 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultivalent glycoconjugates as vaccines and potential drug candidates
Sumati
Bhatia
,
Mathias
Dimde
and
Rainer
Haag
*
Freie Universität Berlin, Institut für Chemie und Biochemie, Takustrasse 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de
First published on 16th May 2014
Abstract
Pathogens adhere to the host cells during the first steps of infection through multivalent interactions which involve protein–glycan recognition. Multivalent interactions are also involved at different stages of immune response. Insights into these multivalent interactions generate a way to use suitable carbohydrate ligands that are attached to a basic scaffold consisting of e.g., dendrimer, polymer, nanoparticle, etc., with a suitable linker. Thus a multivalent architecture can be obtained with controllable spatial and topology parameters which can interfere with pathogen adhesion. Multivalent glycoconjugates bearing natural or unnatural carbohydrate antigen epitopes have also been used as carbohydrate based vaccines to stimulate an innate and adaptive immune response. Designing and synthesizing an efficient multivalent architecture with optimal ligand density and a suitable linker is a challenging task. This review presents a concise report on the endeavors to potentially use multi- and polyvalent glycoconjugates as vaccines as well as anti-infectious and anti-inflammatory drug candidates.
1. Introduction
Multi- and polyvalency play a crucial role in various biological processes like self-organization of matter, cell–cell recognition leading to signal transduction, detection, and immune response.1 Interactions between an m-valent receptor and an n-valent ligand (m, n > 1; also m ≠ n) are considered to be multivalent interactions. The term polyvalency is especially used in the context of ligands binding to receptors on interfaces such as cell membranes, which offer a large number (n » 10) of two-dimensionally distributed binding sites.1 The surface of a cell is covered with various carbohydrates which are usually attached to other biomolecules like proteins or lipids. Different pathogens like bacteria, fungi, viruses, toxins, etc., adhere to the host cells through multivalent interactions between carbohydrates and proteins as a first step in the infection process. The individual interactions between proteins and carbohydrates are weak but multiple simultaneous interactions lead to strong functionally useful avidities. The term avidity is defined in biochemistry as the accumulated strength of the multiple affinities of individual non-covalent binding interactions. Avidity was originally introduced to describe the binding behavior of immunoglobulins with different (multi) valencies (antibodies, IgD, IgEm, and IgG monomers: divalent; IgA homodimer: tetravalent; IgM homopentamer: decavalent).2 The recent advent of carbohydrate microarray technologies3,4 as well as advances in glycobiology and glycochemistry have paved the way for insights into these multivalent interactions. This new knowledge about protein–glycan interactions between pathogens and mammalian host cells has made multivalent glycoconjugates attractive scaffolds for targeting different pathogens. Therefore, several unnatural glycoconjugates with different valencies and the spatial arrangement of ligands based on scaffolds like proteins,5–7 polymers,8–10 fullerenes,11,12 calixarenes,13–15 dendrimers,16–20 nanoparticles,21,22etc., have been reported. The anti-adhesive or cell targeting properties of these multivalent glycoconjugates can competitively inhibit the pathogens from adhering to the host cells which ideally would prevent or treat the disease caused by these pathogens. For example, a synthetic glycolipid Gb3 (α-D-Gal(1,4)β-D-Gal(1,4) β-D-Glc(1,O-ceramide)) analog that was covalently attached to insoluble silica particle has emerged as an anti-adhesive carbohydrate-based drug candidate for adsorbing shiga-like toxins of E. coli. This synthetic analog of the Shiga toxin (Stx) receptor (Synsorb Pk) is currently undergoing clinical trials.23 Furthermore, it has been found that surface sugar densities, as well as the length and nature of the linker used for conjugation to the basic scaffold, play a significant role in the optimum avidities and selectivities. Therefore, designing an efficient multivalent glycoconjugate remains a challenging task.24,25 Advances in the identification and synthesis of glycan epitopes on different pathogens have made carbohydrates attractive vaccine targets and immunomodulators. The structurally defined glycoconjugates displaying these unique glycan antigen structures can follow a lectin-mediated cellular uptake mechanism to generate an immune defense mechanism in the body against chronic infections. In spite of the existing challenges in the isolation and identification of the carbohydrate antigen itself and in the preparation of structurally similar glycan epitopes, a fruitful effort has been made in this area and a number of conjugate versions of polysaccharides vaccines are now commercially available or in clinical trials.26 An octavalent O-polysaccharide (OPS) with toxin A conjugate, for example, has been found to be immunogenic and clinically effective against Pseudomonas aeruginosa and is currently undergoing phase III clinical trials.27This review is a concise overview on the different attempts to apply multivalent glycoconjugates as drugs in pathological conditions like infections or cancer. It highlights the efforts being made by various research groups to design multivalent glycoconjugates that can be used to inhibit the adhesion of bacteria, bacterial toxins, and viruses to the host cells or to stimulate the immune systems against pathogens like HIV. The endeavor to apply multivalent glycoconjugates as anti-inflammatory drugs by targeting leukocyte trafficking will also be discussed.
2. Vaccines
The unique glycocalyx and its dense surface distribution on different pathogens and malignant cells have made carbohydrates attractive vaccine targets. Glycan antigens are diverse and range from capsular polysaccharides to small monosaccharide antigens. Capsular polysaccharides have been in use for decades for providing immunity against different bacterial infections. The main bottleneck in the development of a carbohydrate vaccine, however, is to identify carbohydrate antigen itself. In recent years, the identification and synthesis of the unique glycan epitope present on the various pathogens and malignant cells has been more efficient. This has paved a way for the development of carbohydrate-based vaccines for eliciting the immune response to the different infections and cancer as well. Another obstacle associated with the development of carbohydrate-based vaccines is the poor antibody response to carbohydrates which is usually due to the T-cell independent immune responses triggered by repetitive carbohydrate antigens.28,29 Carbohydrate specific antibodies have a low affinity with dissociation constants in the micromolar range and glycan interactions mainly rely on the avidity effects of multivalent interactions. Avery and Goebel reported that immunogenicity of carbohydrates can be increased by covalent attachment of glycan to a suitable immunogenic protein which has led to the discovery of conjugate vaccines.30 In the carbohydrate-based vaccines glycan antigens are conjugated with a source of helper T-cell epitope, usually with an immunogenic carrier protein. Taxoids, metalloproteins like keyhole limpet haemocyanin (KLH) or virus capsids are also used as carriers.31 Several strategies can be used for linking the carrier to the sugar but one needs to carefully investigate the immunogenicity of these linkers to the glycan antigens.32,33 Adjuvants are also included in the vaccine to increase the immunogenic response to antigens and/or modulates it towards the desired immune responses. Advantages of adjuvants include the reduction of the antigen amount needed for a successful immunization, the reduction of the frequency of booster immunizations needed, and an improved immune response in elderly and immuno compromised patients. Adjuvants include, for example, mineral salts, oil emulsions, surfactant based formulations, microbial derivatives, and particulate adjuvants (virosomes). With a few exceptions, alum (potassium aluminium sulfate) is still the only adjuvant that has been approved for human use in the majority of countries worldwide.34 Therefore, it is found in numerous vaccines, including diphtheria–tetanus–pertussis, human papillomavirus, and hepatitis vaccines.35 QS-21, a saponin derived from the tree bark of Quillaja saponaria, is also being clinically tested.31 Tripalmitoyl-S-glyceryl-cysteinylserine (Pam3Cys) has also been discovered to have adjuvant properties. Fig. 1 depicts some of the different strategies for preparing glycoconjugate vaccines, i.e., multivalent synthetic conjugates, protein conjugates, and lipid conjugates.31 | ||
| Fig. 1 Different categories of glycoconjugate vaccines. (a) Active glycan unit is conjugated to synthetic multivalent scaffolds, for example, dendron or regioselectively addressable functionalized template, and made immunogenic by conjugation with a helper T-cell epitope. (b) Activated glycan unit or the polyvalent glycan scaffold is conjugated with the immunogenic carrier protein. (c) Lipid conjugates can be prepared by covalent linkage of activated glycans to helper T-cell peptides linked to lipid moieties. | ||
2.1. Multivalent glycoconjugate vaccines against different pathogens
A diverse and dense array of protective capsular and lipopolysaccharides is present on the surface of bacterial pathogens. Heterogeneity and complexity of the capsular polysaccharides on bacterial pathogens have always been a great challenge in the development of multivalent carbohydrate-based vaccines. For example, more than 90 serotypes are known for S. pneumonia. The first polysaccharide based vaccine, Pneumo Vax from Merck and Co. launched in 1984 was composed of unconjugated capsular polysaccharide isolated from 14 pneumonia serotypes; the current development provides active immunization against 23 serotypes.36,37 In order to develop a multivalent conjugate vaccine against different bacterial pathogens, polysaccharides are generally isolated from most clinically relevant serotypes and then degraded into smaller products for activation and conjugation to immunogenic carrier proteins. For example, Prevnar from Wyeth/Pfizer is a heptavalent pneumococcal conjugate.While developing the synthetic carbohydrate-based vaccines, the effect of chain length and saccharide density on the potent antibody response can be addressed. For example, conjugates with different saccharide densities of tetra-, octa-, dodeca-, and hexadecasaccharides were prepared for vaccine against S. dysenteriae. These conjugates were based on a tetrasaccharide repeat unit of O-specific oligosaccharides. In the process it was found that octa-, dodeca-, and hexadecasaccharides were immunogenic and that the chain length determined the optimal loading densities of saccharides.38 Furthermore, the nonreducing end of Shigella dysenteriae type 1 O-specific oligosaccharides had an effect on their immunogenicity as conjugates in mice.39 For many bacterial infections, multivalent glycoconjugate vaccines have been developed based on the fragments of their capsular polysaccharides. Examples include Streptococcus pneumoniae, Neisseria meningitides, and Haemophilus influenza.32Table 1 shows some examples of commercially available multivalent glycoconjugate vaccines.
| Vaccine | Manufacturer (trade name) |
|---|---|
| a The information in the table has been taken from a complete list of vaccines licensed for immunization and distribution in the United States, which is available from the US Food and Drug Administration website (http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm093833.htm). CRM197 is a nontoxic mutant of diphtheria toxin. | |
| Pneumococcal vaccine, polyvalent | Merck & Co, Inc. (Pneumovax 23) |
| Pneumococcal 7-valent conjugate (Diphtheria CRM197 protein) | Wyeth Pharmaceuticals Inc. (Prevnar) |
| Pneumococcal 13-valent conjugate (Diphtheria CRM197 protein) | Wyeth Pharmaceuticals Inc. |
| Meningococcal (groups A, C, Y, and W-135) oligosaccharide diphtheria CRM197 conjugate | Novartis Vaccines and Diagnostics, Inc. (Menveo) |
| Meningococcal (groups C and Y and Haemophilus b) tetanus toxoid conjugate | GlaxoSmithKline Biologicals (MenHibrix) |
| Meningococcal (groups A, C, Y and W-135) polysaccharide diphtheria toxoid conjugate | Sanofi Pasteur, Inc. (Menactra) |
| Meningococcal polysaccharide vaccine, groups A, C, Y and W-135 combined | Sanofi Pasteur, Inc. (Menomune-A/C/Y/W-135) |
Infection by parasitic protozoans and helminths is still a big threat to the human population. Over 15 million deaths occur worldwide per year due to parasitic infections and diseases,40 and no commercial vaccine has been developed as yet for any of the parasitic infections like malaria, leishmaniasis, African trypanosomiasis, amoebiasis, schistosomiasis, and lymphatic filariasis. This is due to the complex immune evasion mechanisms used by the different parasites41 and to the lack of understanding of molecular interactions between host immune cells and parasites.42 The unique glycan antigens are abundant and accessible on the surface of the multiple developmental stages of different parasitic protozoans and helminthes which makes glycans attractive vaccine targets. Difficulties in obtaining enough parasitic material for study also limit the progress in this area. Several efforts are, however, being made to investigate potential vaccine antigens using advanced diagnostic techniques for glycan detection and chemical or chemo-enzymatic methods.43 Different surface glycans, for example, associated with Schistosoma spp. and Leishmania spp. have been identified and are being evaluated as immunogens.31,42,44
It is known that each type of parasitic protozoa produces its own unique and highly complex glycan structure. African trypanosomes, for example, keep genetically switching their highly immunogenic glycosylphosphatidylinositol (GPI)-anchored surface glycoprotein which is called variant surface glycoprotein (VSG).45 Virtually all protozoans, including Plasmodium parasites that cause malaria synthesize GPI. The GPI synthesized by Plasmodium may be a novel target for vaccine production. Preclinical studies reported by Seeberger and coworkers showed that a synthetic version of GPI conjugated to keyhole limpet haemocyanin (KLH) (Fig. 2) elicited high titres of immunoglobulin-γ (IgG) and appeared to reduce the pathology of malaria.46 Although the survival rates in mouse models increased, further infection could not be prevented.47
 | ||
| Fig. 2 Synthetic Plasmodium: glycosylphosphatidylinositol-keyhole limpet haemocyanin (GPI-KLH).46 | ||
The same group also presented a versatile approach based on a synthetic GPI glycan array to correlate anti-GPI antibody levels and protection from severe malaria but the question remains whether an antitoxic immune response to malarial GPI provides protection against malaria.48
Some glycoconjugate biosynthetic studies done on Leishmania protozoans have revealed that they also generate a surface lipophosphoglycan (LPG) with unique phosphodiesters of mannose in the backbone and furanosyl galactoside modifications that might be contained within ether based phospholipids, GPI-anchored glycoproteins, mucin-type membrane, and secreted glycoproteins.44,49–51 The recent automated syntheses of Leishmania related and other antigenic glycans reported by Seeberger and coworkers will give a tremendous boost to the generation of libraries of conjugate vaccine candidates for screening.52
2.2. HIV vaccine development and challenges
The dense conserved cluster of oligomannose glycans on the envelop glycoprotein, gp120 subunit of Human Immunodeficiency Virus type 1 (HIV-1), masks conserved protein epitopes and facilitates virus entry via interaction with glycan binding proteins on susceptible host cells. The broadly neutralizing monoclonal antibody 2G12 neutralizes a wide range of HIV-1 strains in animal models by specifically binding to the cluster of oligomannose glycans on gp120.53–56 New broadly neutralizing antibodies that also recognize and penetrate HIV glycan shield have recently been isolated from patients.57 Highly dense clusters of oligomannose on gp120 are stabilized by a network of intermolecular hydrogen bonding and thus led to a close spacing between the carbohydrates which is unusual in mammalian glycoproteins. These observations provide the basis for immunological discrimination by 2G12.58 Using the information available by structural and modeling studies on how 2G12 interacts within the oligomannose clusters of gp120,59 efforts have been made to design an immunogen that elicits 2G12 like antibodies.54 This is a very challenging task due to the unique recognition mode of 2G12. Several multivalent oligomannoside ligands containing Manα(1,2)Man, including GlcNAcMan9 for 2G12, have been synthesized for mimicking the densely conserved oligomannose clusters in the glycan shield on gp120. The synthetic tetramannoside (Man4) corresponding to the D1 arm of GlcNAcMan9 was found to be the minimum recognition motif and as effective as GlcNAcMan9 itself in binding to gp120.55 Synthetic strategies for the multivalent presentation of oligomannose have involved the use of oligodendrons, Qβ bacteriophage, and generation of cyclic glycopeptides60–63 (Fig. 3). A synthetic Man4 on bovine serum albumin (BSA) molecule by Burton and coworkers,64 and a cyclic glycopeptides conjugate of GlcNAcMan9, further conjugated with Outer Membrane Protein Complex (OMPC) by Danishefsky and coworkers,65,66 could be used to induce a 2G12 like antibody response, but antibodies did not bind to the gp120. In another study by Geng and coworkers,67 a mutant strain of Saccharomyces cerevisiae containing a large number and high density of N-linked glycans elicited gp120 cross-reactive antibodies in rabbit, but these antibodies did not neutralize HIV. The current challenge lies in breaking the immune tolerance against HIV-1 strains by eliciting the antibodies with proper specificity. This is due to an inappropriate mimicry of the glycan shield (flexible glycans) and the unique architecture of the current model antibody, 2G12. Moreover, there is a need for more fundamental studies on B cell immunology to break the immune tolerance towards the HIV-1 carbohydrate antigens. | ||
| Fig. 3 (a) GlcNAc2Man9 divalent glycopeptides outer membrane protein complex (OMPC) from Neisseria meningitidis.66 (b) Man9, glycodendron bovine serum albumin (BSA) conjugate.62 (c) Qβ-Man9 conjugate.68 | ||
2.3. Cancer vaccine
Tumor associated carbohydrate antigens (TACAs) are over-expressed self glycans in cancer cells which occur because of the altered glycosylation.69 There is more glycosyltransferase expression in cancer cells than in normal tissues which leads to an increase in the size and branching of N-glycans, as well as to more sialylation,70,71 and linking terminal residues to glycans which results in an overexpression of certain terminal glycan epitopes on tumors, such as sialyl Lewis X, sialyl Lewis A, sialyl 2-6-α-N-acetylgalactosamine (sTn), sialyl Lewis Y, globo-hexaosylceramide, and PSA.72–74 Although TACAs may potentially serve as vaccine antigens, they are poorly immunogenic and it is difficult to isolate the unique TACA glycan structure from natural sources. A common way to break tolerance towards TACAs is to use a monovalent carbohydrate antigen in multiple copies on an immunogenic carrier such as KLH in the presence of a strong adjuvant like QS21. This strategy describes the first generation monomeric cancer vaccine. Investigations on antigen conjugates have established that KLH and QS21 are the most potent carrier and adjuvant pair for breaking tolerance towards TACAs. There are several studies where natural (glycosphingolipid containing sialic acids like GD2, GD3, GM2, and fucosyl GM1) and unnatural or synthetic (N-propionylated PSA, GD2- and GD3-lactone, globohexaosylceramide and Lewis Y) carbohydrate antigens that have been conjugated with KLH in the presence of QS21 have been found to be immunogenic in cancer patients.75–81 A novel approach to break the immunotolerance to TACAs is to combine cell glycoengineering with vaccines prepared using unnatural TACAs.82 The critical step in this new strategy is metabolic engineering of cancer, namely, to induce expression of an artificial form of a TACA by supplying tumors with an artificial monosaccharide precursor. For this purpose, tumor cells were incubated with different N-acyl-D-mannosamines, and modified forms of GM3 expressed on tumor cells were analyzed by flow cytometry using antigen-specific antisera. Recent reports by Guo and coworkers showed that tumor cells expressed unnatural GM3 analog, i.e., GM3NPhAc instead of natural GM3 when incubated in the presence of N-phenylacetyl-D-mannosamine.83 The GM3NPhAc–protein conjugates are particularly immunogenic and produce strong T-cell dependent antibodies. Therefore, the new glycoengineered melanoma cells expressing unnatural GM3NPhAc were selectively killed by GM3NPhAc-specific antibodies produced by GM3NPhAc–KLH conjugates.84 These results suggest the feasibility of selectively eradicating cancer in vivo without affecting the normal cells. This immunotherapy may be applicable to other tumors as long as they express GM3.The development of multivalent vaccines85,86 has been encouraged by the heterogeneity of TACA expression on malignant cells. Danishefsky and coworkers87 have prepared pentavalent and hexavalent carbohydrate antigenic constructs on a single peptide backbone, conjugated with KLH and Pam3Cys. The early immunogenicity studies in mice showed that pentavalent unimolecular conjugates (on KLH or Pam3Cys) were much more immunogenic than the corresponding pool of monomeric KLH conjugates (Fig. 4). Recent clinical studies, however, showed that lower IgM and IgG antibodies titres were produced against individual antigens in high risk patients who had been immunized with hexavalent vaccines including GM2, Globo H, Lewis Y, glycosylated MUC-1-32mer, Tn, and TF. These vaccines were prepared in a clustered formation, conjugated to KLH, and mixed with QS-21.88 Mucin type glycans such as α-N-acetyl-D-galactosamine (αGalNAc, the Tn antigen glycan) are overexpressed on the surface of breast cancer cells. Cameron and coworkers89 have recently reported the preparation of Tn-glycan antigen-based glycopolymers which were conjugated to gold nanoparticles and yielded ‘multicopy multivalent’ glyconanoparticles with a size range of 5–25 nm. These glycoconjugated gold nanoparticles produced a strong and long-lasting production of antibodies that were selective to the Tn-antigen glycan and cross-reactive towards mucin proteins displaying Tn.
 | ||
| Fig. 4 Structure of uimolecular pentavalent vaccine.87 | ||
3. Multivalent glycoconjugates as antiviral compounds
Multivalent binding interactions between glycoproteins on the surface of different pathogens and cellular receptors/oligosaccharides play a crucial role in the initial stages of cellular infection. For example, Fig. 5 shows how the multivalent interactions between hemagglutinin glycoproteins on influenza virus and sialic acid residues on cells lead to endocytosis and then infection. In the recent past, multivalent glycoconjugates on dendritic/globular or linear scaffolds have emerged as potent inhibitors of infection by different viruses like HIV-1, Ebola, influenza, etc., which will be discussed in the following sections by focusing on the most relevant studies. | ||
| Fig. 5 Multivalent interactions between trimeric haemagglutinin (HA3) on influenza virus and sialic acid (SA) residues on cells leading to endocytosis and infection. | ||
3.1. DC-SIGN mediated infection inhibition
Dendritic cells on mucosal surfaces carry a calcium dependent (C-type) lectin that is a dendritic cell-specific intercellular adhesion molecule (ICAM 3) grabbing non-integrin (DC-SIGN). This lectin can bind to a broad spectrum of pathogens: viruses (HIV-1, HIV-2, SIV, Ebola virus, hepatitis C virus, cytomegalovirus and dengue virus), bacteria (Mycobacterium tuberculosis, Helicobacter pylori and Klebsiella pneumonia), yeasts (Candida albicans), and parasites (Leishmania and Schistosoma).90 The carbohydrate recognition domain (CRD) of DC-SIGN recognizes the highly mannosylated and fucosylated glycoproteins which are found on the surface of certain pathogens. For example, highly mannosylated glycan shields on gp120 of HIV-1 are specifically recognized by DC-SIGN of dendritic cells on mucosal surfaces. The HIV-1 virus is taken by these dendritic cells to the lymph nodes where they effectively transfer the virus by a so-called trans infection of T-lymphocytes where the viral replication occurs. Multivalent glycoconjugate systems have been designed to mimic the surface of the virus to compete with natural ligands for binding with receptors for the development of carbohydrate-based antiviral drugs against HIV and Ebola virus infections. Recent investigations by Bernardi, Clerici, and coworkers have shown that a tetravalent dendron with four copies of linear trimannoside inhibits DC-SIGN mediated trans HIV infection process of CD4+ T lymphocytes in the presence of elevated viral loads in cellular and cervical explants models (Fig. 6).91,92 They found that multivalent presentation of linear di- and trimannosides on G3 Boltron-type dendrimers (32 copies) strongly inhibited the cell infection by Ebola pseudotyped viral particles by blocking DC-SIGN receptor showing IC50 values in the nanomolar range (Fig. 6).93 In an another effort to mimic dense mannose clusters on HIV-1 envelop, Penades and coworkers prepared a series of gold nanoparticles with different spacers and different densities of oligomannoside. The manno-GNPs containing the disaccharide Manα1-2Manα were found to be the best inhibitors, because they increase inhibition 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold (100% inhibition at 115 nM) compared to the corresponding monomeric disaccharide (100% inhibition at 2.2 mM).94 Furthermore, these multivalent manno–glyco nanoparticles inhibited DC-SIGN-mediated HIV-1 trans-infection of human T cells at nanomolar concentrations in experimental setting that mimicked the natural route of virus transmission.95
000-fold (100% inhibition at 115 nM) compared to the corresponding monomeric disaccharide (100% inhibition at 2.2 mM).94 Furthermore, these multivalent manno–glyco nanoparticles inhibited DC-SIGN-mediated HIV-1 trans-infection of human T cells at nanomolar concentrations in experimental setting that mimicked the natural route of virus transmission.95
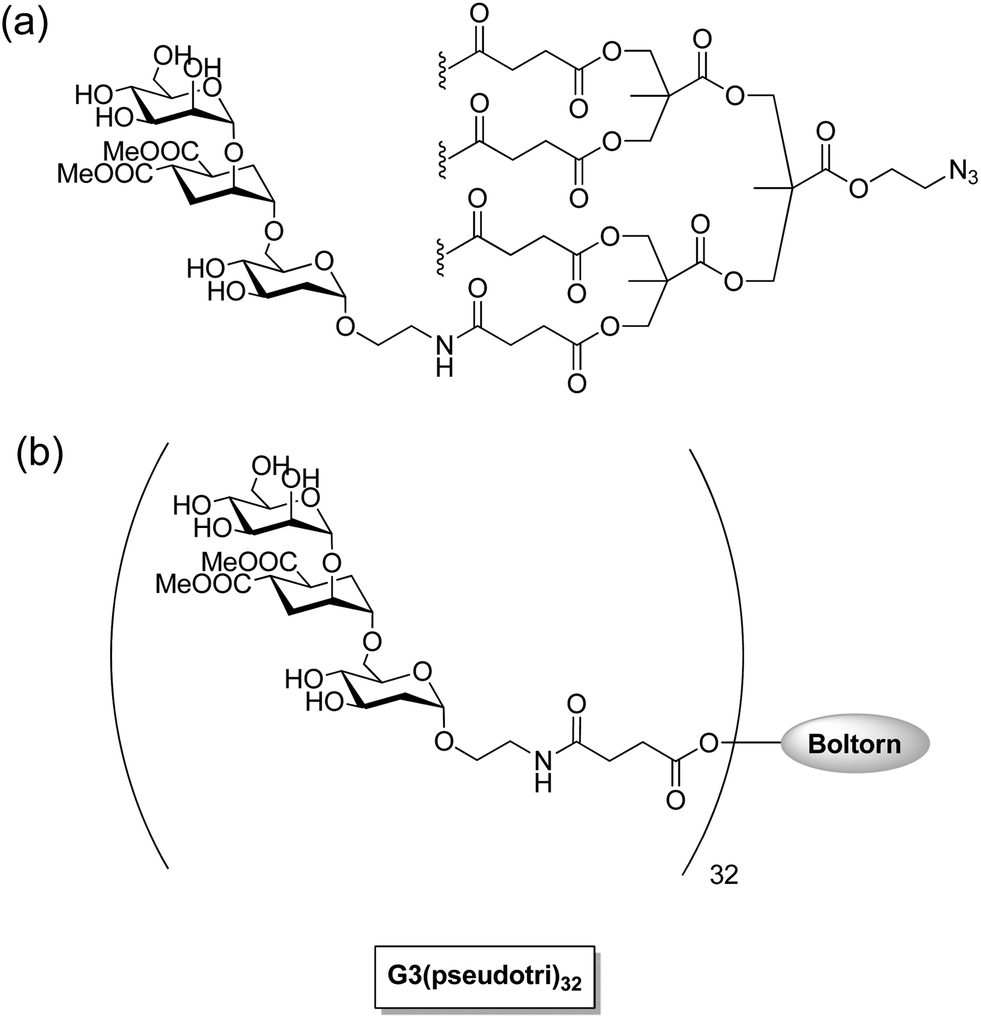 | ||
| Fig. 6 (a) Tetravalent linear trimannoside conjugate for HIV infection inhibition.92 (b) Trimannoside conjugates of G3 Boltorn type dendrimers for Ebola peudotyped virus infection inhibition.93 | ||
3.2. Influenza virus inhibition
Influenza viral infection is one of the biggest challenges for medicinal science and a big threat to the human population. It often causes epidemics and pandemics. Three glycoproteins present on the virus surface, i.e., neuraminidase (NA), M2 ion channel protein, and hemagglutinin (HA) are important targets for fighting influenza. HA glycoprotein is trimeric and most abundant (∼600 copies per virus particle) on the surface of influenza virus.96 There is no multivalent drug available till now which can target HA glycoprotein. The two glycoproteins HA and NA determine the viral subtype. Commercially available unimolecular drugs target either NA (zanamivir/Relenza, Oseltamivir/Tamiflu) or M2 ion channel protein (amantadine and rimantadine) activity. However, the emergence of stable and transmissible drug-resistant influenza strains can render these drugs ineffective. The three-dimensional structure of HA complexed with SA reported by Wiley and coworkers96 led to the discovery that HA glycoproteins on the influenza virus play a significant role in adhering virus to the susceptible cells in a multivalent interaction between three sialic acid binding sites on its globular domain with sialyloligosaccharide moieties of cellular glycoconjugates. Therefore, macromolecular scaffolds bearing multiple copies of sialic acid residues can be of great use for a competitive inhibition of the virus attachment to the cells. Efforts are in progress for the design, synthesis, and investigation of such multivalent glycoconjugates which may lead to the discovery of anti-influenza drugs.So far the synthetic HA inhibitors mainly consisted of low molecular weight scaffolds that form clusters of mono-,97,98 bi-,99 tetra-,100 and octavalent sialosides.101 The octavalent sialosides were based on the polyazido-calix[4]arene scaffold but they showed only a moderate glycoside cluster effect and minimum inhibitory concentration in the millimolar range. Both natural and synthetic multivalent carriers have been explored to prepare multivalent sialosides conjugate. Natural carriers include the usage of self-assembling sialo-glycopeptides,102 proteins (specifically equine α2-macroglobulin and guinea pig α2-macroglobulin),100 and polysaccharides.103 Synthetic carriers that have been typically used are spherical macromolecules like dendrimers104 and liposomes,105 linear polymers,106 and nanostructures.105 Sialylglycopolymers have been studied the most in detail. Whitesides and coworkers have extensively studied α-sialoside-polyacrylamide copolymers to inhibit the agglutination of erythrocytes by influenza A virus. The best copolymer in the series inhibited hemagglutination 104 to 105 times more strongly than did similar concentrations of α-methyl sialoside.107 They also prepared C-glycoside by coupling aminosialosides with preactivated esters, i.e., poly[N-(acryoyloxy)succinimide] (pNAS).108 Using the C-glycoside of sialic acid made it possible to investigate hemagglutinin inhibition at different temperatures from 4–36 °C because C-glycosides cannot be hydrolyzed by the neuraminidase glycoproteins present on influenza virus. Extensive studies were performed to optimize sialylglycopolymers by varying the chemical structure and content of the ligand in the polymer and by altering the structure of the side chains in the carrier which has led to inhibitors with inhibitory concentrations in the nanomolar range and were thousand-fold more effective than the most potent natural inhibitor, equine α2-macroglobulin. These polymeric sialosides were quite effective but they failed to become drugs due to the severe toxicity of their polyacrylamide backbone. Whiteside and coworkers only tested these polymeric inhibitors for a single influenza virus strain X31. Other groups, however, observed a significant variation in the sensitivity of these inhibitors when tested against different virus strains.109 Haag and coworkers chemically functionalized gold nanoparticles sized between 2 nm and 14 nm with sialic acid-terminated glycerol dendron. The 14 nm sialic acid conjugated AuNPs were nontoxic to cells and had a high affinity for HA on the influenza virus surface as observed by electron microscopy techniques and biochemical analysis (Fig. 7).110 The infection inhibition amounted to 30% which led to the hypothesis that virus-sized particles could further improve the inhibition of the infection.111 Recently, Haag and coworkers investigated a new class of biocompatible dendritic multivalent sialo conjugates based on appropriately functionalized polyglycerol nanogels (nPG) which were varied in size between 40–100 nm, respectively (Fig. 8). The size and ligand density on the nanoparticles were optimized, whereby the 50 nm sized sialic acid conjugated nPG with only 12% ligand density was found to be the best conjugate showing 80% inhibition of the influenza virus A virus (strainA/X31).112 Chen and coworkers recently reported that poly-L-glutamine conjugated with multiple copies of drug zanamivir (PGN-ZA) via a flexible linker inhibited influenza viral fusion and release at subnM concentrations of zanamivir.113 These PGN-ZA polymers were still effective against isolated ZA- and oseltamivir-resistant influenza virus.114
 | ||
| Fig. 7 (a) Synthesis of sialic acid functionalized gold nanoparticles (AuNPs). (b) Electron microscopic visualization of 14 nm sized AuNPs with a sialic acid corona.110 | ||
 | ||
| Fig. 8 Sialic acid functionalized polyglycerol nanogels (nPG) with hemagglutinin receptors on the virus surface. The nPG consists of a number of chemically cross-linked dendritic polyglycerol (dPG) units. | ||
4. Antibacterials
Bacteria and bacterial toxins infect host cells via multivalent interactions involving protein–glycan recognition in the first steps of infection. Bacteria constitute a large domain of prokaryotic microorganisms which can be typically a few micrometers in length, whereas bacterial toxins are small poisonous substances secreted by living bacteria during exponential growth. Millions of deaths per year are caused by shiga and cholera toxin infections. Both these toxins belong to the clinically significant subset of bacterial AB5 toxins, where A subunit is enzymatically cleavable and B5 homopentamer has receptors for oligosaccharide recognition on the cellular surface. Fig. 9 shows, for example, an inhibited cellular uptake of a pentameric bacterial toxin by a sugar-conjugated pentacyclen core. Bundle and co-workers designed and improved some oligovalent, water soluble carbohydrate ligands called “Starfish” and “Daisy” to inhibit Shiga toxin (Stx) 1 and Stx2 which were generated by E. coli strains. The inhibition potential was confirmed with in vivo experiments.115,24 Multivalent glycoconjugates based on different scaffolds like pentacyclen, dendrimers, polymers, fullerenes, cyclodextrins, cyclopeptides, and gold nanoparticles have been developed which can interfere with the pathogen (bacteria or bacterial toxin) adhesion process.116 However, the resulting sugar density at the glycoconjugate surface as well as the chemical structure of the linker groups are important factors for an effective inhibitor.20 To determine the corresponding partner for the glycan-binding protein (GBP) of the pathogenic microorganisms, glycan microarrays were also developed to scan a great number of glycan candidates in a short time.4,25,117 The combination of active glycans covalently linked to various carrier platforms resulted in powerful compounds which should be able to treat infectious diseases. These multivalent glycoconjugates may play a role in fighting antibiotic resistant bacteria strains as well.118 | ||
| Fig. 9 Inhibition of the cellular uptake of a pentameric bacterial toxin by a sugar-conjugated pentacyclen core. (a) Cellular uptake of a pentameric bacterial toxin. The whole toxin binds to cellular receptor, followed by endocytosis. (b) Inhibition of a toxin binding to cellular receptors using pentavalent pentacyclen based glycoconjugate. | ||
4.1. Cholera toxin inhibition
Cholera is an intestinal infection caused by the cholera toxin (CT) produced from Vibrio cholera119,120 results in fast dehydration and shock caused by watery diarrhea. The seven cholera pandemics over the past 185 years affected nearly the whole world. Cholera deaths are still common in remote areas where effective treatment is unavailable.121 Conjugative-plasmid mediated multiply antibiotic-resistant (including to tetracycline) V. cholerae O1 (MARV) has also emerged as a major problem, first in Tanzania then in Bangladesh. Antibiotic strains that are resistant to tetracycline, ampicillin, kanamycin, streptomycin, sulphonamides, trimethoprim, and gentamicin have appeared in several cholera endemic countries during the past two decades.121CT belongs to a family of bacterial AB5 toxins with an enzymatically active A subunit which is uptaken by the mammalian cells via an oligosaccharide recognition of the B5 subunit. CT's first mode of action is to strongly bind to the receptor cells on the cell surface. The receptor for the CT is a specific glycolipid, the ganglioside GM1, a regular component in the outer leaflet of many cell membranes. The A subunit is connected to the B-pentamer via a disulfide bridge. After cellular uptake, the A subunit is activated and released in the cytosol by reduction of the disulfide bond.24,122,123 Several multivalent glycoconjugates have been explored to target the B5 subunit of cholera toxin to prevent its cellular uptake.
Different multivalent glycoconjugates, bearing pseudotrisaccharides and four to eight oligo-GM1 units, have been prepared based on calixarenes, poly(propylene imine), and PAMAM dendrimers. The B5 pentamer unit of CT had a much higher affinity for these conjugates than native GM1.124–126 Pieters and coworkers synthesized multivalent versions of the GM1os using glycodendrimers using the click chemistry approach. These multivalent versions strongly inhibited the CTB5-subunit. For example, the most complex multivalent GM1os conjugate, which was an octavalent glycodendrimer, was 380000-fold more potent with IC50 = 50 pM than the monovalent GM1-mimic127 (Fig. 10). This GM1-mimic was compared with natural GM1 gangliosides and showed a similar binding to the CTB5-subunit in an ELISA-type assay.128 Multivalent galactose inhibitors were prepared as simplified versions of multivalent GM1 derivatives. Multivalent binding interactions compensated the low binding affinity of galactose for CTB5-subunit. Tetravalent and octavalent galactose inhibitors with long spacer arms, for example, competed well with natural GM1os.129
 | ||
| Fig. 10 Octavalent GM1os inhibitor of CTB5-subunit.127 | ||
Fan and coworkers explored a series of multivalent ligands based on different glycoside moieties with linkers of various lengths for CT inhibition. In the first approach, a semi-rigid pentacyclen core was used to bind 5 copies of m-nitrophenyl-α-D-galactoside (MNPG) via a semiflexible linker. The MNPG unit represents the active finger which can be bound to the CT receptor. The monovalent MNPG finger showed 100-fold greater inhibition in comparison to the weaker binding monovalent galactose finger. In terms of multivalency, the pentavalent MNPG ligand exhibited a 260-fold better affinity enhancement (IC50 = 0.9 μM) in comparison to the monovalent MNPG finger.130 The second approach involved the design of branched multivalent ligands with 5-fold symmetry. The branched bivalent galactose ligands were attached to the pentacyclen core via flexible arms which led to branched decavalent conjugates. A million-fold improvement in inhibition power (IC50 = 0.04 μM) was observed with the best branched decavalent ligand with a repeating unit of the linker n = 4 in comparison to the monovalent galactose. The compound with a lower number of linker units showed less inhibition affinity. DLS studies proved that the bivalent ligand can complex the CT in a 1:2 manner. The results of Fan and coworkers showed that the rational design of these multivalent ligand and their linker size is very important for CT inhibition131 (Fig. 11).
 | ||
| Fig. 11 Multivalent (single and branched) galactose binders for CT.130,131 | ||
Richards and coworkers examined several glycopolymers based on a poly(pentafluorophenyl methacrylate) precursor with varying saccharide density, linker length, and chain length due to a 1,3 dipolar cycloaddition. Compounds with a longer linker length showed higher CT inhibition. Polymers with the highest and lowest sugar density (100% and 10%) showed the highest activity, which demonstrates the complexity of the glycoconjugate design for CT inhibition.132
4.2. Escherichia coli (E. coli) inhibition
E. coli is a Gram-negative bacterium that easily adheres to the target cells in the urinary tract through a mannose-binding lectin FimH. The FimH lectin is located at the tip of type 1 fimbriae, also called pili, which are the most abundant structures on the surface of Gram-negative bacteria. The uropathogenic E. coli strains can bind to the carbohydrate receptors in the urinary tract through FimH, colonize, and then cause bladder infections. This bacterial adhesion can be prevented by competitive mannosidic inhibitors. Effective inhibitions have been shown by compounds with terminal α-D-mannosyl residues.133–135Lindhorst and coworkers developed various saccharide modified nanodiamond (ND) conjugates to detect and remove E. coli bacteria from aqueous solutions. The surface of the thermally annealed ND was functionalized via a Diels–Alder cycloaddition of the π-bonds on the diamond surface (dienophil) and an in situ generated o-quinodimethane (diene). The covalently arylated ND can easily be sulfonated and the sulfonic acid groups can be subsequently partially reduced to target thiol groups for more flexible functionalization of the ND.136 These modified NDs were used as platforms and were conjugated via a “thiol–ene reaction” with different monosaccharides.
These NDs are also applied to inhibit biofilm formation on biotic surfaces or medical devices due to E. coli attachment. These biofilms result in an increasing source of infections in clinical settings. Barras and coworkers developed ND that were modified with mannose moieties via an azide–alkyne Huisgen cycloaddition. These multivalent glycol–nanodiamonds can inhibit the E. coli type 1 fimbriae-mediated adhesion to eukaryotic cells (T24 bladder cells) and reduce the E. coli biofilm formation.137
A second carbon-based platform inhibiting pathogens with a lower valency is provided by multivalent fullerenes.138 Vincent and co-workers developed “fullerene sugar balls” with twelve mannose moieties per fullerene. Different sugar units were grafted on the fullerene core via a copper-catalyzed 1,3-dipolar cycloaddition to achieve a C60 core which was completely surrounded by sugar residues.12 Durka and coworkers used this method to couple twelve mannose moieties with different linker lengths to the fullerene core to inhibit the FimH of E. coli. The affinity of the mannoside fullerenes to the FimH was determined by ITC and SPR. Those experiments showed that the increased distance between the active mannose moieties and the C60 core can considerably improve the binding affinity to FimH. However, the nanomolar affinity values were measured for both methods. The mannoside fullerene had a benzylic aromatic ring at the anomeric position of the sugar as well as an amide group in the linker structure, which seems to be beneficial for lectin binding139 and can accommodate up to seven FimH molecules.140
A third class of glycoconjugates which can inhibit E. coli bacteria are multivalent sugar functionalized pillar[5]arenes. Nierengarten and coworkers developed a pillar[5]arene glycocluster with ten peripheral acetylated mannosyl groups via a 1,3 dipolar cycloaddition. The final product, which is a promising compound for FimH inhibition, was achieved by deacetylation. The inhibition capacity was determined in a hemagglutination inhibition assay. The assay determines the concentration of the glycoconjugate, which is necessary to prevent adhesion of uropathogenic E. coli with guinea pig's red blood cells. The inhibition titre was in a low μM range (5.91 μM).141
Huang and coworkers used a similar approach and “clicked” galactose groups (hydrophilic compartment) and alkyl chains (hydrophobic compartment) via an azide–alkyne Huisgen cycloaddition to a pillar[5]arene core. Self-assembled nanotubes were formed in water due to van der Waals interactions of the hydrophobic and hydrophilic parts of the glycoconjugate and intermolecular hydrogen bonds after one week. TEM and SEM measurements as well as fluorescence microscopy confirmed the structure. Furthermore, TEM and fluorescence microscope measurements exhibited a high agglutination rate of the multivalent nanotubes with E. coli, which confirms the high affinity of these glycoconjugates to the pathogenic bacteria.142 The length of the self-assemblies here also played a key role in the formation of bacterial clusters and was a crucial factor in controlling agglutination. Because the self-assembled nanotubes were several micrometers long, they could interconnect more bacteria, which resulted in an enhanced agglutination ability.
Other scaffolds of multivalent glycoconjugates which can inhibit E. coli were also established. Gouin and co-workers designed heptavalent glycoconjugates where n-heptyl α-D-mannoside was covalently linked to β-cyclodextrin core via a “click” procedure. 2 μg of the multivalent FimH antagonist instilled in the mouse bladder significantly reduced the urinary tract infection.143 Furthermore, recent advances in functional supramolecular/polymeric structures like bigger nanogels, graphene sheets, micrometer sized fibres, etc. could be promising scaffolds for effective inhibition of large bacteria particles.144
4.3. Inhibition of the lipopolysaccharide synthesis
Lipopolysaccharides (LPS) are thermostable compounds that protect and stabilize the bacterial membrane can be divided into three parts. The core region of LPS consists of oligosaccharides with a 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) and two L-glycero-α-D-manno-heptose (heptose) units in the inner core and hexoses in the outer core.145E. coli with only a few heptose units are significantly more permeable to certain hydrophobic antibiotics.146 The inhibition of the heptose biosynthetic pathway is just one approach which can be used to circumvent the conventional methods of targeting the bacterial receptors.147Vincent and coworkers developed some multivalent glycosylated fullerenes that can inhibit the LPS heptosyltransferase WaaC. This glycotransferase catalyzes the incorporation of the first L-heptose into LPS. A series of fullerene hexa-adducts with 12 copies of peripheral sugar units displaying the mannopyranose core structure of bacterial L,D-heptosides was synthesized via a copper-catalyzed 1,3 dipolar cycloaddition (Fig. 12). The inhibition of WaaC was determined by an assay which measures the IC50 values of ADP-2-heptose. The multivalent fullerene glycoconjugate of 1, 2, and 3 showed an inhibition potential in a low micromolar range (IC50 = 11; 47 and 6.7 μM). The monomeric glycosides, on the other hand, displayed IC50 values above 400 μM. Another effect of the “fullerene sugar balls” is that the inhibition data evaluated on a “per-sugar” basis showed an increase of the multivalent single glycosides in comparison to the monovalent ones.148
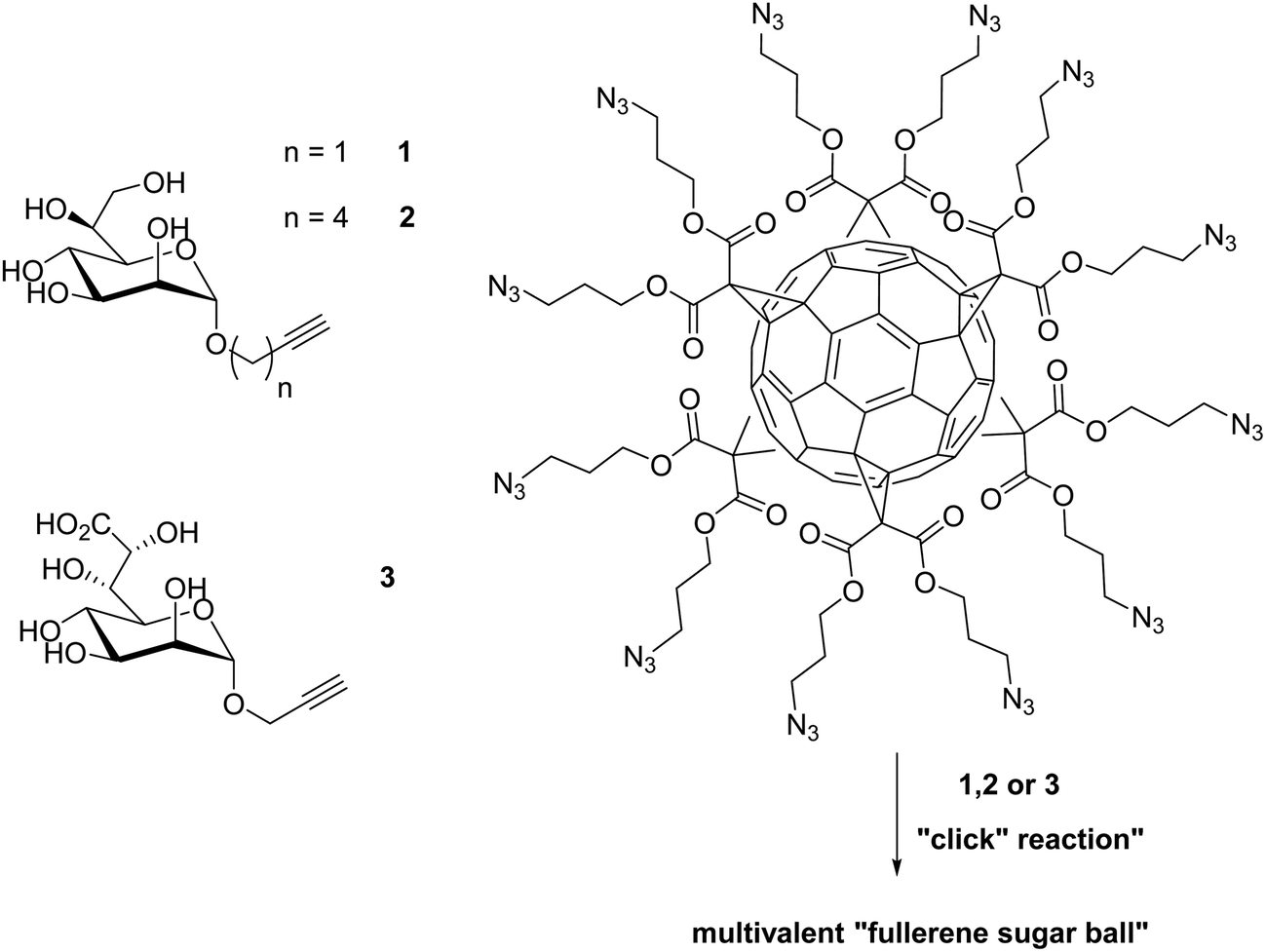 | ||
| Fig. 12 Preparation of three multivalent glycocluster via copper-catalyzed 1,3 dipolar cycloaddition with a series of fullerene hexa-adducts to inhibit the LPS heptosyl-transferase WaaC.148 | ||
5. Anti-inflammatory drugs
The recruitment of circulating leukocytes is crucial at the sites of inflammatory diseases including hypersensitivity reaction as well as autoimmune diseases. The leukocyte recruitment from the bloodstream occurs through a multistep process in which the leukocytes captured on the endothelium, roll along, and interact with the endothelium in postcapilary venules.1 The transmigration of the leukocytes through the vessel wall and further migration into the extracellular tissue takes place subsequently. Leukocyte migration is initiated by interactions between E-, L-, and P-selectins (cell adhesion molecules) and their associated carbohydrate ligands. L-selectin is expressed by the most leukocytes. E-selectin and P-selectin are expressed by inflammed endothelia cells. However, P-selectin is also expressed by activated platelets. One approach for an anti-inflammatory therapy is to target the leukocyte trafficking. Therefore multivalent glycoconjugate systems have been developed that mimic the naturally occurring SLexa ligands and inhibit the selectin-moieties.149–151Patton and coworkers developed a series of multivalent polylysine conjugates to inhibit the interactions of E-selectin. The polylysine was conjugated with tetrasaccharide Lewis X and showed a highly active E-selectin inhibition and reduced the neutrophil rolling on activated endothelia cells with IC50 values in the nanomolar range. Furthermore, the increased size of the multivalent molecule as well as the ligand loading (35% and 50%) at the surface yielded the best inhibition data and a 700-fold increase in inhibition compared to the monovalent compound.152 Haag and coworkers synthesized several multivalent glyco-conjugates to inhibit L- and P-selectin binding.153 These functionalized galactose moieties were then coupled to a dentritic polyglycerol (dPG) scaffold via a 1,3 dipolar cycloaddition. The inhibition rate of the multivalent compound was compared to a tetravalent one based on a pentaerythritol core. The multivalent glycoconjugates showed a significant increase in the inhibition of L- and P-selectin binding compared to the tetravalent ones. The multivalent compound with the sulfated sugar moieties exhibited the highest inhibition rate.153 These studies were expanded and other carbohydrates were coupled to the PE and dPG cores. The multivalent dPG-conjugates with sulfated sugar-moieties showed the best inhibition results with IC50 values in the high picomolar to the low nanomolar range and confirmed the previous results.154 Other scaffolds of multivalent glycoconjugates which can inhibit L-selectin binding were also established. A series of multivalent polyacrylamide-based conjugates155 as well as some neoglycopolymers156 confirms the multivalent effect. Furthermore, Haag and coworkers demonstrated that single dendritic polyglycerol sulfates (lacking the sugar unit) are also excellent candidates to inhibit L-, P-selectin binding and damp inflammation in vivo.151
6. Conclusion
In this article we have reviewed the current applications of multivalent glycoconjugates for a diverse set of diseases and discussed the challenges involved and endeavors made to address them. The development of multivalent glycoconjugate based vaccines has been fruitful, and a number of carbohydrate-based vaccines for various bacterial infections are already commercially available or under clinical trial. Vaccines against Shigella dysenteriae, Streptococcus pneumoniae, Neisseria meningitides, and Haemophilus influenza are commercial examples. However, significant issues remain to be addressed. Development of vaccines against parasitic infections is still a big challenge due to the lack of understanding of complex immune evasion mechanisms used by different parasites. The key difficulty in developing HIV vaccines is mimicking the complex, dense cluster of glycans that are capable of eliciting neutralizing antibodies, specifically against HIV. On the other hand, researchers have been successful in identifying tumor associated carbohydrate antigens (TACAs) and then using them in multivalent forms, along with immunogenic carrier proteins, like keyhole limpet haemocyanin (KLH) to elicit strong T-cell dependent antibodies. Several synthetic antigen candidates have reached clinical trials, although more precise criteria for their efficacy and clinical impact remain to be defined. Besides using carbohydrates as vaccines, several multivalent glycoconjugates with controlled topology and valency have been explored to better understand multivalency, and their potential as anti-infectious agents for several viral and bacterial infections has been demonstrated in multiple examples. However, very few approaches consider biocompatible scaffolds in the appropriate dimension of the biological pathogen for the most effective shielding. Developing compatible polyvalent glycoconjugates with strong binding and infection inhibition efficiencies to reach the market as drugs still remains a challenging goal.Abbreviation
| ADP-2-heptose | Adenosine diphosphate-2-heptose |
| Alum | Potassium aluminium sulfate |
| AuNPs | Gold nanoparticles |
| BSA | Bovine serum albumin |
| CRD | Carbohydrate recognition domain |
| CD4+ | Cluster of differentiation 4+ |
| CT | Cholera toxin |
| DLS | Dynamic light scattering |
| DC-SIGN | Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin |
| dPG | Dendritic polyglycerol |
| E. coli | Escherichia coli |
| E-, L-, and P-selectins | Endothelial-, leukocytes-, and platelets-selectins |
| FimH | Type 1 fimbriae |
| GBP | Glycan-binding protein |
| α-GalNAc | α-N-Acetyl-D-galactosamine |
| GM1 | Monosialotetrahexosyl ganglioside |
| GPI | Glycosylphosphatidylinositol |
| Globo H | Globohexaosylceramide |
| HAs | Hemagglutinin |
| HIV | Human immune deficiency virus type |
| ITC | Isothermal titration calorimetry |
| ICAM 3 | Intercellular adhesion molecule 3 |
| IC50 | Half maximal inhibitory concentration |
| KLH | Keyhole limpet haemocyanin |
| Kdo | 3-Deoxy-D-manno-oct-2-ulosonic acid |
| LPG | Lipophosphoglycan |
| Man4 | Tetramannoside |
| manno-GNPs | Mannoside gold nanoparticles |
| MNPG | m-Nitrophenyl-α-D-galactoside |
| NA | Neuraminidase |
| ND | Nanodiamond |
| nPG | Polyglycerol nanogels |
| OMPC | Outer membrane protein complex |
| PAMAM | Poly(amidoamine) |
| PSA | Polysialic acid |
| Pam3Cys | Tripalmitoyl-S-glyceryl-cysteinylserine |
| pNAS | Poly[N-(acryoyloxysuccinimide)] |
| PGN-ZA | Poly-L-glutamine zanamivir |
| QS21 | Quillaja saponaria 21 |
| S. pneumonia | Streptococcus pneumoniae |
| S. dysenteriae | Shigella dysenteriae |
| sTn | Sialyl 2-6-α-N-acetylgalactosamine |
| SA | Sialic acid |
| SIV | Simian immunodeficiency viruses |
| Stx | Shigatoxin |
| SEM | Scanning electron microscope |
| SLex | Sialyl Lewis x |
| SPR | Surface plasmon resonance |
| Tn | 2-6-α-N-Acetylgalactosaminyl |
| TF | Thomsen–Friedenreich |
| T-lymphocytes | Thymus lymphocyte |
| TEM | Transmission electron microscopy |
| TACAs | Tumor associated carbohydrate antigens |
| VSG | Variant surface glycoprotein |
Acknowledgements
We thank Dr Pamela Winchester for careful proofreading.References
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., 1998, 110, 2908–2953 ( Angew. Chem., Int. Ed. , 1998 , 37 , 2754–2794 ) CrossRef.
- T. Horlacher and P. H. Seeberger, Chem. Soc. Rev., 2008, 37, 1414–1422 RSC.
- J. C. Paulson, O. Blixt and B. E. Collins, Nat. Chem. Biol., 2006, 2, 238–248 CrossRef CAS PubMed.
- B. G. Davis, Chem. Rev., 2002, 102, 579–602 CrossRef CAS PubMed.
- R. J. Payne and C. H. Wong, Chem. Commun., 2010, 46, 21–43 RSC.
- P. M. Rendle, A. Seger, J. Rodrigues, N. J. Oldham, R. R. Bott, J. B. Jones, M. M. Cowan and B. G. Davis, J. Am. Chem. Soc., 2004, 126, 4750–4751 CrossRef CAS PubMed.
- I. Otsuka, B. Blanchard, R. Borsali, A. Imberty and T. Kakuchi, ChemBioChem, 2010, 11, 2399–2408 CrossRef CAS PubMed.
- D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13, 1845–1852 CrossRef CAS PubMed.
- J. Rieger, F. Stoffelbach, D. Cui, A. Imberty, E. Lameignere, J. L. Putaux, R. Jérôme, C. Jérôme and R. Auzély-Velty, Biomacromolecules, 2007, 8, 2717–2725 CrossRef CAS PubMed.
- S. Cecioni, V. Oerthel, J. Iehl, M. Holler, D. Goyard, J.-P. Praly, A. Imberty, J.-F. Nierengarten and S. Vidal, Chem.–Eur. J., 2011, 17, 3252–3261 CrossRef CAS PubMed.
- J.-F. Nierengarten, J. Iehl, V. Oerthel, M. Holler, B. M. Illescas, A. Munoz, N. Martin, J. Rojo, M. Sanchez-Navarro, S. Cecioni, S. Vidal, K. Buffet, M. Durka and S. P. Vincent, Chem. Commun., 2010, 46, 3860–3862 RSC.
- S. Cecioni, R. Lalor, B. Blanchard, J.-P. Praly, A. Imberty, S. E. Matthews and S. Vidal, Chem.–Eur. J., 2009, 15, 13232–13240 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Chem. Rev., 2010, 110, 4949–4977 CrossRef CAS PubMed.
- S. Andre, C. Grandjean, F. M. Gautier, S. Bernardi, F. Sansone, H. J. Gabius and R. Ungaro, Chem. Commun., 2011, 47, 6126–6128 RSC.
- S. Andre, R. J. Pieters, I. Vrasidas, H. Kaltner, I. Kuwabara, F. T. Liu, R. M. Liskamp and H. J. Gabius, ChemBioChem, 2001, 2, 822–830 CrossRef CAS.
- C. Heidecke and T. K. Lindhorst, Chem.–Eur. J., 2007, 13, 9056–9067 CrossRef CAS PubMed.
- M. A. Mintzer, E. L. Dane, G. A. O'Toole and M. W. Grinstaff, Mol. Pharm., 2012, 9, 342–354 CrossRef CAS PubMed.
- M. Touaibia and R. Roy, Mini-Rev. Med. Chem., 2007, 7, 1270–1283 CrossRef CAS.
- W. B. Turnbull and J. F. Stoddart, J. Biotechnol., 2002, 90, 231–255 CAS.
- M. Marradi, M. Martin-Lomas and S. Penades, Adv. Carbohydr. Chem. Biochem., 2010, 64, 211–290 CrossRef CAS.
- X. Wang, E. Matei, L. Deng, O. Ramstrom, A. M. Gronenborn and M. Yan, Chem. Commun., 2011, 47, 8620–8622 RSC.
- G. D. Armstrong, E. Fodor and R. Vanmaele, J. Infect. Dis., 1991, 164, 1160–1167 CrossRef CAS PubMed.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS PubMed.
- A. Imberty, Y. M. Chabre and R. Roy, Chem.–Eur. J., 2008, 14, 7490–7499 CrossRef CAS PubMed.
- C. Jones, An. Acad. Bras. Cienc., 2005, 77, 293–324 CAS.
- A. W. Zuercher, M. P. Horn, J. U. Que, A. Ruedeberg, M. H. Schoeni, U. B. Schaad, P. Marcus and A. B. Lang, FEMS Immunol. Med. Microbiol., 2006, 47, 302–308 CrossRef CAS PubMed.
- J. J. Mond, A. Lees and C. M. Snapper, Annu. Rev. Immunol., 1995, 13, 655–692 CrossRef CAS PubMed.
- C. M. Snapper and J. J. Mond, J. Immunol., 1996, 157, 2229–2233 CAS.
- O. T. Avery and W. F. Goebel, J. Exp. Med., 1931, 54, 437–447 CrossRef CAS.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discov., 2010, 9, 308–324 CrossRef CAS PubMed.
- T. Buskas, Y. Li and G. J. Boons, Chem.–Eur. J., 2004, 10, 3517–3524 CrossRef CAS PubMed.
- J. Ni, H. Song, Y. Wang, N. M. Stamatos and L.-X. Wang, Bioconjugate Chem., 2006, 17, 493–500 CrossRef CAS PubMed.
- N. Petrovsky and J. C. Aguilar, Immunol. Cell Biol., 2004, 82, 488–496 CrossRef CAS PubMed.
- P. Marrack, A. S. McKee and M. W. Munks, Nature review, 2009, 9, 287–293 CAS.
- Merck & Co., Inc., Pneumovax 23 (pneumococcal vaccine polyvalent). Merck website, 1986, http://www.merck.com/product/usa/pi_circulars/p/pneumovax_23/pneumovax_pi.pdf.
- J. B. Robins, R. Austrian, C. J. Lee, S. C. Rastogi, G. Schiffman, J. Henrichsen, P. H. Mäkelä, C. V. Broome, R. R. Facklam and R. H. Tiesjema, et al. , J. Infect. Dis., 1983, 148, 1136–1159 CrossRef PubMed.
- V. Pozsgay, C. Chu, L. Pannell, J. Wolfe, J. B. Robbins and R. Schneerson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5194–5197 CrossRef CAS.
- V. Pozsgay, J. Kubler-Kielb, R. Schneerson and J. B. Robbins, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14478–14482 CrossRef CAS PubMed.
- A. D. Lopez and C. C. Murray, Nat. Med., 1998, 4, 1241–1243 CrossRef CAS PubMed.
- V. L. Price and M. P. Kieny, Curr. Drug Targets Infect. Disord., 2004, 1, 315–324 CrossRef.
- A. K. Nyame, Z. S. Kawar and R. D. Cummings, Arch. Biochem. Biophys., 2004, 426, 182–200 CrossRef CAS PubMed.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
- A. Guha-Niyogi, D. R. Sullivan and S. J. Turco, Glycobiology, 2001, 11, 45R–59R CrossRef CAS PubMed.
- M. A. J. Ferguson, J. Cell Sci., 1999, 112, 2799–2809 CAS.
- D. B. Werz and P. H. Seeberger, Chem.–Eur. J., 2005, 11, 3194–3206 CrossRef CAS PubMed.
- L. Schofield, M. C. Hewitt, K. Evans, A. Siomos and P. H. Seeberger, Nature, 2002, 418, 785–789 CrossRef CAS PubMed.
- F. Kamena, M. Tamborrini, X. Liu, Y.-U. Kwon, F. Thompson, G. Pluschke and P. H. Seeberger, Nat. Chem. Biol., 2008, 4, 238–240 CrossRef CAS PubMed.
- S. J. Turco and A. Descoteaux, Annu. Rev. Microbiol., 1992, 46, 65–94 CrossRef CAS PubMed.
- S. F. Moody, E. Handman, M. J. McConville and A. Bacic, J. Biol. Chem., 1993, 268, 18457–18466 CAS.
- A. Descoteaux and S. J. Turco, Biochim. Biophys. Acta, 1999, 1455, 341–352 CrossRef CAS.
- M. C. Hewitt and P. H. Seeberger, J. Org. Chem., 2001, 66, 4233–4243 CrossRef CAS PubMed.
- J. R. Mascola, G. Stiegler, T. C. VanCott, H. Katinger, C. B. Carpenter, C. E. Hanson, H. Beary, D. Hayes, S. S. Frankel, D. L. Birx and M. G. Lewis, Nat. Med., 2000, 6, 207–210 CrossRef CAS PubMed.
- D. A. Calarese, H.-K. Lee, C.-Y. Huang, M. D. Best, R. D. Astronomo, R. L. Stanfield, H. Katinger, D. R. Burton, C.-H. Wong and I. A. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13372–13377 CrossRef CAS PubMed.
- C. Scanlan, D. Calarese, H.-K. Lee, O. Blixt, C.-H. Wong, I. Wilson, D. Burton, R. Dwek and P. Rudd, Adv. Exp. Med. Biol., 2005, 564, 7–8 CrossRef.
- J. M. Binley, T. Wrin, B. Korber, M. B. Zwick, M. Wang, C. Chappey, G. Stiegler, R. Kunert, S. Zolla-Pazner, H. Katinger, C. J. Petropoulos and D. R. Burton, J. Virol., 2004, 78, 13232–13252 CrossRef CAS PubMed.
- R. Pejchal, K. J. Doores, L. M. Walker, R. Khayat, P. S. Huang, S. K. Wang, R. L. Stanfield, J. P. Julien, A. Ramos and M. Crispin, et al. , Science, 2011, 334, 1097–1103 CrossRef CAS PubMed.
- C. N. Scanlan, J. Offer, N. Zitzmann and R. A. Dwek, Nature, 2007, 446, 1038–1045 CrossRef CAS PubMed.
- D. M. Ratner and P. H. Seeberger, Curr. Pharm. Des., 2007, 13, 173–183 CrossRef CAS.
- A. Kabanova, R. Adamo, D. Proietti, F. Berti, M. Tontini, R. Rappuoli and P. Costantino, Glycoconj. J., 2010, 27, 501–513 CrossRef CAS PubMed.
- J. Wang, H. Li, G. Zou and L. X. Wang, Org. Biomol. Chem., 2007, 5, 1529–1540 CAS.
- S.-K. Wang, P.-H. Liang, R. D. Astronomo, T.-L. Hsu, S.-L. Hsieh, D. R. Burton and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3690–3695 CrossRef CAS PubMed.
- R. D. Astronomo, E. Kaltgrad, A. K. Udit, S. Wang, J. Doores, C. Huang, R. Pantophlet, J. C. Paulson, M. G. Finn and D. R. Burton, Chem. Biol., 2011, 17, 357–370 CrossRef PubMed.
- R. D. Astronomo, H.-K. Lee, C. N. Scanlan, R. Pantophlet, C.-Y. Huang, I. A. Wilson, O. Blixt, R. A. Dwek, C.-H. Wong and D. R. Burton, J. Virol., 2008, 82, 6359–6368 CrossRef CAS PubMed.
- J. G. Joyce, I. J. Krauss, H. C. Song, D. W. Opalka, K. M. Grimm, D. D. Nahas, M. T. Esser, R. Hrin, M. Feng, V. Y. Dudkin, M. Chastain, J. W. Shiver and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15684–15689 CrossRef CAS PubMed.
- I. J. Krauss, J. G. Joyce, A. C. Finnefrock, H. C. Song, V. Y. Dudkin, X. Geng, J. D. Warren, M. Chastain, J. W. Shiver and S. J. Danishefsky, J. Am. Chem. Soc., 2007, 129, 11042–11044 CrossRef CAS PubMed.
- R. J. Luallen, J. Lin, H. Fu, K. K. Cai, C. Agrawal, I. Mboudjeka, F.-H. Lee, D. Montefiori, D. F. Smith, R. W. Doms and Y. Geng, J. Virol., 2008, 82, 6447–6457 CrossRef CAS PubMed.
- R. D. Astronomo, E. Kaltgrad, A. K. Udit, S.-K. Wang, K. J. Doores, C.-Y. Huang, R. Pantophlet, J. C. Paulson, C.-H. Wong, M. G. Finn and D. R. Burton, Chem. Biol., 2010, 17, 357–370 CrossRef CAS PubMed.
- E. Meezan, H. C. Wu, P. H. Black and P. W. Robbins, Biochemistry, 1969, 8, 2518–2524 CrossRef CAS.
- J. W. Dennis, S. Laferte, C. Waghorne, M. L. Breitman and R. S. Kerbel, Science, 1987, 236, 582–585 CAS.
- Y. J. Kim and A. Varki, Glycoconj. J., 1997, 14, 569–576 CrossRef CAS.
- H. J. Gabius, Acta Histochem. Suppl., 1988, 36, 209–216 CAS.
- S. Sell, Hum. Pathol., 1990, 21, 1003–1019 CrossRef CAS.
- J. Taylor-Papadimitriou and A. A. Epenetos, Trends Biotechnol., 1994, 12, 227–233 CrossRef CAS.
- F. Helling, S. Zhang, A. Shang, S. Adluri, M. Calves, R. Koganty, B. M. Longenecker, T. Yao, H. F. Oettgen and P. Livingston, Cancer Res., 1995, 55, 2783–2788 CAS.
- F. Helling, A. Shang, M. Calves and P. O. Livingston, Cancer Res., 1994, 54, 197–203 CAS.
- G. Ritter, E. Boosfeld, R. Adluri, M. Calves, H. F. Oettgen, L. J. Old and P. Livingston, Int. J. Cancer, 1991, 48, 379–385 CrossRef CAS.
- G. Ritter, E. Boosfeld, M. J. Calves, H. F. Oettgen, L. J. Old and P. O. Livingston, Immunobiology, 1990, 182, 32–43 CrossRef CAS.
- L. M. Krug, G. Ragupathi, K. K. Ng, C. Hood, H. J. Jennings, Z. Guo, M. G. Kris, V. Miller, B. Pizzo, L. Tyson, V. Baez and P. O. Livingston, Clin. Cancer Res., 2004, 10, 916–923 CrossRef CAS.
- C. Musselli, P. O. Livingston and G. Ragupathi, J. Cancer Res. Clin. Oncol., 2001, 127(suppl. 2), R20–R26 CrossRef CAS.
- S. Ingale, M. A. Wolfert, J. Gaekwad, T. Buskas and G. J. Boons, Nat. Chem. Biol., 2007, 3, 663–667 CrossRef CAS PubMed.
- Y. Pan, P. Chefalo, N. Nagy, C. Harding and Z. Guo, J. Med. Chem., 2005, 48, 875–883 CrossRef CAS PubMed.
- P. Chefalo, Y. Pan, N. Nagy, Z. Guo and C. V. Harding, Biochemistry, 2006, 45, 3733–3739 CrossRef CAS PubMed.
- Q. Wang, J. Zhang and Z. Guo, Bioorg. Med. Chem., 2007, 15, 7561–7567 CrossRef CAS PubMed.
- G. Ragupathi, S. Cappello, S. S. Yi, D. Canter, M. Spassova, W. G. Bornmann, S. J. Danishefsky and P. O. Livingston, Vaccine, 2002, 20, 1030–1038 CrossRef CAS.
- G. Ragupathi, F. Koide, N. Sathyan, E. Kagan, M. Spassova, W. Bornmann, P. Gregor, C. A. Reis, H. Clausen, S. J. Danishefsky and P. O. Livingston, Cancer Immunol. Immunother., 2003, 52, 608–616 CrossRef CAS PubMed.
- G. Ragupathi, F. Koide, P. O. Livingston, Y. S. Cho, A. Endo, Q. Wan, M. K. Spassova, S. J. Keding, J. Allen, O. Ouerfelli, R. M. Wilson and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 2715–2725 CrossRef CAS PubMed.
- S. F. Slovin, G. Ragupathi, C. Fernandez, M. Diani, M. P. Jefferson, A. Wilton, W. K. Kelly, M. Morris, D. Solit, H. Clausen, P. Livingston and H. I. Scher, Cancer Immunol. Immunother., 2007, 56, 1921–1930 CrossRef CAS PubMed.
- A. L. Parry, N. Clemson, J. Ellis, S. S. R. Bernhard, B. G. Davis and N. R. Cameron, J. Am. Chem. Soc., 2013, 135, 9362–9365 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed , and references therein.
- A. Berzi, J. J. Reina, R. Ottria, I. Sutkeviciute, P. Antonazzo, M. Sanchez-Navarro, E. Chabrol, M. Biasin, D. Trabattoni, I. Cetin, J. Rojo, F. Fieschi, A. Bernardi and M. Clerici, AIDS, 2012, 26, 127–137 CrossRef CAS PubMed.
- S. Sattin, A. Daghetti, M. Thépaut, A. Berzi, M. Sánchez-Navarro, G. Tabarani, J. Rojo, F. Fieschi, M. Clerici and A. Bernardi, ACS Chem. Biol., 2010, 5, 301–312 CrossRef CAS PubMed.
- J. Luczkowiak, S. Sattin, I. Sutkevičiūtė, J. J. Reina, M. Sánchez-Navarro, M. Thépaut, L. Martínez-Prats, A. Daghetti, F. Fieschi, R. Delgado, A. Bernardi and J. Rojo, Bioconjug. Chem., 2011, 22, 1354–1365 CrossRef CAS PubMed.
- O. Martínez-Avila, K. Hijazi, M. Marradi, C. Clavel, C. Campion, C. Kelly and S. Penadés, Chem.–Eur. J., 2009, 15, 9874–9888 CrossRef PubMed.
- O. Martínez-Avila, L. M. Bedoya, M. Marradi, C. Clavel, J. Alcamí and S. Penadés, ChemBioChem, 2009, 10, 1806–1809 CrossRef PubMed.
- W. Weis, J. H. Brown, S. Cusack, J. C. Paulson, J. J. Skehel and D. C. Wiley, Nature, 1988, 333, 426–431 CrossRef CAS PubMed.
- T. J. Pritchett and J. C. Paulson, J. Biol. Chem., 1989, 264, 9850–9858 CAS.
- E. G. Weinhold and J. R. Knowles, J. Am. Chem. Soc., 1992, 114, 9270–9275 CrossRef CAS.
- (a) G. D. Glick and J. R. Knowles, J. Am. Chem. Soc., 1991, 113, 4701–4703 CrossRef CAS; (b) G. D. Glick, P. L. Toogood, D. C. Wiley, J. J. Skehel and J. R. Knowles, J. Biol. Chem., 1991, 266, 23660–23669 CAS; (c) S. Sabesan, J. O. Duus, S. Neira, P. Domaille, S. Kelm, J. C. Paulson and K. Bock, J. Am. Chem. Soc., 1992, 114, 8363–8375 CrossRef CAS; (d) T. Ohta, N. Miura, N. Fujitani, F. Nakajima, K. Niikura, R. Sadamoto, C.-T. Guo, T. Suzuki, Y. Suzuki, K. Monde and S.-I. Nishimura, Angew. Chem., 2003, 115, 5344–5347 ( Angew. Chem., Int. Ed. , 2003 , 42 , 5186–5189 ) CrossRef.
- (a) S. J. Meunier and R. Roy, Tetrahedron Lett., 1996, 37, 5469–5472 CrossRef CAS; (b) A. Marra, M.-C. Scherrmann, A. Dondoni, A. Casnati, P. Minari and R. Ungaro, Angew. Chem., 1994, 106, 2533–2535 ( Angew. Chem., Int. Ed. Engl. , 1994 , 33 , 2479–2481 ) CrossRef CAS; (c) Z. Gan and R. Roy, Can. J. Chem., 2002, 80, 908–916 CrossRef CAS; (d) S. A. Kalovidouris, O. Blixt, A. Nelson, S. Vidal, W. B. Turnbull, J. C. Paulson and J. F. Stoddart, J. Org. Chem., 2003, 68, 8485–8493 CrossRef CAS PubMed.
- A. Marra, L. Moni, D. Pazzi, A. Corallini, D. Bridi and A. Dondoni, Org. Biomol. Chem., 2008, 6, 1396–1409 CAS.
- A. B. Tuzikov, A. A. Chinarev, A. S. Gambaryan, V. A. Oleinikov, D. V. Klinov, N. B. Matsko, V. A. Kadykov, M. A. Ermishov, I. V. Demin, V. V. Demin, P. D. Rye and N. V. Bovin, ChemBioChem, 2003, 4, 147–154 CrossRef CAS PubMed.
- Y. Makimura, S. Watanabe, T. Suzuki, Y. Suzuki, H. Ishida, M. Kiso, T. Katayama, H. Kumagai and K. Yamamoto, Carbohydr. Res., 2006, 341, 1803–1808 CrossRef CAS PubMed.
- (a) M. Llinares and R. Roy, Chem. Commun., 1997, 2119–2120 RSC; (b) K. Matsuoka, H. Oka, T. Koyama, Y. Esumi and D. Terunuma, Tetrahedron Lett., 2001, 42, 3327–3330 CrossRef CAS; (c) S. J. Meunier, Q. Wu, S.-N. Wang and R. Roy, Can. J. Chem., 1997, 75, 1472–1482 CrossRef; (d) R. Roy, D. Zanini, S. J. Meunier and A. Romanowska, J. Chem. Soc., Chem. Commun., 1993, 1869–1872 RSC; (e) D. Zanini and R. Roy, J. Org. Chem., 1996, 61, 7348–7354 CrossRef CAS PubMed; (f) J. J. Landers, Z. Cao, I. Lee, L. T. Piehler, P. P. Myc, A. Myc, T. Hamouda, A. T. Galecki and J. R. Baker Jr, J. Infect. Dis., 2002, 186, 1222–1230 CrossRef CAS PubMed; (g) J. D. Reuter, A. Myc, M. M. Hayes, Z. Gan, R. Roy, D. Qin, R. Yin, L. T. Piehler, R. Esfand, D. A. Tomalia and J. R. Baker Jr, Bioconjugate Chem., 1999, 10, 271–278 CrossRef CAS PubMed; (h) D. Zanini and R. Roy, J. Am. Chem. Soc., 1997, 119, 2088–2095 CrossRef CAS.
- (a) C.-T. Guo, X.-L. Sun, O. Kanie, K. F. Shortridge, T. Suzuki, D. Miyamoto, K. I.-P. J. Hidari, C.-H. Wong and Y. Suzuki, Glycobiology, 2002, 12, 183–190 CrossRef CAS PubMed; (b) J. E. Kingery-Wood, K. W. Williams, G. B. Sigal and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 7303–7305 CrossRef CAS; (c) A. Reichert, J. O. Nagy, W. Spevak and D. Charych, J. Am. Chem. Soc., 1995, 117, 829–830 CrossRef CAS; (d) W. Spevak, J. O. Nagy, D. H. Charych, M. E. Schaefer, J. H. Gilbert and M. D. Bednarski, J. Am. Soc. Chem., 1993, 115, 1146–1147 CrossRef CAS.
- (a) N. V. Bovin, A. B. Tuzikov, A. A. Chinarev and A. S. Gambaryan, Glycoconj. J., 2004, 21, 471–478 CrossRef CAS PubMed; (b) L. Bondioli, L. Costantino, A. Ballestrazzi, D. Lucchesi, D. Boraschi, F. Pellati, S. Benvenuti, G. Tosi and M. A. Vandelli, Biomaterials, 2010, 31, 3395–3403 CrossRef CAS PubMed; (c) D. Lemoine and V. Préat, J. Controlled Release, 1998, 54, 15–27 CrossRef CAS.
- W. J. Lees, A. Spaltenstein, J. E. Kingery-wood and G. M. Whitesides, J. Med. Chem., 1994, 37, 3419–3433 CrossRef CAS.
- M. Mammen, G. Dahmann and G. M. Whitesides, J. Med. Chem., 1995, 38, 4179–4190 CrossRef CAS.
- L. V. Mochalova, A. B. Tuzikov, V. P. Marinina, A. S. Gambaryan, N. E. Byramova, N. V. Bovin and M. N. Matrosovich, Antiviral Res., 1994, 23, 179–190 CrossRef CAS.
- I. Papp, C. Sieben, K. Ludwig, M. Roskamp, C. Böttcher, S. Schlecht, A. Herrmann and R. Haag, Small, 2010, 6, 2900–2906 CrossRef CAS PubMed.
- J. Vonnemann, C. Sieben, C. Wolff, K. Ludwig, C. Böttcher, A. Herrmann and R. Haag, Nanoscale, 2014, 6, 2353–2360 RSC.
- I. Papp, C. Sieben, A. L. Sisson, J. Kostka, C. Böttcher, K. Ludwig, A. Herrmann and R. Haag, ChemBioChem, 2011, 12, 887–895 CrossRef CAS PubMed.
- C. M. Lee, A. K. Weight, J. Haldar, L. Wang, A. M. Klibanov and J. Chen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20385–20390 CrossRef CAS PubMed.
- A. K. Weight, J. Haldar, L. Álvarez De Cienfuegos, L. V. Gubareva, T. M. Tumpey, J. Chen and A. M. Klibanov, J. Pharm. Sci., 2011, 100, 831–835 CrossRef CAS PubMed.
- G. L. Mulvey, P. Marcato, P. I. Kitov, J. Sadowska, D. R. Bundle and G. D. Armstrong, J. Infect. Dis., 2003, 187, 640–649 CrossRef CAS PubMed.
- A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann and T. K. Lindhorst, et al. , Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- T. Feizi, Ann. N. Y. Acad. Sci., 2013, 1292, 33–44 CrossRef CAS PubMed.
- C.-L. Schengrund, Biochem. Pharmacol., 2003, 65, 699–707 CrossRef CAS.
- S. N. De and D. N. Chatterje, J. Pathol. Bacteriol., 1953, 66, 559–562 CrossRef CAS.
- N. K. Dutta, M. V. Panse and D. R. Kulkarni, J. Bacteriol., 1959, 78, 594–595 CAS.
- (a) D. A. Sack, R. B. Sack, G. B. Nair and A. K. Siddique, Lancet, 2004, 363, 223–233 CrossRef CAS; (b) F. S. Mhalu, P. W. Mmari and J. Ijumba, Lancet, 1979, 363, 345–347 CrossRef; (c) R. I. Glass, I. Huq, A. R. Alim and M. Yunus, J. Infect. Dis., 1980, 142, 939–942 CrossRef CAS PubMed.
- J. Holmgren, Nature, 1981, 292, 413–417 CrossRef CAS.
- E. A. Merritt and W. G. J. Hol, Curr. Opin. Struct. Biol., 1995, 5, 165–171 CrossRef CAS.
- D. Arosio, M. Fontanella, L. Baldini, L. Mauri, A. Bernardi, A. Casnati, F. Sansone and R. Ungaro, J. Am. Chem. Soc., 2005, 127, 3660–3661 CrossRef CAS PubMed.
- J. Thompson and C.-L. Schengrund, Glycoconj. J., 1997, 14, 837–845 CrossRef CAS.
- J. P. Thompson and C.-L. Schengrund, Biochem. Pharmacol., 1998, 56, 591–597 CrossRef CAS.
- A. V. Pukin, H. M. Branderhorst, C. Sisu, C. A. Weijers, M. Gilbert, R. M. Liskamp, G. M. Visser, H. Zuilhof and R. J. Pieters, ChemBioChem, 2007, 8, 1500–1503 CrossRef CAS PubMed.
- A. V. Pukin, C. A. G. M. Weijers, B. van Lagen, R. Wechselberger, B. Sun, M. Gilbert, M.-F. Karwaski, D. E. A. Florack, B. C. Jacobs, A. P. Tio-Gillen, A. van Belkum, H. P. Endtz, G. M. Visser and H. Zuilhof, Carbohydr. Res., 2008, 343, 636–650 CrossRef CAS PubMed.
- H. M. Branderhorst, R. M. Liskamp, G. M. Visser and R. J. Pieters, Chem. Commun., 2007, 5043–5045 RSC.
- E. A. Merritt, Z. Zhang, J. C. Pickens, M. Ahn, W. G. J. Hol and E. Fan, J. Am. Chem. Soc., 2002, 124, 8818–8824 CrossRef CAS PubMed.
- Z. Zhang, E. A. Merritt, M. Ahn, C. Roach, Z. Hou, C. L. M. J. Verlinde, W. G. J. Hol and E. Fan, J. Am. Chem. Soc., 2002, 124, 12991–12998 CrossRef CAS PubMed.
- S.-J. Richards, M. W. Jones, M. Hunaban, D. M. Haddleton and M. I. Gibson, Angew. Chem., 2012, 124, 7932–7936 CrossRef.
- D. I. Kisiela, V. B. Rodriguez, V. Tchesnokova, H. Avagyan, P. Aprikian, Y. Liu, X.-R. Wu, W. E. Thomas and E. V. Sokurenko, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19089–19094 CrossRef CAS PubMed.
- S. Knight and J. Bouckaert, in Glycoscience and Microbial Adhesion, ed. T. K. Lindhorst and S. Oscarson, Springer Berlin Heidelberg, 2009, vol. 288, pp. 67–107 Search PubMed.
- C. Grabosch, M. Hartmann, J. Schmidt-Lassen and T. K. Lindhorst, ChemBioChem, 2011, 12, 1066–1074 CrossRef CAS PubMed.
- G. Jarre, Y. Liang, P. Betz, D. Lang and A. Krueger, Chem. Commun., 2011, 47, 544–546 RSC.
- A. Barras, F. A. Martin, O. Bande, J.-S. Baumann, J.-M. Ghigo, R. Boukherroub, C. Beloin, A. Siriwardena and S. Szunerits, Nanoscale, 2013, 5, 2307–2316 RSC.
- N. C. Reichardt, M. Martin-Lomas and S. Penades, Chem. Soc. Rev., 2013, 42, 4358–4376 RSC.
- M. Touaibia, A. Wellens, T. C. Shiao, Q. Wang, S. Sirois, J. Bouckaert and R. Roy, ChemMedChem, 2007, 2, 1190–1201 CrossRef CAS PubMed.
- M. Durka, K. Buffet, J. Iehl, M. Holler, J.-F. Nierengarten, J. Taganna, J. Bouckaert and S. P. Vincent, Chem. Commun., 2011, 47, 1321–1323 RSC.
- I. Nierengarten, K. Buffet, M. Holler, S. P. Vincent and J.-F. Nierengarten, Tetrahedron Lett., 2013, 54, 2398–2402 CrossRef CAS PubMed.
- G. Yu, Y. Ma, C. Han, Y. Yao, G. Tang, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2013, 135, 10310–10313 CrossRef CAS PubMed.
- J. Bouckaert, Z. Li, C. Xavier, M. Almant, V. Caveliers, T. Lahoutte, S. D. Weeks, J. Kovensky and S. G. Gouin, Chem.–Eur. J., 2013, 19, 7847–7855 CrossRef CAS PubMed.
- (a) A. L. Sisson and R. Haag, Soft Matter, 2010, 6, 4968–4975 RSC; (b) D. Steinhilber, A. L. Sisson, D. Mangoldt, P. Welker, K. Licha and R. Haag, Adv. Funct. Mater., 2010, 20, 4133–4138 CrossRef CAS; (c) A. Takagi, A. Hirose, T. Nishimura, N. Fukumori, A. Ogata, N. Ohashi, S. Kitajima and J. Kanno, J. Toxicol. Sci., 2008, 33, 105–116 CrossRef CAS; (d) T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. P. Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS PubMed.
- C. R. H. Raetz and C. Whitfield, Ann. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS PubMed.
- W. G. Coleman and L. Leive, J. Bacteriol., 1979, 139, 899–910 CAS.
- M. Durka, A. Tikad, R. Périon, M. Bosco, M. Andaloussi, S. Floquet, E. Malacain, F. Moreau, M. Oxoby, V. Gerusz and S. P. Vincent, Chem.–Eur. J., 2011, 17, 11305–11313 CrossRef CAS PubMed.
- M. Durka, K. Buffet, J. Iehl, M. Holler, J.-F. Nierengarten and S. P. Vincent, Chem.–Eur. J., 2012, 18, 641–651 CrossRef CAS PubMed.
- K. Ley, C. Laudanna, M. I. Cybulsky and S. Nourshargh, Nat. Rev. Immunol., 2007, 7, 678–689 CrossRef CAS PubMed.
- H. Ulbrich, E. E. Eriksson and L. Lindbom, Trends Pharmacol. Sci., 2003, 24, 640–647 CrossRef CAS PubMed.
- J. Dernedde, A. Rausch, M. Weinhart, S. Enders, R. Tauber, K. Licha, M. Schirner, U. Zügel, A. von Bonin and R. Haag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19679–19684 CrossRef CAS PubMed.
- G. Thoma, R. O. Duthaler, J. L. Magnani and J. T. Patton, J. Am. Chem. Soc., 2001, 123, 10113–10114 CrossRef CAS.
- I. Papp, J. Dernedde, S. Enders and R. Haag, Chem. Commun., 2008, 5851–5853 RSC.
- J. Dernedde, I. Papp, S. Enders, S. Wedepohl, F. Paulus and R. Haag, J. Carbohydr. Chem., 2011, 30, 347–360 CrossRef CAS.
- S. Enders, G. Bernhard, A. Zakrzewicz and R. Tauber, Biochim. Biophys. Acta, 2007, 1770, 1441–1449 CrossRef CAS PubMed.
- P. Mowery, Z.-Q. Yang, E. J. Gordon, O. Dwir, A. G. Spencer, R. Alon and L. L. Kiessling, Chem. Biol., 2004, 11, 725–732 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |