Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis†
Alaitz
Rekondo
,
Roberto
Martin
,
Alaitz
Ruiz de Luzuriaga
,
Germán
Cabañero
,
Hans J.
Grande
and
Ibon
Odriozola
*
Materials Division, IK4-CIDETEC Research Centre, Paseo Miramón 196, 20009 Donostia-San Sebastián, Spain. E-mail: iodriozola@cidetec.es; Fax: +34 943309136; Tel: +34 943309022
First published on 13th September 2013
Abstract
Aromatic disulfide metathesis has been reported as one of the very few dynamic covalent chemistries undergone at room-temperature. Here, bis(4-aminophenyl) disulfide is effectively used as a dynamic crosslinker for the design of self-healing poly(urea–urethane) elastomers, which show quantitative healing efficiency at room-temperature, without the need for any catalyst or external intervention.
Perhaps inspired by nature, or maybe triggered by the need for more durable materials in many industrial applications, self-healing polymeric materials, which are able to autonomously repair damage inflicted on them, are currently the subject of active research.1–7 A particularly useful approach to generate self-healable polymers has been the introduction of reversible or exchangeable bonds into the polymer network. The idea behind this is to reconnect the chemical crosslinks which are broken when a material fractures, restoring the integrity of the material. This is expected to provide polymers with enhanced lifetime and resistance to fatigue. Self-healing approaches based on such dynamic crosslinks have been carried out using both reversible covalent chemistries8,9 and supramolecular interactions.10 One representative example is the supramolecular self-healable elastomeric material developed by Leibler and colleagues,11 based on H-bonding interactions. However, the stronger nature of dynamic covalent bonds compared to non-covalent ones offers the possibility to obtain self-healing polymer networks with superior mechanical strength. Diels–Alder reaction,12 transesterification,13,14 olefin metathesis,15,16 radical reshuffling,17–20 imine21 or hydrazone22 formation, siloxane equilibration,23 thiol–nanoparticle exchange24 and aliphatic disulfide exchange25,26 are some examples of reversible covalent chemistries used for the design of self-healing polymers. In the majority of these cases, an external stimulus such as pH,21,22 or a source of energy such as heat13,14,19,23,25,26 or light17,20,27 is required, in order to promote reshuffling of such reversible chemical bonds. Moreover, to the best of our knowledge spontaneously self-healing thermoset elastomers presenting a quantitative healing efficiency without the addition of a specific catalyst have not yet been described. Here we describe a permanently cross-linked material, based on a covalently cured poly(urea–urethane) elastomeric network which, after being cut in half with a razor blade, is able to self-mend by simple contact at room-temperature. The system is based on the metathesis reaction of aromatic disulfides,28,29 which are known to exchange at room temperature, unlike their aliphatic counterparts, which require heat25 or light30 for the metathesis to occur. A 97% healing efficiency of the material, as calculated by tensile strength measurements, and its easy preparation from commercially available starting materials make this new system broadly applicable in a wide number of industrial sectors where poly(urea–urethane)s are already used.
Among covalent bonds that are susceptible to undergo reversible exchange at room temperature, metathesis of aromatic disulfides offers unique opportunities (Scheme 1a), due to its simplicity and availability. Metathesis of aromatic disulfides has been reported to occur at room temperature both in solution29 and in the solid state28 using a tertiary amine as the catalyst. However, to the best of our knowledge this reaction has not been exploited so far for the preparation of self-healing polymers. Thus, we chose a diamine molecule containing an aromatic disulfide unit on its structure (bis(4-aminophenyl) disulfide 1a, Scheme 1) to be used as a dynamic crosslinker in poly(urea–urethane) systems.
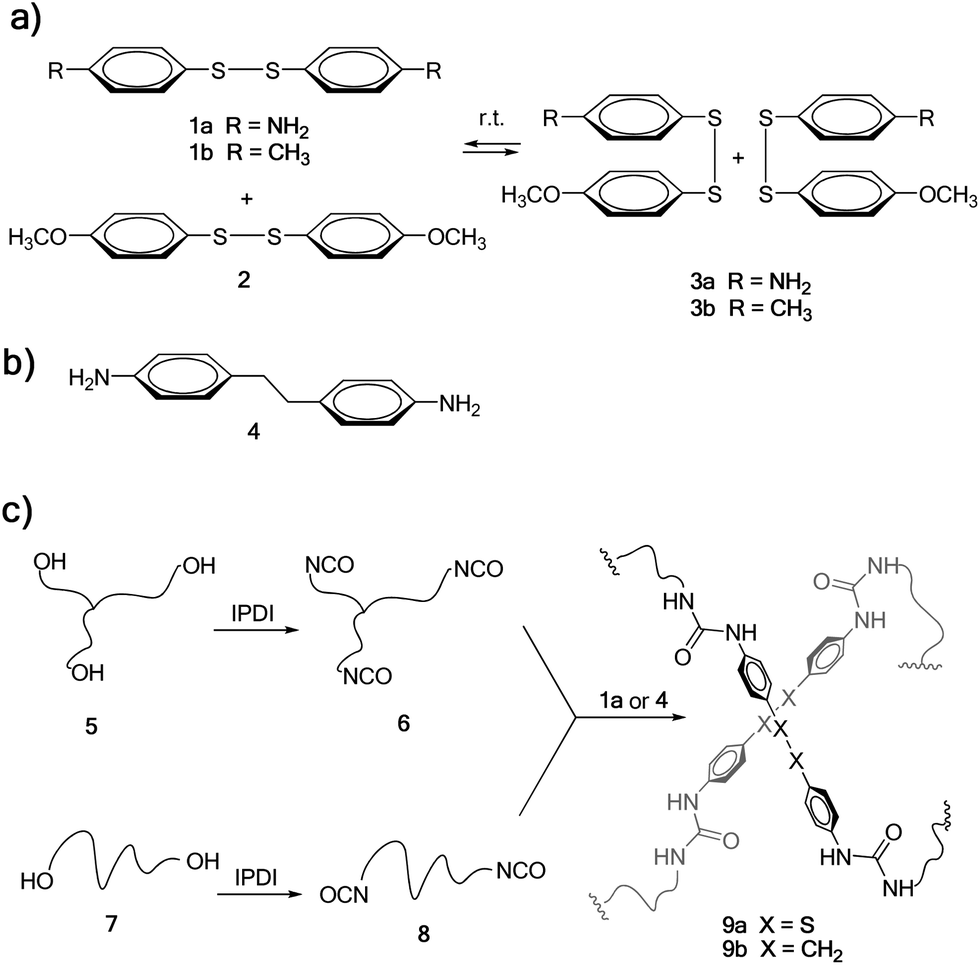 | ||
| Scheme 1 (a) Reversible metathesis reaction of aromatic disulfides 1a–b and 2. (b) Reference aromatic diamine crosslinker without disulfide bonds. (c) Synthetic procedure for obtaining disulfide-containing poly(urea–urethane) elastomer 9a and reference material 9b. | ||
As a model metathesis reaction, we studied the equilibration of equimolar amounts of 1a and bis(4-methoxyphenyl) disulfide 2 in deuterated DMSO (see Scheme 1a). When the reaction was performed in the presence of 0.1 equivalents of NEt3, the equilibrium was reached in less than 1 hour. However, without the addition of any NEt3, metathesis started in less than 1 hour, achieving the equilibrium in 22 hours, as shown by 1H NMR (see Fig. S5† in the ESI), where a mixture of 1a (25 mol%), 2 (25 mol%) and 3a (50 mol%) was obtained. In order to corroborate that the reaction did not occur by a self-catalysis effect of primary aromatic amines present in 1a, the same experiment was performed using 1b and 2, without NEt3. Interestingly, when mixing equimolar amounts of 1b and 2, equilibration was also achieved after 24 hours, corroborating that the exchange reaction occurs without the need for any catalyst (see Fig. S6†).
Poly(urea–urethane)s are widely used in industrial applications such as sealants, adhesives, paints and coatings, insulating foams, etc.31 These can be formulated as monocomponent or bicomponent systems, where a polyol resin is crosslinked with a polyisocyanate, or vice versa, an isocyanate-functionalised resin is crosslinked with polyols or polyamines by ambient humidity (monocomponent systems).
The synthesis of the self-healing material studied in this work was carried out starting from commercially available poly(propylene glycol) (PPG) resins 5 and 7 (Scheme 1c), having an average molecular weight of 6 and 2 kDa, respectively. These were first treated with isophorone diisocyanate (IPDI)32 in the presence of dibutyltin dilaurate (DBTDL) as a catalyst, to obtain tris- and bis-isocyanate-terminated urethanes 6 and 8, respectively. The reaction was followed by FTIR spectroscopy (see Fig. S1 and S2† in the ESI). Then, based on our previous experience, a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of 6 and 8 was used in order to obtain a material with good elastomeric properties. Such a mixture was cured by the addition of diamine crosslinker 1a, typically in a mould at 60 °C for 16 hours. The curing process could be easily monitored by FTIR spectroscopy, where the isocyanate stretching band at 2264 cm−1 completely disappeared and a new band in the form of a shoulder corresponding to the urea appeared at 1650 cm−1 (see Fig. S3† in the ESI). This afforded 9a as a transparent elastomeric material, resembling conventional polyurethanes commercially used as sealants or adhesives. Elastomer 9b, where the disulfide bond was replaced by an ethylene group, was also synthesised as a reference material, using diamine 4 as the crosslinking agent (Scheme 1b) and following an identical synthetic protocol.
:30 mixture of 6 and 8 was used in order to obtain a material with good elastomeric properties. Such a mixture was cured by the addition of diamine crosslinker 1a, typically in a mould at 60 °C for 16 hours. The curing process could be easily monitored by FTIR spectroscopy, where the isocyanate stretching band at 2264 cm−1 completely disappeared and a new band in the form of a shoulder corresponding to the urea appeared at 1650 cm−1 (see Fig. S3† in the ESI). This afforded 9a as a transparent elastomeric material, resembling conventional polyurethanes commercially used as sealants or adhesives. Elastomer 9b, where the disulfide bond was replaced by an ethylene group, was also synthesised as a reference material, using diamine 4 as the crosslinking agent (Scheme 1b) and following an identical synthetic protocol.
Poly(urea–urethane) 9a showed a remarkable self-mending ability at room temperature, without the need for applying any catalyst or external stimulus for the healing to occur. Fig. 1 shows a photographic sequence of a typical healing process. First, a pristine cylinder made from 9a was cut in half with a knife. Then the two halves were put in contact and allowed to stand at room-temperature, without applying any pressure. After 2 hours it was already not possible to separate the two pieces by stretching manually (see Movie S1 in the ESI†). In another experiment, an identical cylinder was “chopped” very slowly by the gravitational force of a copper filament attached to a weight. By the time the filament had completely run through the cylinder, the latter had self-welded entirely (see Movie S2 in the ESI†).
 | ||
| Fig. 1 Photographic sequence of a pristine cylindrical sample of 9a (a), which was cut in half (b and c). The two halves were then allowed to stand for 2 hours by simple contact (d). After that time the material could be manually stretched without rupture (e and f). | ||
The self-healing efficiency of 9a was quantified by tensile strength measurements. For that aim, a 2 mm thick film of 9a was prepared in a mould, which was then cut in the form of dumbbell-shaped specimens, in order to perform tensile strength measurements. Some of the specimens were mechanically tested as pristine samples. The rest of them were cut in half and then mended by simple contact at room temperature for different periods of time. Fig. 2a shows stress vs. strain curves obtained for pristine and self-mended samples at different healing times. The original material exhibited a tensile strength of 0.81 ± 0.05 MPa and an elongation at breaking point of 3100 ± 50%. After 1 hour of contact, the mended samples recovered 62% of their initial mechanical properties. At 2 h, the recovery was already 80%. The mended samples after 24 h showed a tensile strength of 0.77 ± 0.05 MPa and an elongation at a breaking point of 3015 ± 50% (Fig. 1a). This means that a healing efficiency of 97% was achieved, which can be considered quite a remarkable result for a thermoset elastomeric material.
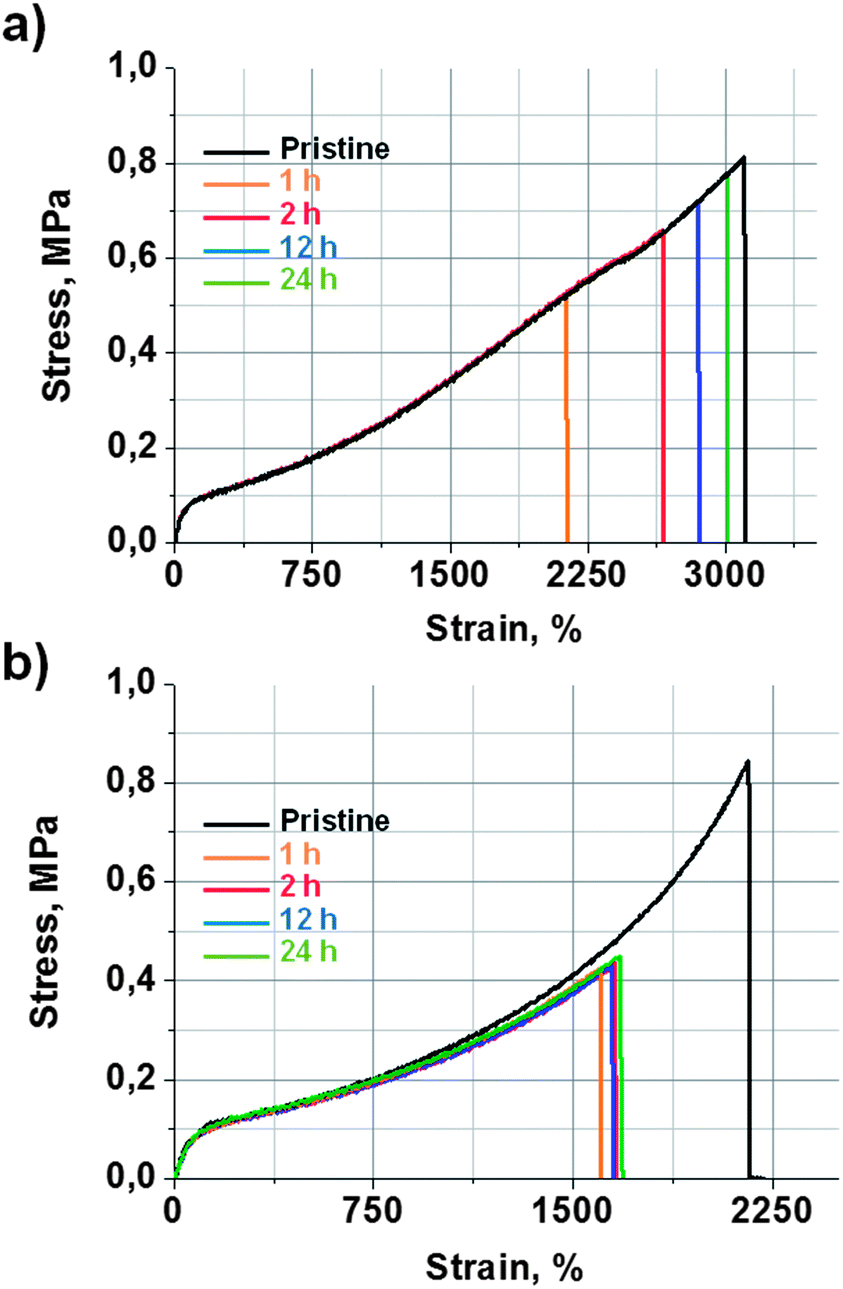 | ||
| Fig. 2 Strain vs. stress curves at different healing times obtained for (a) self-healing elastomer 9a and (b) reference material 9b. Mending was performed by simple contact at r.t. | ||
The remarkable self-healing ability of this system could be attributed to two structural features, which are present in this unique crosslinking unit: (i) the aromatic disulfide which is in constant exchange at room temperature and (ii) two urea groups, capable of forming a quadruple H-bond (Fig. 3).
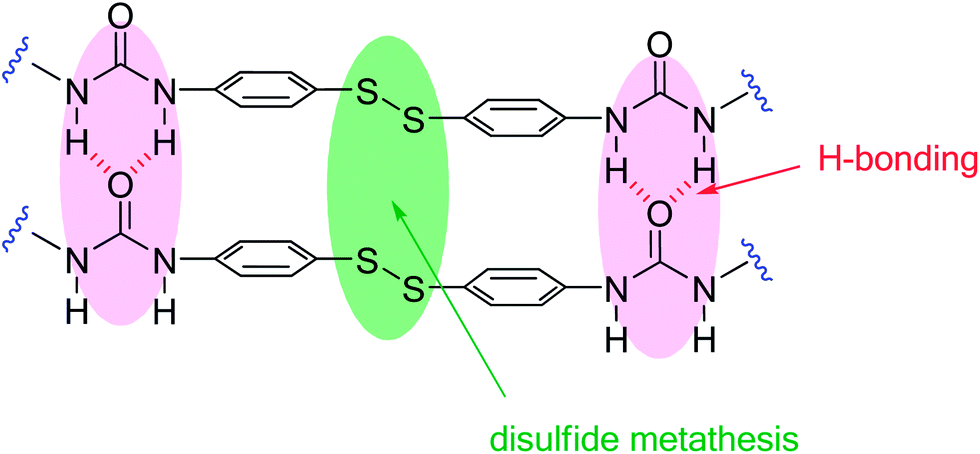 | ||
| Fig. 3 Proposed interactions involved in the self-healing process of 9a. | ||
In order to study these two effects, the self-healing efficiency of 9b was studied considering it as a reference material with no disulfide bonds. Pristine samples of reference material 9b exhibited a tensile strength of 0.84 ± 0.05 MPa and an elongation at breaking point of 2156 ± 50% (Fig. 2b). The mended samples of 9b (r.t., 1, 2, 12 and 24 h) showed a maximum tensile strength of 0.43 ± 0.05 MPa and an elongation at breaking point of 1657 ± 50%. Such values were already achieved after 1 hour, and did not improve with longer healing times. This indicates a maximum healing efficiency of 51%, which must be attributed to the contribution of the quadruple H-bond between the urea groups. On the other hand, 9a recovered 62% of its initial tensile strength at 1 h, but already achieved 80% after 2 hours. After 24 hours, the healing was practically quantitative. These results suggest that H-bonds would give rise to a healing efficiency of around 50% in a short period of time, which is common for both systems. Thus, the further quantitative healing shown by 9a would be attributed to the effect of the aromatic disulfide metathesis.
In summary, a new poly(urea–urethane) thermoset elastomer with aromatic disulfide crosslinks has been designed, which is able to undergo catalyst-free disulfide metathesis. Such a material presents near quantitative self-healing efficiency at room-temperature, without the need for any external intervention such as heat or light. Finally, the fact that poly(urea–urethane)s with similar chemical composition and mechanical properties are already used in a wide range of commercial products makes this system very attractive for a fast and easy implementation in real industrial applications.
Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7-NMP-2012-SMALL-6) under grant agreement no. 309450.Notes and references
- S. J. Garcia, H. R. Fischer and S. van der Zwaag, Prog. Org. Coat., 2011, 72, 211–221 CrossRef CAS.
- T. C. Mauldin and M. R. Kessler, Int. Mater. Rev., 2010, 55, 317–346 CrossRef CAS.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987 RSC.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- N. K. Guimard, K. K. Oehlenschlaeger, J. Zhou, S. Hilf, F. G. Schmidt and C. Barner-Kowollik, Macromol. Chem. Phys., 2012, 213, 131–143 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, Polym. Chem., 2013, 4, 4860–4870 RSC.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. o. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- Y.-X. Lu, F. o. Tournilhac, L. Leibler and Z. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS PubMed.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- R. Nicolaÿ, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules, 2010, 43, 4355–4361 CrossRef.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- Y. L. Zhang, B. Yang, X. Y. Zhang, L. X. Xu, L. Tao, S. X. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- F. Y. Liu, F. Y. Li, G. H. Deng, Y. M. Chen, B. Q. Zhang, J. Zhang and C. Y. Liu, Macromolecules, 2012, 45, 1636–1645 CrossRef CAS.
- P. W. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- R. Martin, A. Rekondo, J. Echeberria, G. Cabanero, H. J. Grande and I. Odriozola, Chem. Commun., 2012, 48, 8255–8257 RSC.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- U. Lafont, H. van Zeijl and S. van der Zwaag, ACS Appl. Mater. Interfaces, 2012, 4, 6280–6288 CAS.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278–7288 RSC.
- A. M. Belenguer, T. Friscic, G. M. Day and J. K. M. Sanders, Chem. Sci., 2011, 2, 696–700 RSC.
- R. J. Sarma, S. Otto and J. R. Nitschke, Chem.–Eur. J., 2007, 13, 9542–9546 CrossRef CAS PubMed.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152 RSC.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2012, 113, 80–118 CrossRef PubMed.
- H.-K. Ono, F. N. Jones and S. P. Pappas, J. Polym. Sci., Polym. Lett. Ed., 1985, 23, 509–515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, synthesis and characterization data for prepolymers, crosslinked elastomers and aromatic disulfide metathesis experiments. Movies S1 and S2. See DOI: 10.1039/c3mh00061c |
| This journal is © The Royal Society of Chemistry 2014 |