 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Branched α-D-mannopyranosides: a new class of potent FimH antagonists†
Tihomir
Tomašić
a,
Said
Rabbani
b,
Martina
Gobec
a,
Irena Mlinarič
Raščan
a,
Črtomir
Podlipnik
c,
Beat
Ernst
*b and
Marko
Anderluh
*a
aUniversity of Ljubljana, Faculty of Pharmacy, Aškerčeva 7, SI-1000 Ljubljana, Slovenia. E-mail: marko.anderluh@ffa.uni-lj.si; Fax: +386 14258031; Tel: +386 14769639
bInstitute of Molecular Pharmacy, Pharmacenter, University of Basel, Klingelbergstrasse 50, CH-4056 Basel, Switzerland. E-mail: beat.ernst@unibas.ch; Fax: +41 612671552; Tel: +41 612671551
cUniversity of Ljubljana, Faculty of Chemistry and Chemical Technology, Aškerčeva 5, SI-1000 Ljubljana, Slovenia
First published on 11th June 2014
Abstract
FimH is a type I fimbrial lectin located at the tip of type-1 pili of uropathogenic Escherichia coli guiding its ability to adhere and infect urothelial cells. Accordingly, blocking FimH with small-molecule antagonists is considered as a promising new therapeutic alternative to treat infections caused by uropathogenic Escherichia coli (UPEC). Herein we report that our recently disclosed α-D-mannopyranosides bearing diaryl substituted 1,3-diaminopropanol or glycerol moieties act as potent FimH antagonists in vitro, as determined by a competitive binding assay on isolated FimH lectin. Most of the assayed compounds display FimH antagonistic activity in the range of 58–1000 nM. Based on the promising results of the first series of compounds, we have designed and synthesized a new series of asymmetrically disubstituted glyceryl α-D-mannopyranosides with improved physicochemical properties. Molecular docking calculations were employed to predict compounds' binding poses leading to two possible binding modes; so-called in- and out-docking modes in the “tyrosine gate” (formed by Tyr48 and Tyr137) for one aromatic moiety, which is in accordance with previous findings, while the second aromatic moiety reaches the previously unexplored lipophilic region formed by Phe142 and Ile13. Furthermore, compounds were found to be non-cytotoxic on HepG2 cells in concentrations up to 10 μM pointing to their selective toxicity, which is one of the key features of potential therapeutics for the treatment of urinary tract infections.
Introduction
The discovery of antibacterial drugs in the past century is undoubtedly one of the key medical achievements that have profoundly changed the therapy of bacterial diseases.1 However, temporary illusion of the final victory over bacteria has vanished over the years due to the growing incidence of bacterial resistance. The latter is probably the major driving force towards novel antibacterials.2 While in the past, the focus of the antibacterial therapy was on the selective toxicity with the clear aim of direct bactericidal or bacteriostatic activity, one emerging strategy to fight bacterial infections is the so-called anti-adhesion therapy.3 Because bacteria's ability to infect host tissues is governed by its adhesion to a specific tissue, the inhibition of this process is considered an efficient way to stop infection and biofilm formation. The anti-adhesion agents are expected to have several advantages over the existing antimicrobial agents. They are less likely to provoke bacterial resistance than bactericidal agents, as (a) anti-adhesion does not induce a selection pressure, and (b) mutations of the adhesin would directly affect the pathogen's ability to bind to the host receptor thereby diminishing its virulence.4 Unfortunately, no antibacterial anti-adhesion agent is currently registered and we still await for the first therapeutic application of the anti-adhesion therapy.5Bacterial surface lectins that adhere to glycosylated host proteins are the most common adhesion molecules and virulence factors that guide bacteria's tissue selectivity.6 For example, uropathogenic Escherichia coli (UPEC) that provokes the vast majority of uncomplicated urinary tract infections invades the urinary tract by adhering via several fimbrial lectins, among which type I fimbrial lectin FimH is probably the most important one.6,7 It is a D-mannose selective adhesin that allows UPEC binding to the luminal surface of urothelial cells.8,9 Furthermore, UPEC invades epithelial cells by FimH-mediated aggregation into intracellular bacterial communities.10 These facts justify the therapeutic use of FimH antagonists; the inhibition of FimH by a small-molecule FimH antagonist might be used to treat UPEC infections as a standalone therapy or in combination with known antibacterial agents.
D-Mannose and methyl α-D-mannopyranoside bind to FimH with Kd values of 2.3 and 2.2 μM, and inhibit the yeast agglutination by E. coli with IC50 of 0.56 and 0.45 mM respectively.11,12 This unusual high affinity of the interaction of a monosaccharide with a lectin is due to a well-defined, tight FimH mannose-binding pocket forming an extended hydrogen bonding network (Fig. 1).
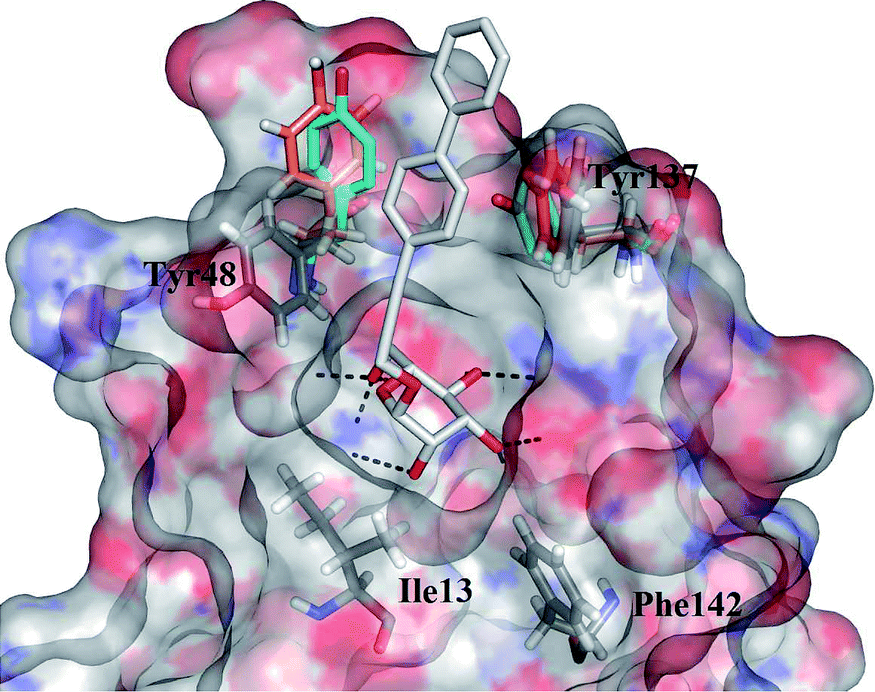 | ||
| Fig. 1 Overlay of the FimH binding sites from several crystal structures (PDB entries: 3MCY in closed,214AUY in half open, 4AV5 in open conformation,14 the latter rendered as a transparent surface coloured according to the electrostatic potential). Tyr48 and Tyr137 of the “tyrosine gate” are rendered in solid sticks coloured according to the different PDB entries (3MCY in cyan, 4AUY in orange): both show substantial flexibility. The ligand from 4AV5, 3-biphenylpropynyl α-D-mannopyranoside, is rendered in solid grey sticks: the mannose residue is buried in the specific binding pocket while the aromatic residue forms contacts with both tyrosine residues of the “tyrosine gate”. Hydrogen bonds are presented as black dashed lines. | ||
This is easily disrupted by a “wrong” configuration of a hydroxyl group at position 2, and consequently, glucose and other monosaccharides with equatorial hydroxyl group (instead of an axial one) at position 2 bind to FimH with negligible affinity.11 A systematic study of Bouckaert et al. reported simple α-D-mannopyranosides 1 with elongated alkyl moieties displaying 1000-fold higher affinities, with heptyl α-D-mannopyranoside being the most potent representative (up to 5 nM, Fig. 2).11 The latter was found to efficiently block UPEC adhesion to 5673 bladder cells, antagonize its invasion, biofilm formation, and finally to reduce bacterial load in a murine cystitis model.13
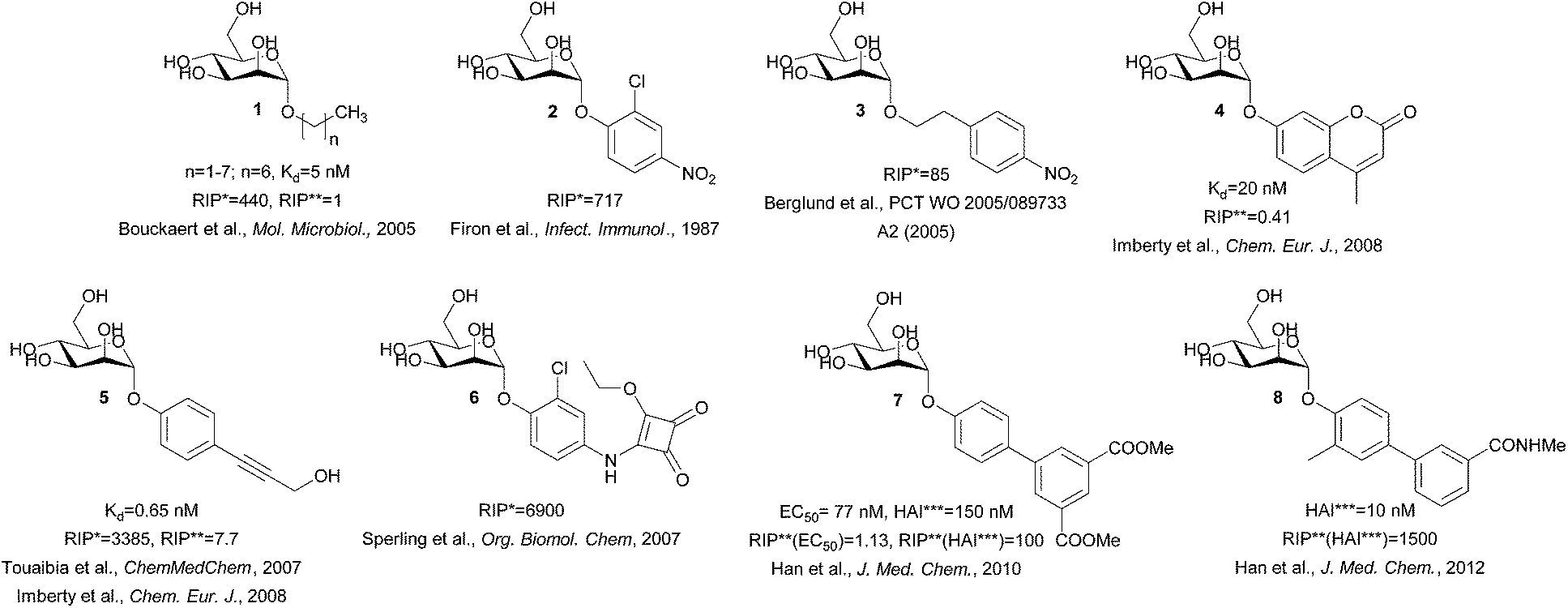 | ||
| Fig. 2 Structures of the potent FimH antagonists. RIP* = relative inhibitory potency compared to the methyl α-D-mannopyranoside, RIP** = relative inhibitory potency compared to the heptyl α-D-mannopyranoside, HAI*** = hemagglutination inhibition.11,15–22 | ||
The crystal structure of the heptyl α-D-mannopyranoside-FimH complex (PDB entry: 4LOV) revealed that the alkyl residue protrudes towards the so-called “tyrosine gate” formed by 2 tyrosine residues (Tyr48 and Tyr137, Fig. 1).11 These hydrophobic interactions with the FimH binding site residues were witnessed by favourable entropic changes, and accordingly, the free binding energy is improved in a linear correlation with each additional methylene group from methyl to heptyl α-D-mannopyranoside (Fig. 2).11,14 The “tyrosine gate” compensates for the entropic loss caused by the loss of conformational and rotational freedom of the alkyl and aryl ligands,15 and explains the potent activity of aromatic α-D-mannopyranoside 2 as reported by Firon et al.16 almost 3 decades ago. Since then, a number of aromatic α-D-mannopyranosides (3–8, and their analogues) have been designed and synthesized (Fig. 2 and 3).17–28 In terms of binding affinity, these molecules were in the range of heptyl α-D-mannopyranoside, but in some cases were far more potent as measured with the cell-based assays.
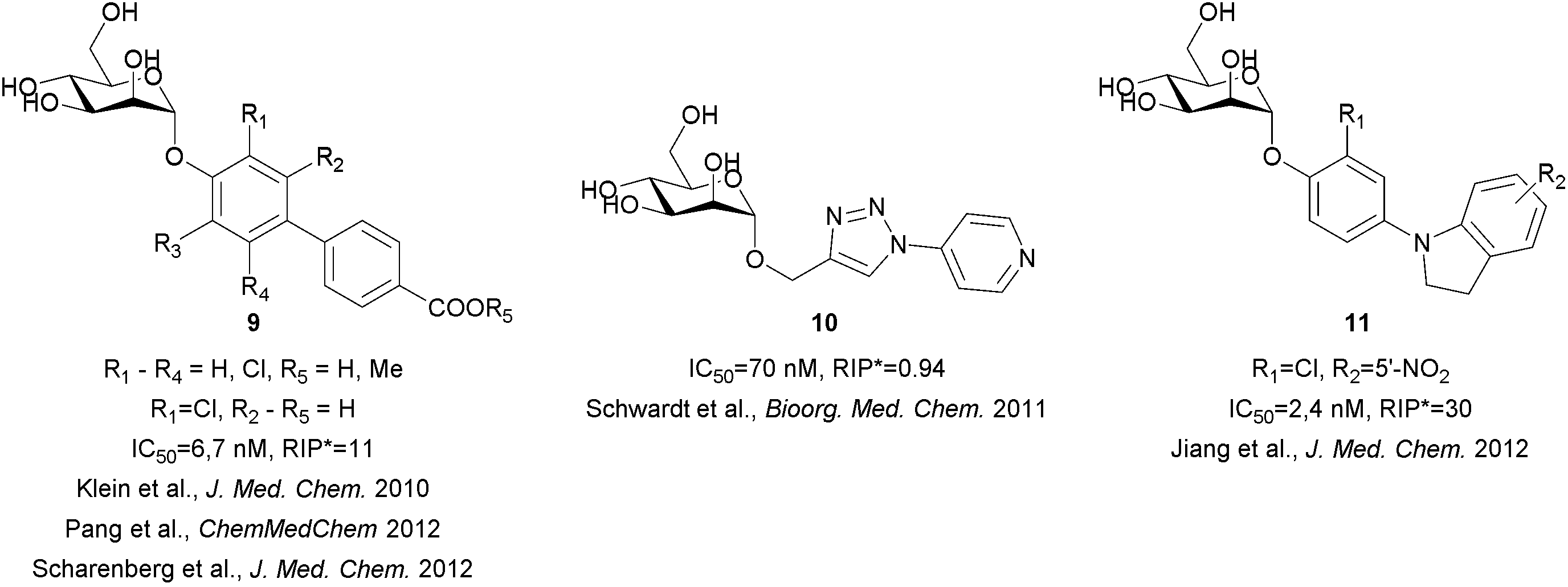 | ||
| Fig. 3 Various types of α-D-mannopyranoside-based FimH antagonists, as reported by the B. Ernst group.23–28 RIP* = relative inhibitory potency compared to the heptyl α-D-mannopyranoside. | ||
All these compounds follow the same paradigm of glycomimetic design, where the “core monosaccharide” is utilized to selectively anchor the ligand in the lectin binding site, while the aglycon moiety attached to the anomeric center boosts the binding affinity.29
Extensive efforts have been made not only to improve the affinity and selectivity of FimH antagonists, but also to ameliorate their pharmacokinetic properties.23–28 In particular, biphenyl α-D-mannopyranoside derivatives were proven to offer excellent solubility as free acids, while ester prodrugs may be used to accomplish sufficient level of absorption, good selectivity and potent in vivo activity in a mouse model (Fig. 3).23,26 However, these compounds suffer from low solubility (in the case of the prodrugs) and/or fast renal clearance (in the case of free acids). Recently, novel classes of α-D-mannopyranosides, with aryl triazole, (aza)indolylphenyl and indolinylphenyl aglycones have been reported.24,28 The latter showed optimal in silico fit to FimH, higher lipophilicity that halted rapid renal clearance and high plasma protein binding, with compound 11 (Fig. 3) being one of the most potent FimH antagonists in an in vivo assay (UTI mouse model).28
Recently, we have reported the design and synthesis of D-mannose-based DC-SIGN antagonists bearing diaryl substituted 1,3-diaminopropanol or glycerol moieties.30 Some of the compounds were shown to inhibit HIV-1 gp120 binding to DC-SIGN in the medium micromolar range, and were proven to act as functional antagonists of DC-SIGN-mediated DC adhesion. In the present study, we have assayed these compounds on the FimH lectin domain using a competitive binding assay.31 Their potency and selectivity towards FimH prompted us to design a second generation of disubstituted 2-glyceryl α-D-mannopyranosides with improved physicochemical properties. Selection of target molecules for the synthesis was supported by molecular modelling. Furthermore, to assess the potential cytotoxicity, we have performed the metabolic activity assay on HepG2 human cells.
Results and discussion
Design and results of biological assays
The first series of branched α-D-mannopyranosides 12, 13 and 14 (Table 1) were designed to fit the DC-SIGN binding site.30 A careful examination of the FimH binding site revealed that the aromatic moieties of these molecules might occupy the “tyrosine gate”, and also the opposing lipophilic region formed by Phe142 and Ile13 (Fig. 1).|
|
||||
|---|---|---|---|---|
| Entry | Ar | DC-SIGN IC50 [mM] | FimH IC50 [nM] | RIP |
| a IC50 of compounds in the DC-SIGN-CRD competitive binding assay.30 b IC50 of compounds in the FimH-CRD competitive binding assay.31 c RIP = relative inhibitory potency on FimH compared to the heptyl α-D-mannopyranoside. d Not active below 1 μM concentration. | ||||
| 12a |
|
8.79 | 472 | 0.12 |
| 12b |
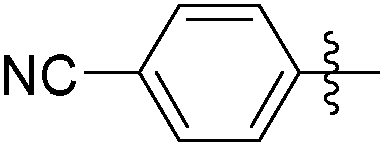
|
4.37 | 978 | 0.06 |
| 12c |
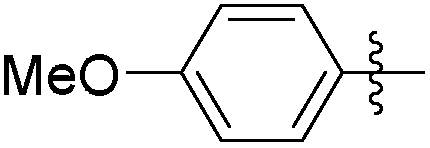
|
3.89 | 536 | 0.11 |
| 12d |
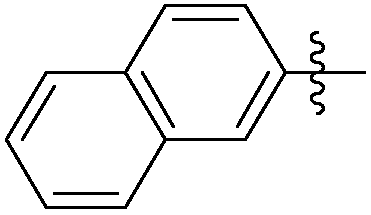
|
1.34 | 763 | 0.08 |
| 12e |
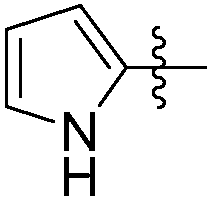
|
13.25 | 607 | 0.10 |
| 12f |

|
14.94 | NAd | / |
| 12g |
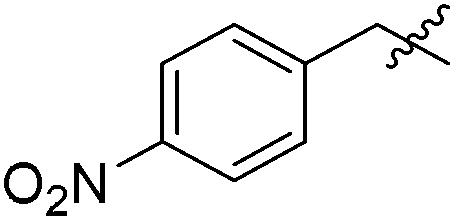
|
6.49 | NAd | / |
| 13a |

|
2.42 | 450 | 0.13 |
| 13b |
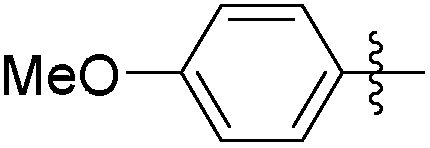
|
5.15 | NAd | / |
| 13c |
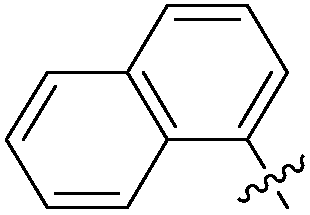
|
0.042 | 58 | 1.02 |
| 13d |
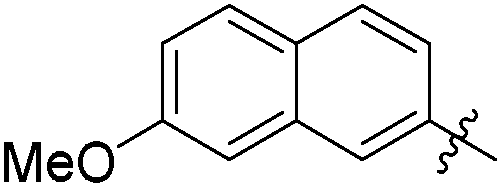
|
0.053 | 593 | 0.10 |
| 13e |
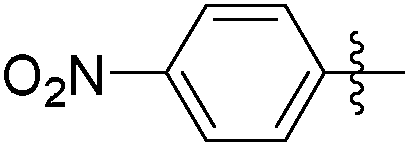
|
>0.5 | 804 | 0.07 |
| 13f |
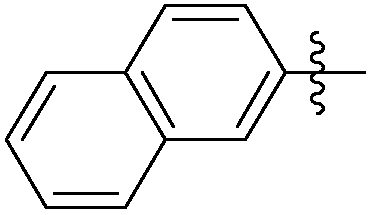
|
0.034 | NAd | / |
| 14a |
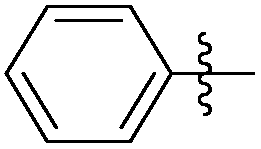
|
8.64 | 144 | 0.41 |
| 14b |
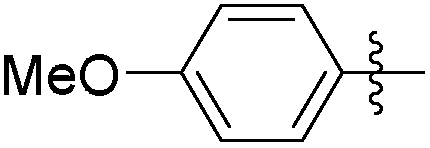
|
1.01 | 208 | 0.28 |
| 14c |
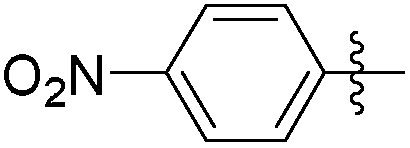
|
5.73 | 105 | 0.56 |
| 14d |
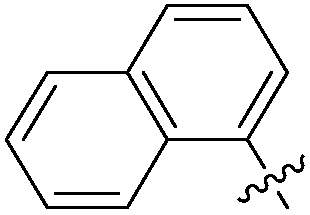
|
>0.5 | NAd | / |
| 14e |
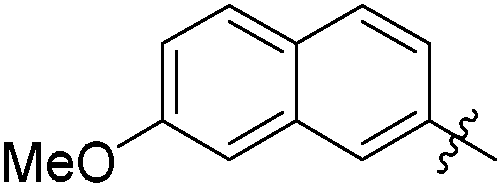
|
>0.5 | 875 | 0.07 |
| 14f |
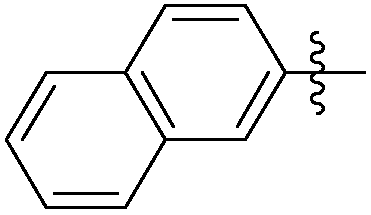
|
0.395 | NAd | / |
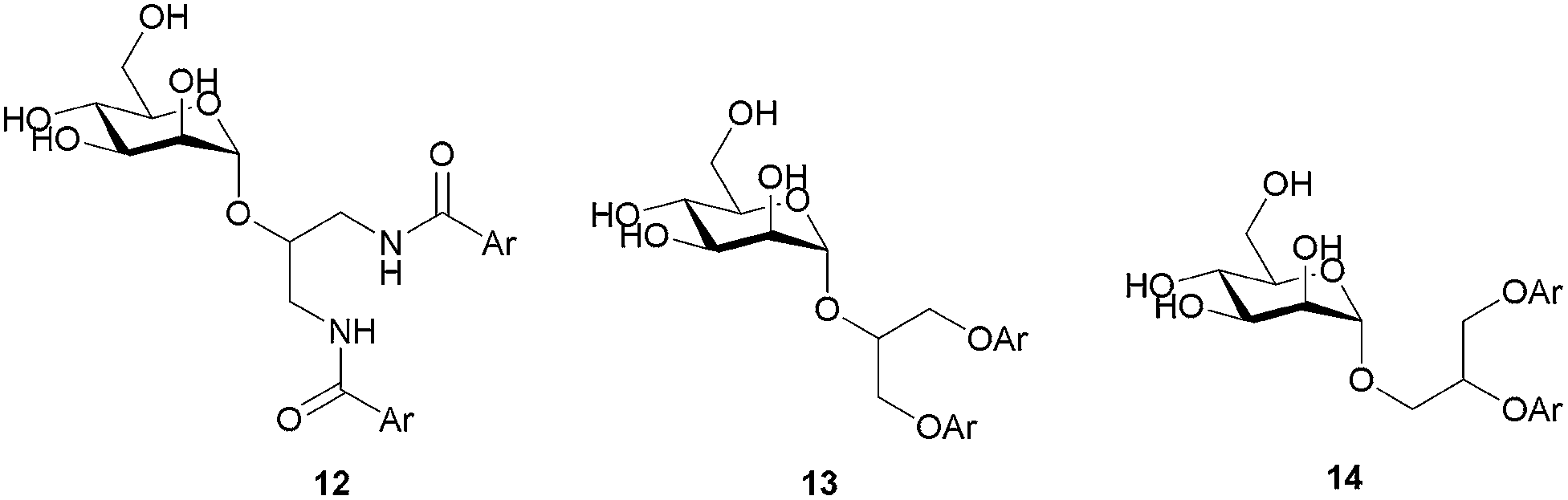
Although all representatives of the first generation include a large number of rotatable bonds, and therefore are expected to suffer from entropic loss upon binding, the additional lipophilic region formed by Phe142 and Ile13 could compensate for the entropic penalty. The binding mode of the compounds was studied by their docking to the binding site of FimH in its closed, half open and open conformations (PDB entries: 3MCY, 4AUY, 4AV5, respectively)14,21 using ligand docking program GOLD32 (GOLD Suite v5.2). The estimated binding poses have confirmed our assumption that aromatic residues of the compounds 12–14 could accommodate in both the “tyrosine gate” and the lipophilic region formed by Phe142 and Ile13, as exemplified by the binding pose of 13c in the open conformation of the FimH binding site (Fig. 4).
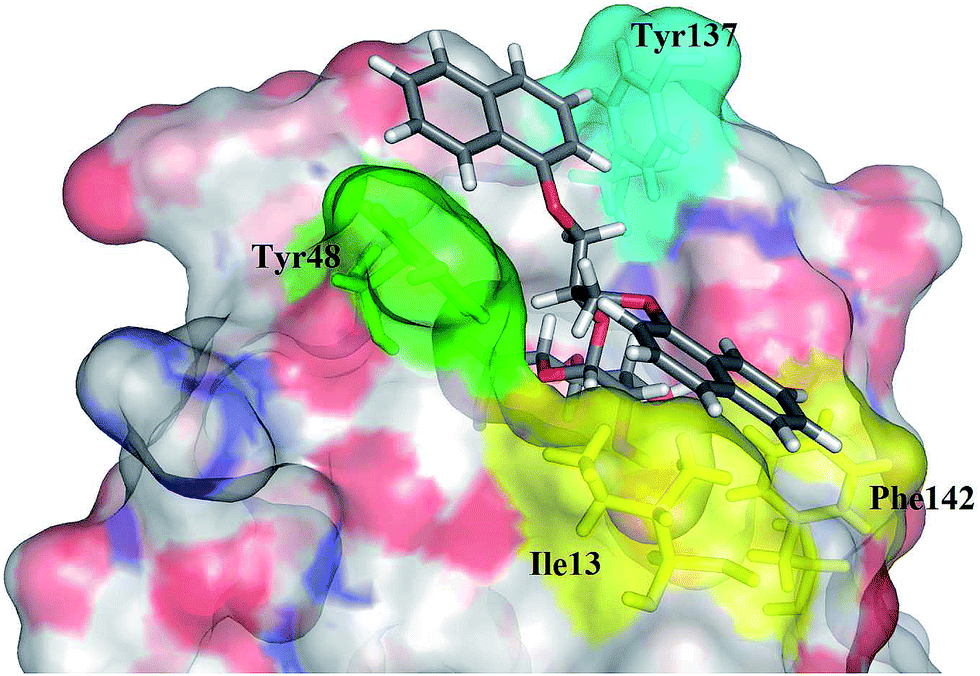 | ||
| Fig. 4 GOLD-calculated binding pose of 13c (in grey sticks) in the FimH binding site in its open conformation (PDB entry: 4AV5). | ||
Analysis of the docking poses of the ligands 12–14 in different FimH conformations revealed that the aromatic moieties of the ligands are in a similar orientation in closed, half open and open FimH conformations. However, more detailed analyses indicated that the best fit can be observed in the FimH open conformation (PDB entry: 4AV5), since the open “tyrosine gate” enables the in-docking mode of one of the aromatic moieties, while the other is predicted to form hydrophobic interactions with Ile13 and Phe142 (Fig. 4). When the “tyrosine gate” is in its closed conformation, the ligand aromatic ring can only be accommodated in the out-docking mode (Fig. S1†). This observation is also in agreement with the scores obtained by the GOLDscore33 scoring function, which favours the in-docking mode to the open FimH conformation over the out-docking mode to half open and closed conformations.
The results of the competitive binding assay with the lectin domain of FimH exhibited that most of the antagonists reach nanomolar affinities, with compound 13c being the most potent one with an IC50 of 58 nM (Table 1). For compounds with the 1,3-diamino-2-propanol linker (12) slightly lower affinities were obtained in general. We presume that the amide bond that links the aromatic moiety and the linker is rigid, more bulky and polar, and may not allow optimal orientation of the aromatic moieties in the “tyrosine gate”, as well as the interaction complementarity with the binding site. The glycerol linker in compounds with general formulae 13 and 14 presumably allows better steric complementarity in some cases, and hence higher affinity (e.g.13c and 14a–c), which correlates with higher scores found in docking studies. The most potent compound 13c bears two 1-naphthyl residues pointing to the conclusion that this residue contacts a large proportion of the “tyrosine gate” and the opposing lipophilic region. This is in agreement with the predicted binding pose of 13c (Fig. 4).
Although primarily designed to bind DC-SIGN-CRD, the majority of compounds exhibited quite high affinities towards FimH. This is in accordance with a previous report in which α-D-mannopyranosides were found to bind to FimH-CRD with relatively high affinity and in a selective manner as compared to DC-SIGN-CRD.26
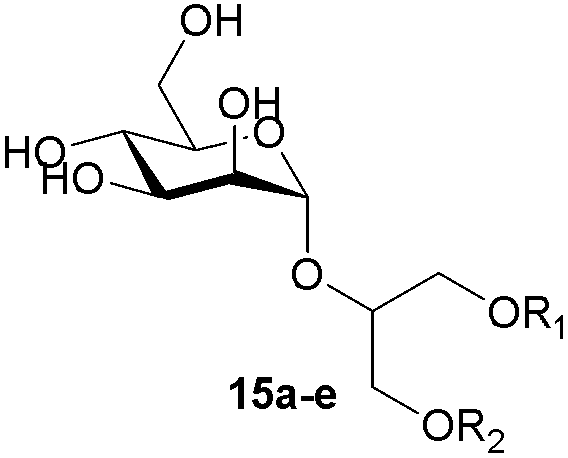
The second generation of “asymmetrical” branched α-D-mannopyranosides (15) was designed by utilizing 13c and 13d as the basis. Their synthesis (Fig. 5) was achieved as reported,30 with some modifications as described in the ESI.† The naphthalene-1-yl and 7-methoxynaphthalene-2-yl moieties of 13c and 13d were not modified to maximize the interaction with the “tyrosine gate” (R1, Table 2), while the other aryl moiety attached to the glyceryl linker was containing more hydrophilic and possibly ionisable functional groups at physiological pH (R2, Table 2).
 | ||
| Fig. 5 General synthetic scheme for compounds 15b–e. Reagents and conditions. (a) (i) Methyl 2-(4-hydroxyphenyl)acetate or methyl 6-hydroxy-2-naphthoate or methyl 4-hydroxybenzoate, KOH, MeOH, r.t., 20 min; (ii) tetrabutylammonium bromide, toluene/N,N-dimethylformamide, 90–120 °C, 2–20 h; (b) 2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl trichloroacetimidate, TMSOTf, CH2Cl2, 0 °C, then r.t., 24 h; (c) (i) NaOMe, MeOH, 1 h, then Amberlite® IR120 H, 15 min; (ii) 2 M NaOH, 1,4-dioxane/MeOH, r.t., 24 h. | ||
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | FimH IC50 [nM] | RIP |
| a RIP = relative inhibitory potency on FimH compared to the heptyl α-D-mannopyranoside. | ||||
| 15a |

|
Et- | 746 | 0.08 |
| 15b |
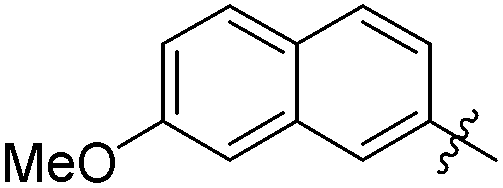
|
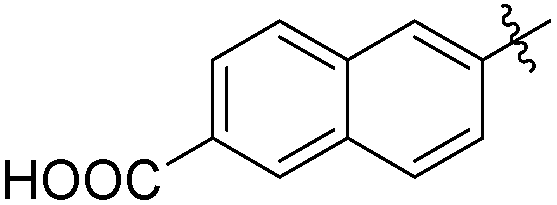
|
158 | 0.37 |
| 15c |
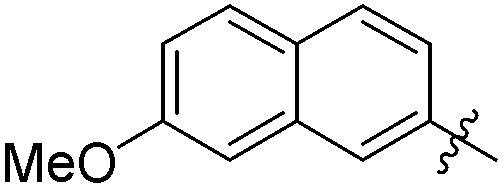
|
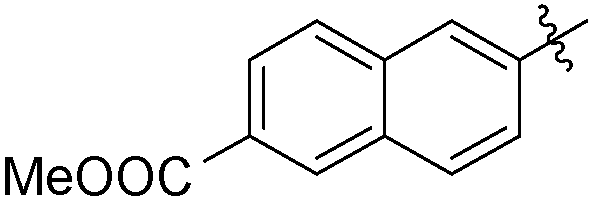
|
212 | 0.28 |
| 15d |
|

|
346 | 0.17 |
| 15e |
|
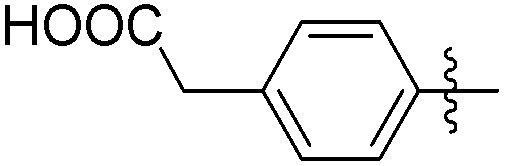
|
232 | 0.25 |
| Heptyl α-D-mannopyranoside | 58.9 | 1 | ||
The impact of these changes on the lipophilicity/hydrophilicity was proven by their solubility in aqueous media during the metabolic activity assay (see below), where the majority of compounds with the general formula 15 (except 15a, lacking the ionisable moiety) were proven to be soluble at 10 μM concentration. The IC50s of these compounds were in the medium nanomolar range pointing to the fact that, along with optimized solubility, the affinity of the branched α-D-mannopyranosides was maintained in the same range. The affinity was either slightly reduced for compounds containing one 1-naphthyl moiety (13cvs.15d and e) or improved for the 7-methoxy-2-naphthyl-based compounds (13dvs.15b and c). This may be explained by two alternative hypotheses. The first hypothesis relies on the additional interactions within the binding site of FimH. Our docking calculations of compounds 15b–e revealed that the carboxylic acid (15b, d and e) or methyl ester (15c) moiety can form additional hydrogen bonds with FimH binding site residues. Docking of the ligands 15b–e in the open FimH conformation revealed the in-docking mode of the carboxyl-substituted naphthyl (15b, c) or phenyl (15d, e) moiety with a possible hydrogen bond with the hydroxyl group of the Tyr137 side chain (Fig. 6), while the other aromatic ring contacts the lipophilic region formed by Phe142 and Ile13. In contrast, docking poses of 15b–e in the closed FimH conformation predicted an out-docking mode of the naphthyl moiety, while the aromatic ring bearing the carboxyl or ester group is suggested to form additional ionic and hydrogen bonds with the Arg98 side chain (Fig. 7). The second hypothesis is based on the electron–donor effect of the ionized carboxylate and its strengthening of π–π stacking with either the phenyl residues of the “tyrosine gate” or Phe142. In the latter case, the carboxylate would point towards the solvent thus diminishing the hydrophobic surface in contact with water, which would otherwise cause entropic penalties during binding. However, which binding mode is more reliable still remains to be determined.
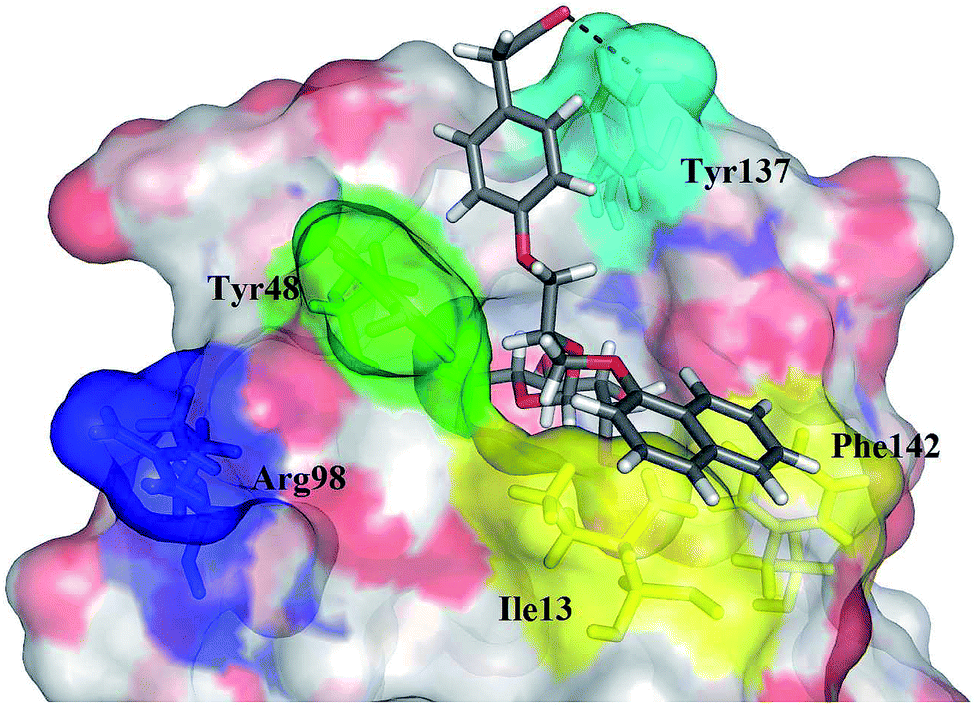 | ||
| Fig. 6 The calculated binding pose of (R)-15e (in grey sticks) in FimH binding site in its open conformation (PDB entry: 4AV5). Plausible hydrogen bond with the Tyr137 side chain is presented as a black dashed line. | ||
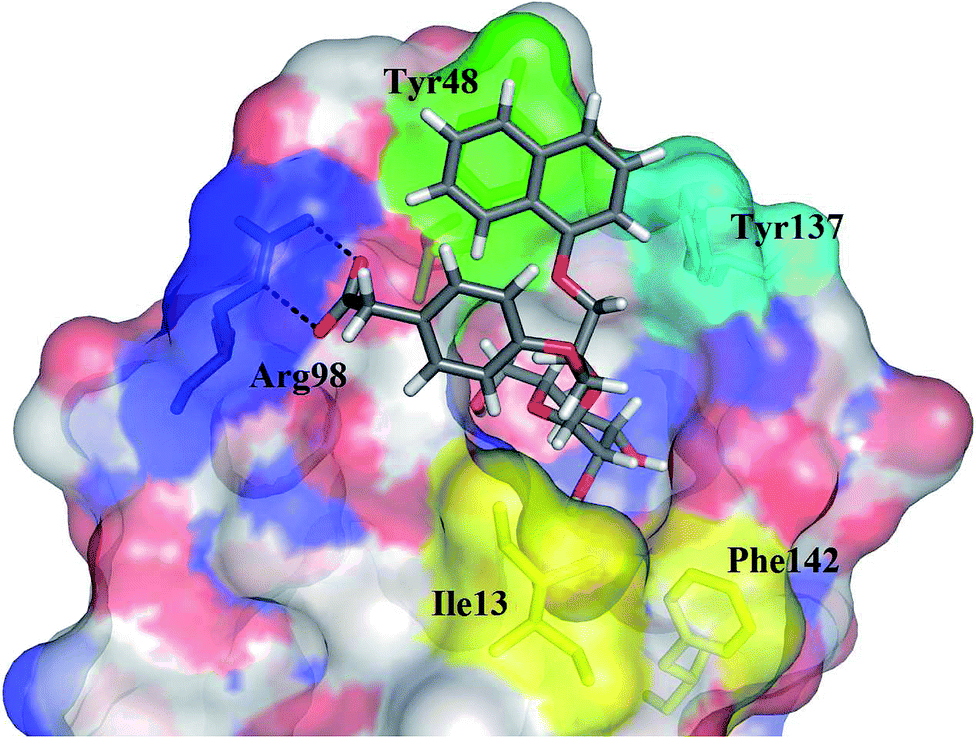 | ||
| Fig. 7 The calculated binding pose of (R)-15e (in grey sticks) in the FimH binding site in its closed conformation (PDB entry: 3MCY). Plausible hydrogen bonds with an Arg98 side chain are presented as black dashed lines. | ||
For the assessment of cytotoxicity, we have performed a metabolic activity assay with HepG2 cells. The cells were treated with 1 and 10 μM of the compound of interest, and the metabolic activity was assessed after 24 h treatment. The results clearly demonstrate that the majority of the compounds assayed did not exhibit any significant cytotoxicity (Fig. 8). Despite their notable potency and lack of cytotoxicity, the first series of compounds was characterized by scarce solubility in water media. Since only negligible quantities of DMSO (<1%) are allowed in the metabolic activity assay, some of the compounds were not sufficiently soluble at 10 μM concentration (as judged by microscopic inspection) and their cytotoxicity was therefore determined only at 1 μM concentration. The compounds with the general formula 12 were not assayed due to low solubility in the media containing low percentage of DMSO.
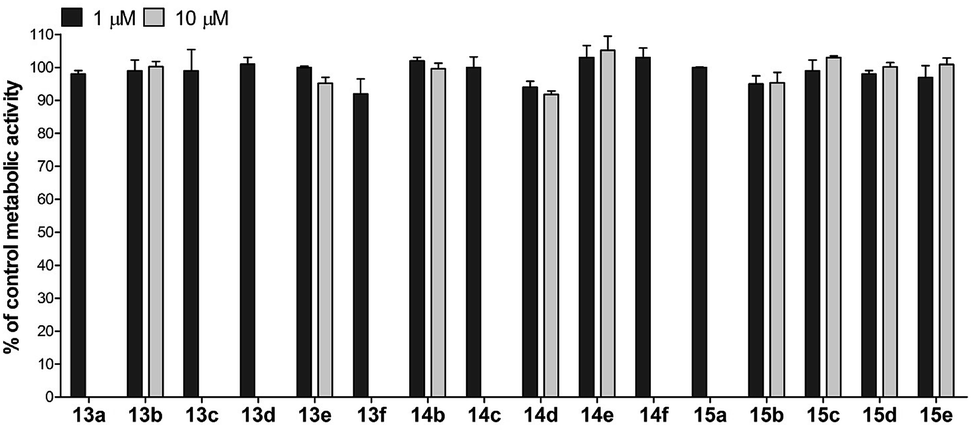 | ||
| Fig. 8 Metabolic activity of the HepG2 cell line treated for 24 h with 1 and/or 10 μM of the assayed compounds. Results are presented as percentage of metabolic activity of control cells stimulated with vehicle (mean ± SD) from three independent experiments, each conducted in triplicate. | ||
Conclusions
In the present work we report the FimH antagonistic activity of our recently disclosed D-mannose-based DC-SIGN antagonists bearing diaryl substituted 1,3-diaminopropanol (12) or glycerol (13 and 14) moieties. Most of the tested compounds displayed FimH antagonistic activity in the range of 58–1000 nM and thus showed selectivity for FimH. The most potent compound 13c exhibited a IC50 value of 58 nM, which was proven as equipotent to the heptyl α-D-mannopyranoside in our hands. Inspired by the promising activity of the first generation of compounds, we designed and synthesized a novel series (15) possessing improved solubility, while maintaining affinity in the same range. Moreover, all compounds were found to be non-cytotoxic on HepG2 cells in concentrations up to 10 μM and thus represent an interesting potential for further improvement toward novel therapeutics for the treatment of UTI.Experimental
All the detailed experimental procedures, materials and methods can be found in the ESI.†Acknowledgements
This work was supported by the Slovenian Research Agency (Grant no. P1-0208).References
- I. Chopra, J. Antimicrob. Chemother., 2013, 68, 496 CrossRef CAS PubMed.
- R. Wise, J. Antimicrob. Chemother., 2011, 66, 1939 CrossRef CAS PubMed.
- (a) B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661 CrossRef CAS PubMed; (b) I. Ofek, D. L. Hasty and N. Sharon, FEMS Immunol. Med. Microbiol., 2003, 38, 181 CrossRef CAS; (c) M. Touaibia and R. Roy, Mini-Rev. Med. Chem., 2007, 7, 1270 CrossRef CAS.
- A. M. Krachler and K. Orth, Virulence, 2013, 4, 284 CrossRef PubMed.
- D. Cozens and R. C. Read, Expert Rev. Anti Infect. Ther., 2012, 10, 1457 CrossRef CAS PubMed.
- (a) N. Sharon, Biochim. Biophys. Acta, 2006, 1760, 527 CrossRef CAS PubMed; (b) H. De Greve, L. Wyns and J. Bouckaert, Curr. Opin. Struct. Biol., 2007, 17, 506 CrossRef CAS PubMed.
- N. Sharon, FEBS Lett., 1987, 217, 145 CrossRef CAS.
- J. Yu, J. H. Lin, X. R. Wu and T. T. Sun, J. Cell Biol., 1994, 125, 171 CrossRef CAS.
- D. S. Eto, T. A. Jones, J. L. Sundsbak and M. A. Mulvey, PLoS Pathog., 2007, 3, e100 CrossRef PubMed.
- (a) S. S. Justice, C. Hung, J. A. Theriot, D. A. Fletcher, G. G. Anderson and M. J. Footer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1333 CrossRef CAS PubMed; (b) K. J. Wright, P. C. Seed and S. J. Hultgren, Cell. Microbiol., 2007, 9, 2230 CrossRef CAS PubMed.
- (a) J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C. S. Hung, J. Pinkner, R. Slättegård, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441 CrossRef CAS PubMed; (b) S. Vanwetswinkel, A. N. Volkov, Y. G. J. Sterckx, A. Garcia-Pino, L. Buts, W. F. Vranken, J. Bouckaert, R. Roy, L. Wyns and N. A. J. van Nuland, J. Med. Chem., 2014, 57, 1416 CrossRef CAS PubMed; (c) C. Fessele and T. K. Lindhorst, Biology, 2013, 2, 1135 CrossRef PubMed.
- (a) N. Firon, I. Ofek and N. Sharon, Biochem. Biophys. Res. Commun., 1982, 105, 1426 CrossRef CAS; (b) N. Firon, I. Ofek and N. Sharon, Carbohydr. Res., 1983, 120, 235 CrossRef CAS.
- A. Wellens, C. Garofalo, H. Nguyen, N. Van Gerven, R. Slättegård, J. P. Hernalsteens, L. Wyns, S. Oscarson, H. De Greve, S. Hultgren and J. Bouckaert, PLoS One, 2008, 3, e2040 Search PubMed.
- A. Wellens, M. Lahmann, M. Touaibia, J. Vaucher, S. Oscarson, R. Roy, H. Remaut and J. Bouckaert, Biochemistry, 2012, 51, 4790 CrossRef CAS PubMed.
- G. Roos, A. Wellens, M. Touaibia, N. Yamakawa, P. Geerlings, R. Roy, L. Wyns and J. Bouckaert, ACS Med. Chem. Lett., 2013, 4, 1085 CrossRef CAS PubMed.
- N. Firon, S. Ashkenazi, D. Mirelman, I. Ofek and N. Sharon, Infect. Immun., 1987, 55, 472 CAS.
- J. Berglund, J. Bouckaert, H. De Greve and S. Knight, PCT WO 2005/089733 A2, 2005.
- A. Imberty, Y. M. Chabre and R. Roy, Chem. – Eur. J., 2008, 14, 7490 CrossRef CAS PubMed.
- M. Touaibia, A. Wellens, T. C. Shiao, Q. Wang, S. Sirois, J. Bouckaert and R. Roy, ChemMedChem, 2007, 2, 1190 CrossRef CAS PubMed.
- O. Sperling, A. Fuchs and T. K. Lindhorst, Org. Biomol. Chem., 2006, 4, 3913 CAS.
- Z. Han, J. S. Pinkner, B. Ford, R. Obermann, W. Nolan, S. A. Wildman, D. Hobbs, T. Ellenberger, C. K. Cusumano, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2010, 53, 4779 CrossRef CAS PubMed.
- Z. Han, J. S. Pinkner, B. Ford, E. Chorell, J. M. Crowley, C. K. Cusumano, S. Campbell, J. P. Henderson, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2012, 55, 3945 CrossRef CAS PubMed.
- T. Klein, D. Abgottspon, M. Wittwer, S. Rabbani, J. Herold, X. Jiang, S. Kleeb, C. Lüthi, M. Scharenberg, J. Bezençon, E. Gubler, L. Pang, B. Cutting, O. Schwardt and B. Ernst, J. Med. Chem., 2010, 53, 8627 CrossRef CAS PubMed.
- O. Schwardt, S. Rabbani, M. Hartmann, D. Abgottspon, M. Wittwer, S. Kleeb, A. Zalewski, M. Smiesko, B. Cutting and B. Ernst, Bioorg. Med. Chem., 2011, 19, 6454 CrossRef CAS PubMed.
- L. Pang, S. Kleeb, K. Lemme, S. Rabbani, M. Scharenberg, A. Zalewski, F. Schädler, O. Schwardt and B. Ernst, ChemMedChem, 2012, 8, 1404 CrossRef PubMed.
- M. Scharenberg, O. Schwardt, S. Rabbani and B. Ernst, J. Med. Chem., 2012, 55, 9810 CrossRef CAS PubMed.
- M. Scharenberg, X. Jiang, L. Pang, G. Navarra, S. Rabbani, F. Binder, O. Schwardt and B. Ernst, ChemMedChem, 2014, 9, 78 CrossRef CAS PubMed.
- X. Jiang, D. Abgottspon, S. Kleeb, S. Rabbani, M. Scharenberg, M. Wittwer, M. Haug, O. Schwardt and B. Ernst, J. Med. Chem., 2012, 55, 4700 CrossRef CAS PubMed.
- M. Anderluh, DC-SIGN Antagonists – a Paradigm of C-Type Lectin Inhibition, in Carbohydrates – Comprehensive Studies on Glycobiology and Glycotechnology, ed. Chuan-Fa Chang, InTech, 2012 Search PubMed.
- T. Tomašić, D. Hajšek, U. Švajger, J. Luzar, N. Obermajer, I. Petit-Haertlein, F. Fieschi and M. Anderluh, Eur. J. Med. Chem., 2014, 75, 308 CrossRef PubMed.
- S. Rabbani, X. Jiang, O. Schwardt and B. Ernst, Anal. Biochem., 2010, 407, 188 CrossRef CAS PubMed.
- GOLD Suite, version 5.2.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction schemes, experimental procedures and characterization of intermediate and target compounds, biological assay details, molecular modelling procedure and additional figures. See DOI: 10.1039/c4md00093e |
| This journal is © The Royal Society of Chemistry 2014 |