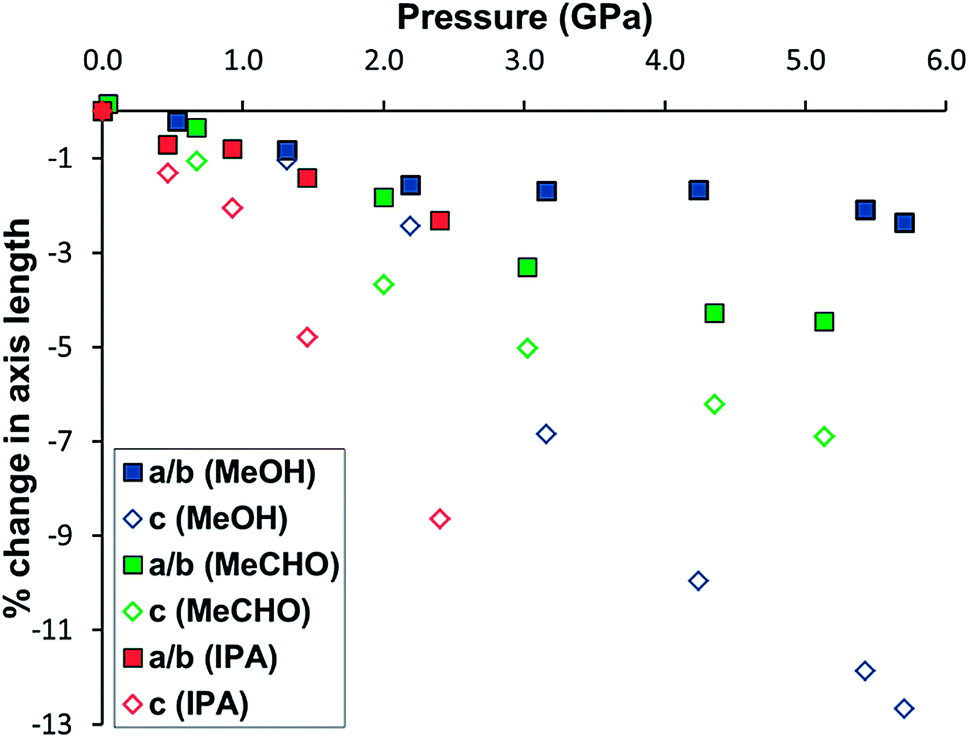 | ||
| Fig. 6 Change in the length of the a/b-(squares) and c-axes (open diamonds) of STAM-1 in methanol (blue), acetaldehyde (green) and IPA (red) as a function of pressure. | ||
| P (GPa) | Unit cell V (Å3) | Hydrophobic pore | Hydrophilic pore | ||
|---|---|---|---|---|---|
| V (Å3) | e − | V (Å3) | e − | ||
| 0 | 2058.2(4) | 209 | 15 | 142 | 33 |
| 0.7 | 2026.9(2) | 224 | 98 | 132 | 50 |
| 2.0 | 1915.2(3) | 236 | 144 | 116 | 66 |
| 3.0 | 1831.8(4) | 218 | 55 | 111 | 25 |
| 4.4 | 1773.1(7) | — | — | — | — |
| 5.1 | 1753.7(12) | — | — | — | — |
At 2.0 GPa, six molecules of acetaldehyde were present in the hydrophobic channel with three acetaldehyde molecules present in the hydrophilic channel. On increasing the pressure to 3.0 GPa, a ∼70% drop in the solvent content of both channels occurred as the acetaldehyde was flushed out of the framework. This is the same behaviour which we believe occurred in the pressure series of STAM-1MeOH, though the gating pressure of STAM-1MeOH occurred above 4.2 GPa; more than twice that of STAM-1 in acetaldehyde. The greater rigidity of STAM-1MeOH caused by the ligand exchange would therefore appear to significantly increase the gating pressure. This enhancement of structural rigidity is also highlighted by comparison of the change in the unit cell axes. Up to 5.4 and 5.1 GPa, respectively, there is an almost identical reduction in the unit cell volume of STAM-1MeOH and STAM-1 in acetaldehyde; by 304.5(4) Å3 and 304.4(4) Å3 (see Fig. 4). However, in acetaldehyde, STAM-1 is significantly more compressible along the a/b axes than STAM-1MeOH, which instead accommodates the pressure increase almost entirely along the c-axis due to the stiffness afforded by the methanol ligands in the a/b face (Fig. 6).
These results show that the compression behaviour and stability of STAM-1 is highly dependent on the choice of solvent used as a hydrostatic medium. When compressed in IPA to 1.3 GPa, the a/b-axes contract to a similar degree as observed for acetaldehyde due to a more flexible substructure at the hydrophilic pore, but since IPA only penetrates the larger of the framework channels, the structure is amorphised at relatively low pressures. When penetrating solvents – acetaldehyde and methanol – are used as pressure-transmitting media, the super-filling of all the framework channels yields a much harder framework stable to significantly higher pressures. Although the hardness of the framework is dependent on the size and thus the penetration of medium into the channels, the rigidity of the framework, and therefore the direction of compression, is influenced by ligand exchange, which is dictated by the functionality of the solvent.
Pressure-induced ligand exchange with acetonitrile
Upon loading of a crystal of STAM-1 in a DAC with acetonitrile to 0.3 GPa, a single-crystal to single-crystal phase transition yielded STAM-1MeCN (Fig. 2c and e), arising from a ligand exchange at the axial position of the CuII paddle wheel, as with methanol. On immersing a crystal of STAM-1 in acetonitrile at ambient temperature and pressure, the ligand exchange reaction did not occur. Ligand exchange can of course take place over a period of days or weeks. Therefore, a small sample of STAM-1 crystals was soaked in a vial of acetonitrile at room temperature for two weeks. Subsequent diffraction data performed at 150 K again showed no evidence of ligand exchange. High pressure, therefore, appears to be required to force the acetonitrile ligand exchange, at room temperature at least, and thus STAM-1MeCN is the first ever example of a pressure-induced PSM of a MOF.Each acetonitrile ligand in the STAM-1MeCN structure has an occupancy of one third and thus only occupies one of the potential three coordination sites protruding into the hydrophilic pore (as shown in Fig. 2). By only part-exchanging with the axial water ligand, steric strain between adjacent ligands is prevented. If full exchange of the water ligands had occurred, this would undoubtedly have resulted in a large change in the framework structure in order to accommodate the larger ligands, most likely resulting with the sample becoming amorphous, as we observed previously with ethanol. This result also implies distinct differences in the CuII affinity for ethanol and acetonitrile, allowing the latter to part-exchange in a sterically hindered environment. As the pressure was applied to the crystal to form STAM-1MeCN, the pore volume of the large hydrophobic channels increased from 209 to 249 Å3 per unit cell, with a corresponding increase in solvent content corresponding to three molecules of acetonitrile. Contrarily, the ligand exchange at the hydrophilic pore causes a reduction in volume from 142 Å3 to 54 Å in the volume of the hydrophilic channels. The solvent content decreased from 33 to 24e− Å3, corresponding to the replacement of three water molecules with one acetonitrile molecule per hydrophilic channel, per unit cell (see Table 2). Like with STAM-1MeOH, the ligand exchange closes the channel openings, which become discrete pores.
The compression of STAM-1 in acetonitrile to 0.3 GPa resulted in an expansion of the unit cell volume by 0.30% and c-axis by 1.53% but a contraction of the a/b-axes by 0.54%. This contrasts sharply with the behaviour observed for STAM-1MeOH, which expanded along the a/b- and c-axes by 0.34 and 0.30%, respectively, when loaded to 0.2 GPa. The structure of the native STAM-1 has a greater flexibility along the c-axis direction than the a/b direction (see Fig. 1), since the c-axis runs parallel to the framework channels which sit between non-bonded two-dimensional layers. The changes in the unit cell dimensions of STAM-1MeCN at high pressure, comparative to those of STAM-1MeOH, suggest that the part-occupancy of the acetonitrile ligands yield a less rigid structure along the a/b plane than the full-occupancy of methanol, allowing a greater compression along the a/b-axes in STAM-1MeCN.
We propose that if the pressure in the DAC had been increased further the ligand part-exchange could have been forced to full occupancy as more liquid would be forced into the channels, possibly causing the collapse of the crystal as observed with ethanol. However, the hydrostatic limit of acetonitrile is low and further data collections were not possible since the solvent froze at 0.5 GPa.
The STAM-1MeCN crystal could be recovered from the DAC and was stable at 150 K under a nitrogen stream at ambient pressure. Crystallographic data is shown in Table 1. Upon returning to ambient pressure, the volume of the hydrophobic channel of STAM-1MeCN decreased from 249 Å3 to 225 Å3 due to the accompanying discharge of acetonitrile, which reduced from three to two molecules of acetonitrile. There was also a slight decrease in the hydrophilic channel volume and solvent content (Table 2). X-ray diffraction data showed that upon heating back to room temperature, the structure reverted back to native STAM-1 as the acetonitrile ligand exchanged with atmospheric water.
Ambient-pressure ligand exchange with amines
Further investigation into the lability of the axial water ligand and the STAM-1 ligand exchange reaction was conducted using amines analogous in size to the solvents used previously as hydrostatic media. Upon addition of excess methylamine solution, ethylamine and n-propylamine to vials of native STAM-1, the pale blue crystals instantly became oil-like and underwent a colour change to dark blue; a characteristic indicator of an amine-water ligand substitution in an octahedral CuII complex. The formation of an oil is symptomatic of a complete loss of long-range order due to a structurally destructive reaction of STAM-1 with amines. This is not surprising given the reactivity of CuII ions with small, weakly basic aliphatic primary amines. When dried, the oil-like samples formed larger aggregates which were largely amorphous. Fortunately however, small crystals were able to be extracted from within the amorphous aggregates in each sample and diffraction data were collected at 150 K. The crystal structure of each confirms that an analogous ligand exchange reaction occurred between STAM-1 and methylamine and ethylamine (henceforth STAM-1MeNH2 and STAM-1EtNH2) (Fig. 7 and S17 and S18†), as was observed previously with methanol and acetonitrile. No exchange reaction occurred with n-propylamine.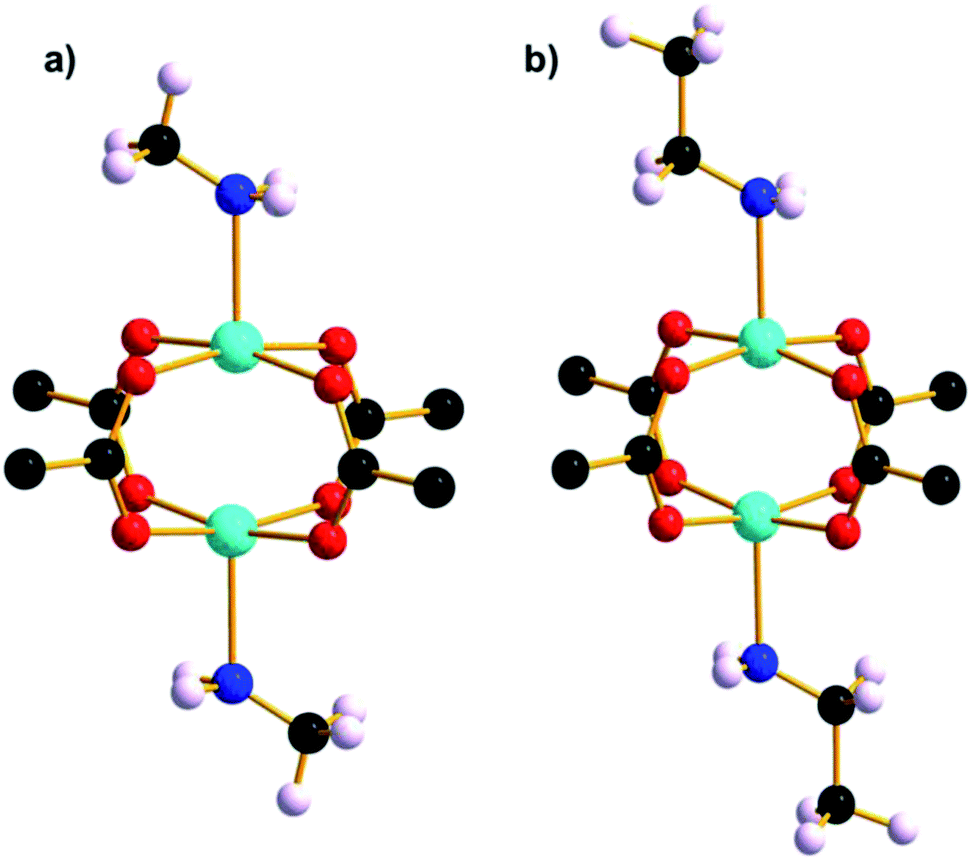 | ||
| Fig. 7 Cu paddle wheel units in (a) STAM-1MeNH2 and (b) STAM-1EtNH2. The part-occupied amine ligands are shown at full occupancy. | ||
Crystallographic data for STAM-1MeNH2 and STAM-1EtNH2 are provided in Table S5.† Upon storage at room temperature for one week, the crystal structures of both STAM-1MeNH2 and STAM-1EtNH2 reverted back to the native STAM-1 form as the amine ligands exchanged with water.
The calculated pore content of STAM-1MeNH2 indicates that one molecule of residual water is present in each hydrophilic channel per unit cell. Methylamine, which is a gas at room temperature, was delivered to STAM-1 as a 2.0 M solution in tetrahydrofuran (THF). THF was not only in excess, but is non-polar and is too large to penetrate the small hydrophilic pore and therefore adsorption by the large hydrophobic pore is likely. In the hydrophobic channel, there was residual electron density (36e−) which could not be modelled reliably but which we attribute to one molecule of THF. Interestingly in this case, the exchange of methylamine at the small pore and uptake of THF to the large pore is a serendipitous example of selective guest uptake in a MOF.
Since the structural integrity of the STAM-1MeNH2 and STAM-1EtNH2 crystals was maintained after the reaction with the amines, it is likely that the few crystals that were able to be extracted from each sample were shielded from exposure to most of the amine due to their encapsulation within the larger amorphous aggregates. Thus the amine-exchanged crystals were not a result of a recrystallisation from the oil, which remained amorphous when dried. It may be the case, therefore, that the STAM-1 framework reacts with amines in two stages; initial ligand exchange at the axial CuII coordination site, followed by a destructive exchange at the equatorial position occupied by the BTC linkers. In the structure of STAM-1EtNH2, the ethylamine ligand has an occupancy of one third, i.e., it occupies one of the potential three coordination sites around the small pore. Since ethylamine is of a similar size to acetonitrile, the part-exchange may be dictated by the size of the small hydrophilic pore, as observed in STAM-1MeCN. This theory is also reinforced by the absence of a ligand exchange with n-propylamine which, like IPA, is too large to fit into the small STAM-1 pore. However, the methylamine ligand in the STAM-1MeNH2 structure also only occupies one of the three sites. Given that methylamine is approximately the same size as methanol, which is fully occupied in STAM-1MeOH, the part-exchange of methylamine was unexpected. An explanation for the part-exchange of methylamine, compared to the full exchange of methanol, may be due to a partial reversion of a fully-exchanged material to native STAM-1, or due to differences in ligand exchange kinetics. The fact that STAM-1MeOH is stable at ambient temperature and pressure, while STAM-1MeNH2 reverts to native STAM-1 at room temperature, may indicate that STAM-1MeOH is a thermodynamic product and STAM-1MeNH2 is a kinetic product, but more work would be required to prove this theory.
Pressure-induced decomposition
The final solvent used to investigate the PSM behaviour of STAM-1 was water, which when used as a hydrostatic medium in a DAC could be squeezed into the hydrophobic channels. At 0.1 GPa there was a 0.63% increase in unit cell volume, a 0.14% contraction in the a/b axes and a 0.94% expansion of the c-axis (see Table S6†). This contrasts with the changes observed with methanol, which increases the rigidity of the structure in the a/b plane due to ligand exchange, but is in keeping with results observed with acetonitrile and acetaldehyde, which have a more flexible substructure in the a/b plane. The volume of the hydrophobic and hydrophilic channels increased slightly – by ∼17 Å3 and ∼27 Å3, respectively – as water was forced into the framework. Void analysis with cell (calculated using the SQUEEZE algorithm19) shows that there are approximately five molecules of water in the hydrophobic channel per unit cell, and four in the hydrophilic channel (see Table S6†). This serves as an effective demonstration of how pressure can be used to force hydrophilic molecules into hydrophobic environments counteracting any ‘chemical’ effects of the pores (Fig. 8a). The structure at 0.1 GPa can therefore be considered as ‘super-filled’ by water molecules. The pressure was then increased to 0.3 GPa, whereupon the STAM-1 crystal in the DAC turned black, while thin clear needles could be seen growing out from the surface of the crystal (Fig. 9b). Given that this occurred far below the freezing/crystallisation pressure of water, the behaviour of STAM-1 indicates a chemical decomposition.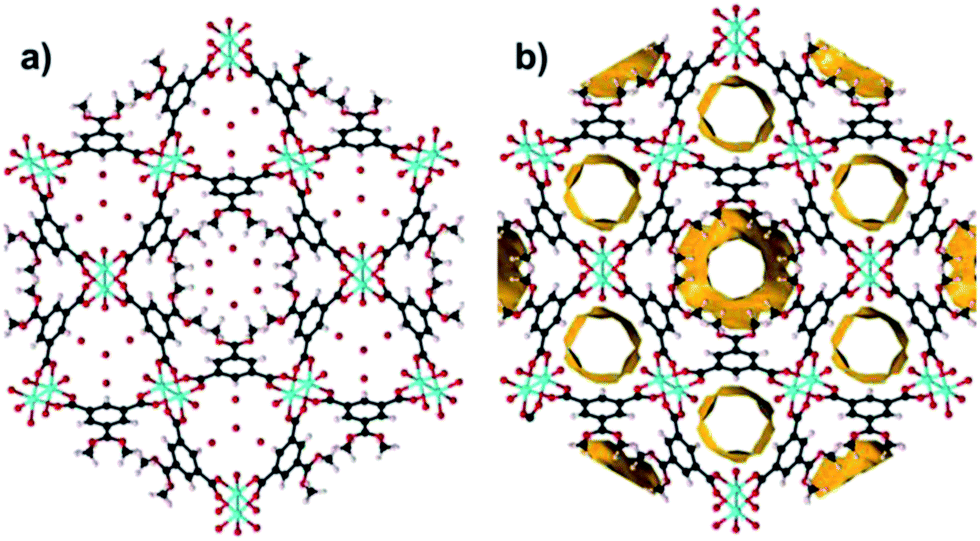 | ||
| Fig. 8 Hydrophobic and hydrophilic channels in STAM-1 showing (a) disordered water in the hydrophobic pore at 0.1 GPa and (b) 1.2 Å contact surface calculated using void analysis in MERCURY v3.1. | ||
 | ||
| Fig. 9 Photomicrographs of (a) a crystal of STAM-1 under ambient conditions and (b) a decomposed STAM-1 crystal in water at 0.3 GPa. A large ruby chip is shown on the left of the decomposed STAM-1, and another at the top of the gasket hole. | ||
The decomposed sample was non-diffracting, so structural changes could not be resolved crystallographically. However, the STAM-1 framework voids show intuitively that as more water is squeezed into the hydrophobic channel with increasing pressure, water molecules will be forced into closer proximity to the disordered ester groups of the hydrophobic pore (Fig. 8b). Considering the previously-observed high-pressure results with alcohols, amines, acetonitrile and acetaldehyde, it is clear that the presence of water is responsible for the crystal decomposition. Given the energetically unfavourable nature of the interaction between water and ester groups, we attribute the crystal decomposition to a pressure-induced hydrolysis of the methyl ester groups within the hydrophobic pores. Pressure-induced ester hydrolysis has previously been reported for peptide derivatives, in a similar pressure range as that used here, with the authors also noting that methyl esters in particular are the most susceptible to this behaviour.26
For comparison with STAM-1 in water, we performed an analogous pressure study on HKUST-1. A crystal was collected at ambient temperature and pressure and then loaded into a DAC with water as a hydrostatic medium. Under ambient conditions, the crystal of HKUST-1 contained approximately 102 residual water molecules per unit cell in the pores. On increasing pressure to 0.2 GPa, there was an initial increase in the pore content to ∼206 water molecules (Table S7†). The crystal quality and data resolution, however, declined substantially on increasing pressure further to 0.4 GPa, allowing only unit cell dimensions to be extracted from these data. With the increase in pore content, there was also an accompanying increase in unit cell volume and axes lengths, all of which is in keeping with previous work performed on HKUST-1. The crystal then became amorphous above 0.4 GPa but no physical evidence of decomposition was observed in the crystal. We believe that an analogous STAM-1 decomposition is prevented in HKUST-1 due to the full Cu paddle wheel connectivity through all three carboxylate groups on each BTC linker (i.e. there is no ester group in HKUST-1 for the water to react with). Further work is required to validate our theory of the nature of STAM-1 decomposition, but our attempts to provide spectroscopic information on the STAM-1 decomposition were unsuccessful. When the pressure in the DAC was returned to ambient pressure, the decomposed STAM-1 broke apart and the needles growing from the STAM-1 surface dissolved in the hydrostatic medium and thus could not be isolated. This behaviour is compatible with the probable decomposition product from an ester hydrolysis – methanol and the free acid derivative of the esterified BTC linker.
Conclusions
In summary, we have investigated the susceptibility of STAM-1 to post-synthetic ligand exchange at the axial CuII coordination site with various solvents. Notably we have presented the first ever example of a pressure-induced PSM of a MOF, demonstrating how the application of pressure is a useful tool to induce PSM and thus facilitate discovery of new framework materials. The high-pressure behaviour of STAM-1 is very sensitive to the size and functionality of the hydrostatic media used as pressure-transmitting liquids. The molecular size of the solvents used as hydrostatic media in compression experiments has shown to be of critical importance in stabilising the STAM-1 structure to compression. However, the functional moiety of the solvent governs the affinity for the CuII coordination site and thus the extent of (or absence of) ligand exchange. This in turn affects the direction of compression within the framework. We have also shown that hydrophilic liquids can be squeezed into hydrophobic environments, with high pressure able to override any energy penalty for this process. In the case of water as a hydrostatic medium, this actually leads to a chemical decomposition of the sample – the first result of its kind reported for a MOF material.Acknowledgements
We thank the Royal Society of Edinburgh and the Scottish Government for a fellowship to Dr Stephen A. Moggach. We also thank the STFC for provision of beamtime and the EPSRC and Leverhulme Trust for financial support. Professor Russell E. Morris is a Royal Society Industry Fellow.References
- W. Morris, B. Leung, H. Furukawa, O. K. Yaghi, N. He, H. Hayashi, Y. Houndonougbo, M. Asta, B. B. Laird and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 11006–11008 CrossRef CAS PubMed.
- X. Lin, J. Jia, P. Hubberstey, M. Schroeder and N. R. Champness, CrystEngComm, 2007, 9, 438–448 RSC.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J.-S. Chang, D.-Y. Hong, Y. K. Hwang, S. H. Jhung and G. Ferey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS PubMed.
- Z. R. Herm, J. A. Swisher, B. Smit, R. Krishna and J. R. Long, J. Am. Chem. Soc., 2011, 133, 5664–5667 CrossRef CAS PubMed.
- R. Matsuda, T. Tsujino, H. Sato, Y. Kubota, K. Morishige, M. Takata and S. Kitagawa, Chem. Sci., 2010, 1, 315–321 RSC.
- K. A. Cychosz, R. Ahmad and A. J. Matzger, Chem. Sci., 2010, 1, 293–302 RSC.
- Z.-Z. Lu, R. Zhang, Y.-Z. Li, Z.-J. Guo and H.-G. Zheng, J. Am. Chem. Soc., 2011, 133, 4172–4174 CrossRef CAS PubMed.
- B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- N. R. Champness, Dalton Trans., 2011, 40, 10311–10315 RSC.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed.
- T. Gadzikwa, O. K. Farha, K. L. Mulfort, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2009, 3720–3722 RSC.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS.
- Y.-S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131–2134 RSC.
- J. An, O. K. Farha, J. T. Hupp, E. Pohl, J. I. Yeh and N. L. Rosi, Nat. Commun., 2012, 3, 604 CrossRef PubMed.
- M. I. H. Mohideen, B. Xiao, P. S. Wheatley, A. C. McKinlay, Y. Li, A. M. Z. Slawin, D. W. Aldous, N. F. Cessford, T. Düren, X. Zhao, R. Gill, K. M. Thomas, J. M. Griffin, S. E. Ashbrook and R. E. Morris, Nat. Chem., 2011, 3, 304–310 CrossRef CAS PubMed.
- B. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS PubMed.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087–7089 CrossRef CAS PubMed.
- A. L. Spek, Utrecht University, Utrecht, The Netherlands, 2004.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. J. Graham, J.-C. Tan, D. R. Allan and S. A. Moggach, Chem. Commun., 2012, 48, 1535–1537 RSC.
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2008, 130, 10524–10526 CrossRef CAS PubMed.
- Y. H. Hu and L. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 1741031–1741035 Search PubMed.
- A. J. Graham, D. R. Allan, A. Muszkiewicz, C. A. Morrison and S. A. Moggach, Angew. Chem., Int. Ed., 2011, 50, 11138–11141 CrossRef CAS.
- P. Serra-Crespo, E. Stavitski, F. Kapteijn and J. Gascon, RSC Adv., 2012, 2, 5051–5053 RSC.
- A. F. Garcia, H. Zoller, P. Butz, J. Starke and B. Tauscher, Food Chem., 2006, 95, 301–306 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic Information Files (CIFs), details of experimental methods and supplementary compression data. See DOI: 10.1039/c3nr04161a |
| This journal is © The Royal Society of Chemistry 2014 |