 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface plasmon resonance for the characterization of bacterial polysaccharide antigens: a review
Barbara
Brogioni
and
Francesco
Berti
*
Novartis Vaccines, Research Center, Via Fiorentina 1, 53100 Siena, Italy. E-mail: francesco.berti@novartis.com
First published on 23rd April 2014
Abstract
Carbohydrates, in the form of oligosaccharides, polysaccharides and lipopolysaccharides, are ubiquitous components of the cell surface of bacteria. In the last 30 years polysaccharide-based vaccines have proven to be highly safe and efficacious against bacterial infections like meningitis and pneumonia. Surface plasmon resonance (SPR) analysers have emerged as powerful tools for the characterization of label-free biomolecular interactions, enabling the dynamics of complex formation and dissociation to be monitored in real-time. A wide variety of sensor chips are currently available for many applications. Nevertheless, biosensors with nucleophilic functionalities, such as amino groups, could be useful to extend the SPR applicability. Using selected examples, this review gives an overview of significant applications of conventional flow SPR to investigate the specific interactions of bacterial polysaccharide antigens. SPR has made significant progress in the last two decades and is now becoming a relevant technology for developing immunological assays for in-depth characterization of carbohydrate antigens.
1. Introduction
Molecular recognition is a central biological process, mediating a wide variety of specific events. For this reason, in the fields of drug discovery and biotherapeutics, the characterization of real-time biomolecular interactions is of particular interest for the study of diverse binding partners from proteins, oligonucleotides, carbohydrates, and lipids to small molecules.Several commercially available instruments exploit the phenomenon of surface plasmon resonance (SPR) to monitor the interaction between label-free molecules in real-time by the use of optical biosensors.1,2 In a biosensor experiment one molecule, called the ligand, is immobilized on the surface of a sensor chip, and the second, called the analyte, is injected over the coated surface in a continuous flow. The binding event between the ligand and the analyte generates a response proportional to the bound mass evidenced by a variation in the refractive index of the solvent near the surface. The formation and dissociation of the biospecific complex can be described using the direct estimation of the association rate constant (ka) and the dissociation rate constant (kd), from which the equilibrium dissociation constant is derived as the ratio KD = kd/ka. By means of SPR analysis, insights into binding affinity and kinetics can be obtained, providing elements useful for investigating, selecting and tailoring target molecules.
Due to the enormous diversity and complexity of structures occurring in nature, carbohydrates are a unique class of biopolymers. This structural complexity originates from the availability of a wide variety of monosaccharide units that can be combined through diverse stereochemical bonds. Nine common monosaccharides found in mammalian cells are glucose, galactose, mannose, xylose, N-acetylglucosamine, N-acetylgalactosamine, fucose, glucuronic acid, and N-acetylneuraminic acid (also called sialic acid). The combination of these nine monomeric units allows for many more different structures than is possible with the natural amino acids or nucleotides.3 Carbohydrate chain length is also a variable parameter, ranging from a single sugar unit, through short oligosaccharide units, to polysaccharide polymers containing hundreds or even thousands of monosaccharide repeating units.
The surface of many human pathogens is covered by oligosaccharides and polysaccharides in the form of capsules, glycoproteins and glycolipids. Lipopolysaccharides (LPS), present in Gram-negative bacteria, are structurally formed by three components: lipid A, the core region (a non-repetitive oligosaccharide of approximately nine sugars) and an O-antigen (OAg) which is the serogroup-specific polysaccharide.4 Surface polysaccharides confer mechanical stability to the bacterial surface and may also be important virulence factors, disrupting phagocytosis and preventing the alternate pathway of complement activation. To escape immunological recognition by a host, some bacterial pathogens have developed molecular mimicry decorating their surface with polysaccharides that share structural homology with molecules produced by human cells.5 In such cases, the immune system can adopt a strategy to target conformational epitopes unique to the pathogenic polysaccharide in order to distinguish carbohydrates of bacterial origin from those of the host.6,7 Capsular polysaccharides of most bacteria are the cause of invasive diseases. They are important targets in the development of vaccines since polysaccharides are the most conserved and accessible molecules on the bacterial surface and can elicit protective antibodies.8–10 To date, vaccines based on bacterial polysaccharides conjugated to a protein, acting as a carrier, have been licensed against diseases caused by Streptococcus pneumoniae, Neisseria meningitidis (Nm) and Haemophilus influenzae type b (Hib).11,12
SPR technology has been successfully applied to the direct detection of carbohydrate specific interactions, and a review comparing SPR with conventional analytic techniques for the detection of glycans and glycoconjugates has recently been published.13
The aim of the present review is to provide an overview of the applications of SPR to the study of specific interactions of bacterial polysaccharide antigens.
2. SPR basic principles and experimental settings
2.1. SPR phenomenon and instrumental configuration
When a beam of light propagating in a material of higher refractive index (i.e. glass) meets the interface of a material having a lower refractive index (i.e. water), at a certain angle of incidence, the light is totally reflected into the material of higher refractive index. This is called total internal reflection (TIR). If the interface between the two materials is coated with a thin layer of a conducting metal (i.e. gold) the reflection is not total. In fact, under TIR conditions, a part of the reflected light is transformed to an electrical field, called the evanescent wave, which penetrates across the interface inside the lower refractive index material. The evanescent wave, which exponentially decreases with distance from the interface, passes the metal film and interacts with the surface free electrons of the metal, called surface plasmons, propagating within the conductor surface in contact with the lower refractive index medium. At a specific incidence energy and angle, the momentum of light photons and surface plasmons is equal in magnitude and direction, and the electrons become excited. The interaction with light photons causes the oscillation (or resonance) of surface plasmons and the formation of an enhanced evanescent wave. Because a part of the light has been transformed to SPR, it is no longer totally reflected. Therefore, SPR is seen as a drop in intensity of the reflected light. The resonance conditions are very sensitive to the refractive index of the lower refractive index medium, within the effective penetration depth of the evanescent field. Penetration depth into the lower refractive index material is about a few hundred nanometers. Changes in concentration, and therefore in the refractive index of the medium, correspond to a shift of the reflected angle which can be measured as an SPR response.SPR instruments can be designed with different setups. Commonly, a glass prism couples the polarized light into the sensor coated with a thin layer of gold. The metal layer is integrated with a flow channel where reactants flow in buffer solution. Both, ligand immobilization and binding events, take place at this interface (Fig. 1). The reflected light is recorded by a detector measuring its intensity with a photodiode. Variations in the refractive index at the sensor surface are recorded in resonance units (RU). For most proteins 1 RU corresponds to a variation in concentration of 1 pg mm−2 on the sensor surface.14 The graph plot of RU versus time is called sensorgram. Although there are several SPR instruments currently available, the most widely used so far are produced by Biacore™ (GE Healthcare). Since they were first released into the market in 1990,15 Biacore instruments have demonstrated excellent quality of sensitivity and versatility, becoming the leading manufacturer of SPR-based instruments.
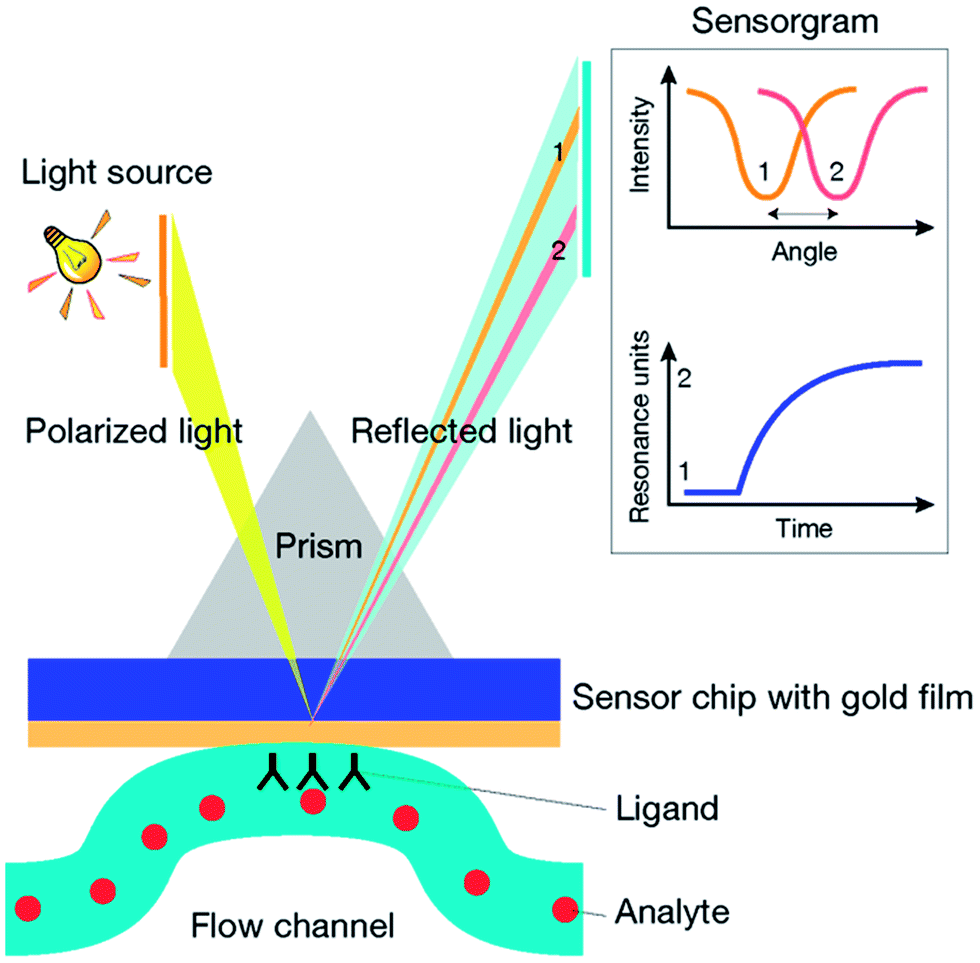 | ||
| Fig. 1 Schematic configuration of an SPR detector. The incident polarized light is coupled by a glass prism on the biosensor chip coated with a thin layer of gold and integrated with a flow channel for continuous flow of buffer. At a defined incidence angle, the SPR phenomenon is seen as a dip in the intensity of the reflected light, characteristic of the specific angle of reflection. The shift of the angle of reflection from position 1 to position 2 reveals a change in the composition of the medium near the gold film as a result of the binding between the ligand and the analyte. The angular variations are recorded in resonance units (RU) and plotted versus time in a sensorgram. | ||
2.2. Sensor chip variability
One indicator of the success of optical biosensor technology is the evidence that biosensor analysers have been established as common tools to study biomolecular interactions. Annual reviews of the most relevant optical biosensor literature have been published for a decade by Rich et al. (latest review Rich and Myszka 2011).16 Biosensor chips are at the heart of this technology because molecules can be immobilized and interact at their surface. There are different types of commercially available sensors, covering a wide range of applications.17,18 Some of the more commonly used biosensors are listed in Table 1 along with an indication of their surface modification and intended use. Technical advances to enhance optical biosensor sensitivity continue to increase the utility of this technology, e.g. a novel Au/ZnO-based biosensor has been employed for the detection of carbohydrate tumor markers exhibiting higher sensitivity than that of the conventional Au/Cr-based sensor.19| Sensor modification | General application |
|---|---|
| Carboxyl groups with diverse immobilization capacity | General purposes |
| Streptavidin derivatization | Biotinylated ligands |
| Nitrilotriacetic acid (NTA) groups | For capturing histidine-tagged ligands |
| Lipophilic modification | Lipids and liposomes |
| Hydrophobic surface with alkyl groups | Lipids |
| Plain gold surface | For custom analyses |
Biomolecules may be attached to the surface of a sensor chip using three different methods. The first is the covalent immobilization, where the ligand is attached to the chip surface through a covalent linkage. Covalent immobilization is performed using standard coupling chemistries involving functional groups such as amino, thiol, and aldehyde groups on the ligand. Among the sensor surfaces suitable for covalent immobilization comprising a carboxylated matrix, the CM5 sensor chip from Biacore™ is widely used because it provides high surface capacity for immobilizing a wide range of ligands, from proteins to carbohydrates. Immobilization over carboxylated surfaces has to be performed in a buffer where the ligand is positively charged otherwise it cannot approach the negatively charged chip surface. The second method is the affinity capture. In this method the ligand is attached by non-covalent interactions to a suitable molecule, which, in turn, is usually covalently immobilized. This is the case for instance for sensor chips having high affinity for biotin or histidine-tagged proteins. High affinity capture is considered the most appropriate method for studying monoclonal antibodies (mAb), in fact an Fc receptor (usually protein A or protein G) can be immobilized by a covalent link to the sensor surface, after which the mAb is captured and interaction with analyte can take place. The last immobilization method involves hydrophobic adsorption to modify the surface of the sensor chip for attachment of a lipid ligand.
2.3. The SPR experimental design
There are several factors to be considered in the design of an SPR experiment. One of the most important is the selection of the ligand. In this choice, as a general rule, one-to-one interactions are preferred so that the formation and dissociation of the complex between the ligand and the analyte can be interpreted in terms of first order interaction. This is the case when the surface is homogeneous and specifically immobilized and the analyte has only a single ligand-binding site. If there are no differences in the avidity between the two molecules, some practical aspects can be taken into account: since the analyte is usually injected several times at different concentrations, and the ligand only needs to be used once for the immobilization, the availability and cost should be considered when deciding which molecule to immobilize. In addition, the molecule selected as the ligand should be the more stable molecule since it should be able to withstand the conditions used for regeneration, i.e., the process of removing the bound analyte from the biosensor among interaction cycles. The choice of sensor chip and immobilization chemistry has then to be driven by the concept that the coupling chemistry should involve groups that are distant from the site of interaction, i.e. glycoconjugates are immobilized by the protein carrier, as the carbohydrate moiety is not affected by the coupling.20The SPR response is directly related to the mass concentration of the material at the chip surface. Therefore, based on the equation Rmax = (MWanalyte/MWligand) × RL × Sm where Rmax is the theoretical analyte binding capacity (RU), MWanalyte and MWligand are the molecular weight of analyte and ligand molecules, RL is the immobilization level of the ligand (RU) and Sm the stoichiometric ratio, the relative sizes of the ligand and the analyte should be considered. The theoretical binding capacity is a useful guideline for the immobilization level and for assessing the activity of the surface. The appropriate immobilization level differs according to the purpose of the analysis. For kinetic experiments low density surfaces are recommended (up to 300 RU, depending on the sensitivity of the instrument). In particular it is recommended to use the lowest amount of immobilized ligand that will give an acceptably measurable response, minimizing bulk and mass transport effects. These effects should be reduced by using appropriate flow rates, based on instrument characteristics, which are generally selected to be as fast as possible.
Kinetic analyses monitor the interaction as a function of time over a range of analyte concentrations. The whole concentration dataset is fitted to a mathematical model describing the interaction, and from which ka and kd are calculated and a derived value, KD, is obtained. Kinetic rate constants can be confirmed by comparing the equilibrium dissociation constant determined from kinetic analysis with the value determined by steady-state analysis.
To study oligosaccharide and polysaccharide interactions by SPR, 10 mM HEPES buffered saline (HBS) in 150 mM NaCl at pH 7.4, containing 3.0 mM EDTA, and 0.005% of a surfactant, such as P-20, is commonly used.
3. Bacterial polysaccharides and SPR
3.1. Bacterial polysaccharides as vaccine targets
Bacterial pathogens can display capsular polysaccharides on their surface unique to the bacterial species but also to the specific strain of the bacteria. The use of bacterial polysaccharides as immune agents is quite a new approach. The first polysaccharide vaccine, Pneumovax® (Merck), was commercially launched in 1983 containing unconjugated glycan antigens from 14 pneumonia serotypes. However, for a bacterial polysaccharide to induce sufficient protective antibodies in infants (neonates and children under 2 years), it is necessary to covalently link the polysaccharide to an immunogenic protein carrier.21 This can be the tetanus toxoid (TT) protein or the diphtheria toxin derivative CRM197 protein, a nontoxic mutant. Only in this conjugated form the bacterial polysaccharide elicits T cells and induces long-lasting protection in infants.8,22–25 A simple physical mixture of polysaccharide and protein does not behave as well as the corresponding glycoconjugate. The efficacy of using glycoconjugates as vaccines was first proven in 1987 in both adults and infants when a vaccine against the Hib bacterium was licensed.26 Before this vaccine was available, Hib was the most common cause of bacterial meningitis in children under five years of age in the United States. The introduction of the Hib conjugate vaccine has had a remarkable impact, decreasing the incidence of disease caused by this bacterium. This highlighted the critical importance of producing efficacious conjugate vaccines for the benefit of public health. In fact, after this success, bacterial polysaccharide conjugate vaccines against meningococcal and pneumococcal bacteria have also been licensed. Manufacturing challenges and crucial analytical requirements for conjugate production have been reported.27 For most vaccines, protective antibodies elicited by glycoconjugates are powerful tools for evaluating the efficacy of a test vaccine. However, carbohydrate-specific antibodies usually have low affinity (KD in the μM range), compared with protein-specific antibodies (KD in the nM range). This has been ascribed mainly to either unfavourable entropy contribution upon complex formation with loss of conformational flexibility28 or solvent re-arrangement upon binding.29 Therefore, given this intrinsic low affinity, carbohydrate-specific antibody interactions are based on avidity effects, due to the high degree of polymerization of glycans, where multivalent interactions reside.93.2. Polysaccharide immobilization on biosensors
The specific interactions of bacterial carbohydrates can be studied by SPR, based on the intrinsic nature and characteristics of these molecules. Bacterial polysaccharides can have a high degree of polymerization and an extensive negative charge; therefore, they are repelled by negatively charged sensor chip surfaces if not placed in a suitable pH buffer. Moreover, carbohydrates lack functional groups appropriate for direct binding to the sensor chip surface. Modifications of the sensing surface or derivatization strategies of the carbohydrate are necessary. Some possible modifications from the literature are reported in this paragraph.MacKenzie and Jennings (2003) reported that carbohydrates can be conveniently immobilized on sensor surfaces as glycoconjugates.20 Polysaccharides conjugated to carrier proteins such as human serum albumin (HSA) or bovine serum albumin (BSA) can be coupled through the amino groups of the carrier to carboxylated sensor chips by standard coupling chemistry. Prior to immobilization, the carboxyl groups of the sensor chip are activated with N-hydroxysuccinimide (NHS) and N-ethyl-N′-(dimethylaminopropyl) carbodiimide (EDC). The immobilization must be performed at a pH below the isoelectric point (pI) of the ligand, where it is positively charged, because when pH >pI both carboxyl groups and glycoconjugate are negatively charged and the ligand is repelled. Typically, 10 mM sodium acetate at pH 4.0 or pH 4.5 is used for conjugated bacterial polysaccharides.20
Another approach for the immobilization of carbohydrates on a carboxylated matrix has been reported by Young and co-workers.30 In this strategy the Brucella A antigen was modified, by the introduction of an amino group, as the diaminopropane derivative, which was generated through reductive amination of its reducing end. Immobilization to the sensor surface was then accomplished by covalent binding using the EDC and NHS chemistry.
The use of a class of maleimide-terminated self-assembled monolayers has been described for the immobilization of thiol-terminated carbohydrates.31 This approach has the advantage of being an efficient and selective reaction chemistry, in particular because glycans do not contain native thiol functionalities. This characteristic combined with advances in solid-phase carbohydrate synthesis could open the possibility of carbohydrate chips with multiple ligands on a single array.
3.3. Bacterial polysaccharide–protein interactions
Defining the best carbohydrate antigen for eliciting highly specific antibodies by active vaccination against a certain pathogen is a difficult process because limited knowledge is available on glycan epitope recognition.32In this section, selected examples of conventional flow SPR applications for monitoring the interactions of bacterial polysaccharides with antibodies are presented. Table 2 lists selected bibliography of SPR biosensing carbohydrates from pathogens which are promising vaccine candidates or already in clinical practice.
| Antigen | Repeating unit | Reference |
|---|---|---|
| Group B Streptococcus type III capsular polysaccharide | →6)-[α-D-NeupNAc-(2→3)-β-D-Galp-(1→4)]-β-D-GlcpNAc-(1→3)-β-D-Galp-(1→4)-β-D-Glcp-(1→ | Conformational epitope of the type III group B Streptococcus capsular polysaccharide33 |
| Salmonella serogroup B O-antigen | →2)-[α-D-Abep-(1→3)]-α-D-Manp-(1→4)-α-L-Rhap-(1→3)-α-D-Galp-(1→ | Analysis by surface plasmon resonance of the influence of valence on the ligand binding affinity and kinetics of an anti-carbohydrate antibody36 |
| Francisella tularensis type A and B O-antigen | →2)-β-D-QuipN4Fm-(1→4)-α-D-GalpNAcAN-(1→4)-α-D-GalpNAcAN-(1→3)-β-D-QuipNAc-(1→54,55 | Characterization of monoclonal antibodies to terminal and internal O-antigen epitopes of Francisella tularensis lipopolysaccharide39 |
| Neisseria meningitidis serogroup C polysaccharide | →9)-α-D-NeupNAc(7/8OAc)-(2→ | Surface plasmon resonance analysis of antipolysaccharide antibody specificity: responses to meningococcal group C conjugate vaccines and bacteria44 |
| Neisseria meningitidis serogroup B capsular polysaccharide | →8)-α-D-NeupNAc-(2→ | High affinity binding of long-chain polysialic acid to antibody, and modulation by divalent cations and polyamines45 |
| Yersinia pestis triheptose motif of LPS inner core | L-α-D-Hepp-(1→7)-L-α-D-Hepp-(1→3)-L-α-D-Hepp | Epitope recognition of antibodies against a Yersinia pestis lipopolysaccharide trisaccharide component46 |
| Bacillus anthracis secondary cell wall polysaccharide | →6)-[α-D-Galp-(1→3)]-[β-D-Galp-(1→4)]-α-D-GlcpNAc-(1→4)-β-D-ManpNAc-(1→4)-[α-D-Galp-(1→3)]-β-D-GlcpNAc-(1→ | Endolysins of Bacillus anthracis bacteriophages recognize unique carbohydrate epitopes of vegetative cell wall polysaccharides with high affinity and selectivity47 |
| The secondary cell wall polysaccharide of Bacillus anthracis provides the specific binding ligand for the C-terminal cell wall-binding domain of two phage endolysins, PlyL and PlyG48 | ||
| Escherichia coli O157 O-antigen | →4)-β-D-Glcp-(1→3)-α-D-GalpNAc-(1→2)-α-D-PerpNAc-(1→3)-α-L-Fucp-(1→56 | Detection of Escherichia coli O157:H7 using a surface plasmon resonance biosensor49 |
SPR technology has been successfully applied to support the identification and characterization of the conformational epitope of the type III polysaccharide from Streptococcus agalactiae, also known as Group B Streptococcus (GBS), recognized by specific protective mAb elicited by vaccination.33 GBS is a Gram-positive encapsulated bacterium which is a major cause of bacterial sepsis, and meningitis in neonates. GBS bacteria express capsular polysaccharides with complex branched structures but having close structural similarities. Variations in the structure correspond to serological differences, which are used for strain classification. At present, ten serotypes (Ia, Ib and II–IX) have been identified, all sharing a common sialic acid unit as the terminal branched chain.34 Zou et al. (1999) reported that the protective epitope of GBS type III was length dependent and conformational.33 SPR experiments were conducted with a large panel of oligosaccharide probes and a specific anti-GBS type III mAb 1B1 and its Fab fragment. The results of this work were that for GBS type III, two repeating units were the minimum required for Fab binding and that, while an increase from 2 to 7 repeating units produced additional optimization of the epitope, it remained monovalent. These data support the hypothesis that the conformational epitope of GBS type III is represented by an extended helical structure and that, although not prevalent and discontinuous, the immune system preferentially selects the helical epitope because it is unique to the polysaccharide. In order to elucidate the binding capacity of different length oligosaccharides by SPR, GBS type III fragments from 1 to 20 repeating units were conjugated to HSA by reductive amination with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 protein loading on oligos. A cross-linked material was produced when longer chains (from 25 to 80 repeating units) were randomly activated. Oligosaccharide–HSA conjugates were immobilized, and the 1B1 Fab fragment showed a good fitting to a first order interaction model. From 2 to 7 repeating units the KD was virtually constant (approximately 4.1 μM). A 3-fold increase in affinity was measured between 7 and 20 repeating units, due to both optimization of the conformational task and the occurrence of epitope multivalency; further increases in chain length did not significantly improve the binding. Epitope multivalency was studied by a saturation experiment which provided the sensor capacity and allowed for the calculation of binding stoichiometry. This experiment indicated a Fab-antigen binding stoichiometry of 1:1 using surfaces immobilized with 2 or 6–7 repeating units, and a binding stoichiometry of 3:1 for the 20 repeating units, suggesting that multivalency occurs above 6–7 repeating units. The variability and uncertainty of the ratio of monovalent and bivalent binding occurring at the double branches of mAb 1B1 provided only a relative measure of the binding avidity. Since avidity effects are associated with an increase of ka rates and slower kd rates due to re-binding events, IgG 1B1 dissociation rates slower than those of Fab dissociation rates were observed in the binding with all conjugates.33
:1 protein loading on oligos. A cross-linked material was produced when longer chains (from 25 to 80 repeating units) were randomly activated. Oligosaccharide–HSA conjugates were immobilized, and the 1B1 Fab fragment showed a good fitting to a first order interaction model. From 2 to 7 repeating units the KD was virtually constant (approximately 4.1 μM). A 3-fold increase in affinity was measured between 7 and 20 repeating units, due to both optimization of the conformational task and the occurrence of epitope multivalency; further increases in chain length did not significantly improve the binding. Epitope multivalency was studied by a saturation experiment which provided the sensor capacity and allowed for the calculation of binding stoichiometry. This experiment indicated a Fab-antigen binding stoichiometry of 1:1 using surfaces immobilized with 2 or 6–7 repeating units, and a binding stoichiometry of 3:1 for the 20 repeating units, suggesting that multivalency occurs above 6–7 repeating units. The variability and uncertainty of the ratio of monovalent and bivalent binding occurring at the double branches of mAb 1B1 provided only a relative measure of the binding avidity. Since avidity effects are associated with an increase of ka rates and slower kd rates due to re-binding events, IgG 1B1 dissociation rates slower than those of Fab dissociation rates were observed in the binding with all conjugates.33
Avidity effects caused by multiple affine anti-carbohydrate antibodies were detected in preparations of single chain antibody variable domains (scFv) containing trace amounts of aggregates. SPR analyses of these preparations showed complex biphasic binding profiles.35 These data were reinterpreted in a subsequent study in which the kinetics of the LPS OAg of Salmonella serogroup B bacteria binding wild type Fab and scFv monomers, as well as dimeric mutant scFv, all derived from the specific antibody Se155–4, were studied in detail by SPR.36 The recognition of Se155–4 was reported to be controlled by a 3,6-dideoxyhexose (abequose moiety) epitope on the O-polysaccharide.37 For SPR analysis the antigen was immobilized as BSA conjugate, and the binding profiles of Se155-4 IgG, Fab and scFv monomers and dimers purified by size-exclusion HPLC were compared. The whole panel of dissociation kinetics was recorded in the presence of the free abequose trisaccharide with the aim of minimizing antibody rebinding by the competition with immobilized OAg for the release of both forms of scFv. The kd values of all monomers in the presence of the free trisaccharide were rapid, approximately 0.25 s−1. Equilibrium binding data were estimated to be approximately 6 μM for monomeric Se155-4 Fab and wild type scFv. The authors reported that divalency decreases the dissociation rates by approximately 20-fold, with the faster kd comparable to pure monomers, whilst increasing the association rates by 5-fold. The scFv dimers showed good curve fitting to a first order interaction model for association phases, while the dissociation phases were biphasic as a result of both monovalent and bivalent attachment of dimers to the surface, in particular at antibody concentrations producing signals near Rmax due to a higher monovalent accessibility. Bivalent interactions are always more difficult to characterize since the total binding energy is not purely the sum of the two single binding components but also includes an interaction energy term.38 The manuscript of MacKenzie et al.36 represented the first comprehensive effort to characterize the kinetics of a protein–carbohydrate complex using SPR as analytical tool, demonstrating its usefulness for investigating low affinity interactions. Additionally, the authors emphasized the need to address particular attention to the interpretation of SPR data for protein–sugar systems. The repetitive structure of most carbohydrate ligands can lead to incorrect data fitting if bivalent and multivalent interactions are not recognized, thus overestimating the intrinsic affinity of the interaction.
Roche et al. (2011) applied SPR to gain information about the specificity and avidity of protective antibodies against Francisella tularensis (Ft) LPS.39 Ft is the Gram negative bacterium that causes tularemia; two subspecies, type A and type B, are responsible for most cases of human disease. Both types A and B share a common LPS structure formed by an OAg with four sugar units, connected at its reducing end to a core oligosaccharide, which in turn is coupled to lipid A at its reducing end. LPS was ascribed as the main protective antigen eliciting protecting anti-Ft LPS IgG2a mAbs in mice following intranasal lethal infection with the Ft live vaccine strain.40 Roche et al.39 compared the binding characteristics of four IgG2a anti-Ft LPS mAbs and showed that all four were specific for the OAg of Ft LPS. In particular several orthogonal techniques (SPR, western blots, ELISA, immunoprecipitation) supported the finding that three of the antibodies were specific for internal repeating epitopes, while the fourth presented higher avidity to a unique terminal epitope of Ft OAg. SPR analysis was performed using two experimental designs. In the first, mAbs were immobilized on anti-mouse IgG coated sensor chips and probed with Ft OAg. In the other, one purified mAb was immobilized as in the first assay and used to capture Ft LPS; the chip was then probed with three out of the four anti-Ft OAg mAbs. The first assay format was suitable for determining the affinity of the end-binding mAb FB11 (KD = 0.4 μM) but not to probe the strongest internal-binding mAb Ab52, due to multivalent interactions with multiple epitopes on each OAg molecule. The KD for this multivalent interaction was determined as 4.4 × 10−13 M, indicating an essentially irreversible reaction. The second format was used to compare the bivalent avidity of the end-binding and internal-binding Ft OAg mAbs. FB11 was determined to have the highest avidity (KD = 5.0 × 10−11 M) and the order of avidity of the other three mAbs correlated with their potency in ELISA and western blot. SPR analysis demonstrated that non-overlapping epitopes of Ft OAg can generate different types of antibodies, i.e., end-binding mAbs had higher bivalent avidity for Ft OAg compared to the internal-binding mAbs. This suggested that antibodies targeting terminal epitopes of Ft OAg were more difficult to induce but might have higher avidity and have greater utility in defending against Ft.39 Since end-binding anti-Ft LPS antibodies recognized shorter sugar chains compared to internal-binding mAbs, the authors inferred that this observation could have a significant role in the design of vaccines and immunotherapies against tularemia.
Nm bacteria are one of the main causes of bacterial meningitis in children and young adults worldwide. There are 13 known Nm serogroups, differing in the capsular polysaccharide structure, five of which are responsible for the majority of cases of infection, namely serogroups A, B, C, W-135, and Y. The meningococcal group C capsular polysaccharide (MenC) is a linear homopolymer of α(2→9)-linked sialic acid residues that are O-acetylated (OAc+) at carbons in position 7 or 8.41 Generally infections occur with OAc+ strains, although fatal infection with de-O-acetylated (OAc−) strains can also occur. It has been seen that the presence or absence of OAc groups generates unique epitopes and specificities of antibodies elicited by vaccination with MenC glycoconjugates, affecting bactericidal activity.42,43 By means of SPR, García-Ojeda et al. (2004) have determined the binding and apparent dissociation half-times for mAbs specific to different MenC epitopes and for polyclonal antibodies (pAb) from sera of mice immunized with different MenC–CRM197 or –TT conjugate vaccines or fixed bacteria.44 MenC OAc− or OAc+–BSA conjugates were immobilized on sensor chips (about 243 RU immobilized in both sensors) and antibodies were injected over the sensor surfaces. It is better to immobilize the polysaccharide linked to a carrier different from that used in the immunization to avoid the risk of detecting binding events specific for the carrier and/or the linker between the carrier and the polysaccharide. In these experiments, in addition to the bivalent nature of mAbs, probing pAbs resulted in an even more complicated modeling of equilibrium and kinetic binding data, since the sensorgrams of polyclonal sera were the sum of a number of different pAb interactions, and the exact concentrations of the pAbs were unknown. The authors of this paper evaluated the global kinetics of antibodies using various models, including those specific for bivalency or heterogeneous ligands. However, the fittings were not overly convincing and the binding valency was ambiguous. Therefore the authors modeled the dissociation step in a concentration-independent exponential decay for both mAb and pAb binding to MenC. The kinetic values were thus evaluated calculating the apparent binding half-life of the pAb using this modeling. The comparison of the apparent half-life values was performed using either the Student t-test or the Tukey–Kramer honestly significant difference (HSD) test for multiple evaluations. The overall SPR apparent binding half-life of anti-MenC mAb calculated using exponential decay modeling correlates with binding patterns seen by using the fluorescence enzyme-linked immunosorbent assay (FELISA). The polyclonal immune sera showed apparent dissociation half-lives similar to or longer than those of the lower-avidity mAbs and shorter half-lives than the high-avidity mAb, suggesting that mAbs may have higher avidity than the conjugate vaccine immune serum pAbs. However, the authors suggested the need to develop more sophisticated modeling methods for pAb binding since this could increase the value derived from evaluation of immune sera. The SPR assay detected a greater avidity of sera from mice immunized with conjugate vaccines than sera from mice immunized with fixed bacteria. In addition, it was observed that conjugate vaccines generated relatively high-avidity pAbs which reacted with both OAc+ and OAc− antigens suggesting the presence of a critical shared or overlapping epitope recognized by all the conjugate vaccine immune sera.
The meningococcal group B capsular polysaccharide (MenB) is similar to the MenC polysaccharide being composed of α(2→8)-linked polysialic acid (PSA). Häyrinen et al. (2002) probed the interactions of PSA of different degrees of polymerization with a mouse anti-MenB mAb using SPR.45 The results showed that antibody binding affinity was dramatically affected by PSA chain length demonstrating a progressively higher affinity for large polymers. Long chain (approximately 200 sialic acid residues) native MenB polysaccharides were found to have a dissociation constant of about 0.9 nM, as the net avidity due to mono- or polyvalent binding, describing the biological effects with all interactions. This paper further suggested that divalent cations, such as Ca2+, could have a positive effect on the stabilization of the PSA conformational epitope.
A recent paper describes the combination of SPR with two complementary techniques, synthetic glycan array screening and STD-NMR (Saturation Transfer Difference-Nuclear Magnetic Resonance) to facilitate rationale design of carbohydrate antigens that generate high affinity antibodies.46 In this work, antibodies against Yersinia pestis LPS were elicited with the triheptose motif representing the LPS inner core of the antigen. Epitope recognition was elucidated using the three orthogonal techniques. For the SPR experiments, mAbs were captured by α-mouse IgG antibody and oligosaccharide haptens were passed over the captured mAbs. These antibodies were specific for the LPS of Y. pestis. No significant binding to LPS of other Gram-negative bacteria was observed suggesting that the epitopes were characteristic of the particular bacterial species.
3.4. Other applications
Some other recently published applications of SPR investigating the selectivity of bacterial carbohydrate interactions are reported in this paragraph.Two recent papers have utilized SPR to monitor binding of two endolysins, PlyL and PlyG, from bacteriophages of Bacillus anthracis to a range of oligosaccharides derived from the secondary cell wall polysaccharides of B. anthracis.47,48 Endolysins are enzymes produced by bacteriophages (viruses that infect and replicate within bacteria) to hydrolase their bacterial host for phage progeny release during the final stage of infection. In both papers, B. anthracis PlyL and PlyG lysins were immobilized on biosensors using standard amine coupling procedures. Titration experiments were performed with low molecular weight oligosaccharides, which made the analysis challenging due to the small refractive index detection that it is possible to achieve in such cases. Both papers agreed that PlyL and PlyG endolysins interacted with high affinity with compounds having a specific galactosydic pattern, binding with dissociation constants in the μM range. These findings demonstrated the specificity of endolysins to bind selectively to secondary cell wall polysaccharide structures.
Traditional methods for the detection of pathogenic bacteria involve time consuming microbiological culture, followed by bacterial isolation and identification. In recent years, much faster methods for pathogen recognition have been developed such as immunological assays and methods based on nucleic acid probes. Escherichia coli is one of the food-borne pathogenic bacteria whose infection is a major concern worldwide. Whole bacterial cells of E. coli strain O157:H7 have been monitored by SPR in antibody binding experiments.49 In addition, Wang and co-authors (2013) recently reported a first attempt to detect E. coli O157:H7 by SPR using lectins.50 These are plant or animal proteins (or glycoproteins), from non-immune origin, which reversibly bind to specific carbohydrate structural epitopes. Lectins are sensitive tools for the recognition and mapping of carbohydrate-containing molecules and for the characterization of carbohydrate specific interactions with proteins.51,52 Wang et al. selected five different lectins from Triticum vulgaris (WGA), Canavalia ensiformis (ConA), Ulex europaeus (UEA), Arachis hypogaea (PNA), and Maackia amurensis (MAL). These were chosen according to their specific affinity to the main classes of sugar structures and employed to screen the optimal complex formation with E. coli O157:H7 strain. Following activation of the sensor chip surface with NHS/EDC chemistry, lectins were immobilized through amide coupling and the different biosensors were tested. The authors reported that, taken together, the five lectins could discriminate the E. coli O157:H7 strain from other bacteria such as E. coli DH 5α and L. monocytogenes. The ability to correlate SPR response with the concentration of E. coli O157:H7 in real food samples was also tested although analyses were complicated by the high complexity of the matrix.
3.5. Why is SPR the method of choice to study protein–polysaccharide interactions?
Conventional analytical techniques for screening and measurements of carbohydrate-specific interactions comprise different approaches like immunoassays, chromatographic methods, glycan microarrays and flow cytometry assays. These techniques are highly sensitive and effective for the characterization of carbohydrate–protein interactions; however, they are often laborious and time-consuming, require labeling of the probe before the detection and provide only limited information about the binding affinity. For instance, the enzyme-linked immunosorbent assay (ELISA) does not require a large amount of reagents and samples and can be used for broad sample screening to discriminate between good and poor binders; however, detailed information regarding binding kinetics is lacking.The SPR technique does not require labeling of the probe in order to carry out the detection and enables real-time analyses of biomolecular events providing accurate kinetic parameters such as ka and kd and relative ranking e.g. of mAbs. In addition, SPR instruments allow simple assay design, require small amount of sample, and can be used for high throughput screening as the ELISA. SPR is a non-destructive technique; therefore, the same sample can be further analyzed by coupling SPR with a complementary technique to provide additional insights into the composition of the analyte.
4. Conclusions
Methods for monitoring specific interactions between bacterial carbohydrate antigens and antibodies are crucial to detect active immunization against selected pathogens. An understanding of the structural features of the conformational epitope recognized by neutralizing antibodies can offer critical insights into the selection and design of polysaccharide antigens.SPR has been revealed as a potent technology for the characterization of the binding of polysaccharide antigens with antibodies and proteins involved in bacterial cell wall recognition and for its capability to measure reaction rate constants and affinity in real-time. In particular, SPR studies have also supported the elucidation of antigenic conformational epitopes.
Careful design and data processing of SPR experiments can dramatically improve the quality of biosensor data. For instance, the molecule and the chemistry used for immobilization should not alter the binding sites, for example, carbohydrates are often immobilized as glycoconjugates. Optical biosensor technology offers a wide variety of sensor chip surfaces with functional groups suitable for an extensive array of applications. However, sensor chips having exposed NH2 groups, and lacking negatively charged matrices, will strongly facilitate SPR characterization of polysaccharide antigens. We believe that such biosensors could have a positive impact in vaccine candidate research and development, since most polysaccharides used for vaccines are negatively charged and are therefore repelled by carboxylated groups exposed on most commonly available sensors.
The quality of carbohydrate-based antigens can be better controlled using their physic/immuno-chemical properties. Indeed, this concept is accepted by the scientific community and explicitly stated in a WHO technical report: ‘Polysaccharide vaccines consist of defined chemical entities and, when prepared to the same specifications, are expected to have comparable potencies, regardless of the manufacturer’.53 Therefore the polysaccharide antigen can be fully described by testing the identity, purity, and molecular size.27 Identity, structural conformity and purity can be verified by a number of methodologies including in vitro immunological tests, colorimetric tests and spectroscopic technologies (i.e. NMR).
Over the recent years, SPR has made significant progress and is becoming an important technology for the development of immunological assays for in-depth carbohydrate antigen characterization.
Notes and references
- Handbook of Surface Plasmon Resonance, ed. R. B. M. Schasfoort and A. J. Tudos, Royal Society of Chemistry, London, 2008 Search PubMed.
- Real-time Analysis of Biomolecular Interactions, ed. K. Nagata and H. Handa, Springer-Verlag, Tokyo, 2000 Search PubMed.
- D. B. Werz and P. H. Seeberger, Chem.–Eur. J., 2005, 11, 3194–3206 CrossRef CAS PubMed.
- J. X. Huang, M. A. K. Azad, E. Yuriev, M. A. Baker, R. L. Nation, J. Li, M. A. Cooper and T. Velkov, J. Lipids, 2012, 475153 Search PubMed.
- C. M. Taylor and I. S. Roberts, Contrib. Microbiol., 2005, 12, 55–66 CAS.
- H. J. Jennings, E. Katzenellenbogen, C. Lugowski, F. Michon, R. Roy and D. L. Kasper, Pure Appl. Chem., 1984, 56, 893–905 CAS.
- J. R. Brisson, S. Uhrinova, R. J. Woods, M. van der Zwan, H. C. Jarrell, L. C. Paoletti, D. L. Kasper and H. J. Jennings, Biochemistry, 1997, 36, 3278–3292 CrossRef CAS PubMed.
- A. J. Pollard, K. P. Perrett and P. C. Beverley, Nat. Rev. Immunol., 2009, 9, 213–220 CrossRef CAS PubMed.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed.
- B. Kuberan and R. J. Linhardt, Curr. Org. Chem., 2000, 4, 653–677 CrossRef CAS.
- H. J. Jennings and R. A. Pon, in Microbial Glycobiology, ed. A. P. Moran, Elsevier Inc., 2009, pp. 933–956 Search PubMed.
- W. Zou and H. J. Jennings, in Carbohydrate-Based Vaccines and Immunotherapies, ed. Z. Guo and G.-J. Boons, John Wiley & Sons Inc., Hoboken, New Jersey, 2009, pp. 55–88 Search PubMed.
- G. Safina, Anal. Chim. Acta, 2012, 712, 9–29 CrossRef CAS PubMed.
- E. Stenberg, B. Persson, H. Roos and C. Urbanizcky, J. Colloid Interface Sci., 1991, 143, 513–526 CrossRef CAS.
- U. Jönsson, L. Fägerstam, B. Ivarsson, B. Johnsson, R. Karlsson, K. Lundh, S. Löfås, B. Persson, H. Roos, I. Rönnberg, S. Sjölander, E. Stenberg, R. Ståhlberg, C. Urbaniczky, H. Östlin and M. Malmqvist, BioTechniques, 1991, 11, 620–627 Search PubMed.
- R. L. Rich and D. G. Myszka, J. Mol. Recognit., 2011, 24, 892–914 CrossRef CAS PubMed.
- R. L. Rich and D. G. Myszka, Curr. Opin. Biotechnol., 2000, 11, 54–61 CrossRef CAS.
- C. L. Baird and D. G. Myszka, J. Mol. Recognit., 2001, 14, 261–268 CrossRef CAS PubMed.
- C.-C. Chang, N.-F. Chiu, D. S. Lin, Y. Chu-Su, Y.-H. Liang and C.-W. Lin, Anal. Chem., 2010, 82, 1207–1212 CrossRef CAS PubMed.
- C. R. MacKenzie and H. J. Jennings, Methods Enzymol., 2003, 363, 340–354 CAS.
- A. Weintraub, Carbohydr. Res., 2003, 338, 2539–2547 CrossRef CAS PubMed.
- D. F. Kelly, A. J. Pollard and E. R. Moxon, J. Am. Med. Assoc., 2005, 294, 3019–3023 CrossRef CAS PubMed.
- P. Costantino, R. Rappuoli and F. Berti, Expert Opin. Drug Discovery, 2011, 6, 1045–1066 CrossRef CAS PubMed.
- S. K. Mazmanian and D. L. Kasper, Nat. Rev. Immunol., 2006, 6, 849–858 CrossRef CAS PubMed.
- G. Ada and D. Isaacs, Clin. Microbiol. Infect., 2003, 9, 79–85 CrossRef CAS.
- J. Eskola, H. Peltola, A. K. Takala, H. Käyhty, M. Hakulinen, V. Karanko, E. Kela, P. Rekola, P.-R. Rönnberg, J. S. Samuelson, L. K. Gordon and P. H. Mäkelä, N. Engl. J. Med., 1987, 317, 717–722 CrossRef CAS PubMed.
- C. E. Frasch, Vaccine, 2009, 27, 6468–6470 CrossRef CAS PubMed.
- J. P. Carver, Pure Appl. Chem., 1993, 65, 763–770 CrossRef CAS.
- R. U. Lemieux, L. T. Delbaere, H. Beierbeck and U. Spohr, Ciba Found. Symp., 1991, 158, 231–248 CAS.
- N. M. Young, M. A. J. Gidney, B.-M. E. Gudmundsson, C. R. MacKenzie, R. To, D. C. Watson and D. R. Bundle, Mol. Immunol., 1999, 36, 339–347 CrossRef CAS.
- B. T. Houseman, E. S. Gawalt and M. Mrksich, Langmuir, 2003, 19, 1522–1531 CrossRef CAS.
- D. Serruto and R. Rappuoli, FEBS Lett., 2006, 580, 2958–2992 CrossRef PubMed.
- W. Zou, C. R. Mackenzie, L. Thérien, T. Hirama, Q. Yang, M. A. Gidney and H. J. Jennings, J. Immunol., 1999, 163, 820–825 CAS.
- M. J. Cieslewicz, D. Chaffin, G. Glusman, D. Kasper, A. Madan, S. Rodrigues, J. Fahey, M. R. Wessels and C. E. Rubens, Infect. Immun., 2005, 73, 3096–3103 CrossRef CAS PubMed.
- S.-J. Deng, C. R. MacKenzie, J. Sadowska, J. Michniewicz, N. M. Young, D. R. Bundle and S. A. Narang, J. Biol. Chem., 1994, 269, 9533–9538 CAS.
- C. R. MacKenzie, T. Hirama, S.-J. Deng, D. R. Bundle, S. A. Narang and N. M. Young, J. Biol. Chem., 1996, 271, 1527–1533 CrossRef CAS PubMed.
- D. R. Bundle, E. Eichler, M. A. J. Gidney, M. Meldal, A. Ragauskas, B. W. Sigurskjold, B. Sinnott, D. C. Watson, M. Yaguchi and N. M. Young, Biochemistry, 1994, 33, 5172–5182 CrossRef CAS.
- W. P. Jencks, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 4046–4050 CrossRef CAS.
- M. I. Roche, Z. Lu, J. H. Hui and J. Sharon, Hybridoma, 2011, 30, 19–28 CrossRef CAS PubMed.
- Z. Lu, M. I. Roche, J. H. Hui, B. Unal, P. L. Felgner, S. Gulati, G. Madico and J. Sharon, Immunol. Lett., 2007, 112, 92–103 CrossRef CAS PubMed.
- A. K. Bhattacharjee, H. J. Jennings, C. P. Kenny, A. Martin and I. C. P. Smith, J. Biol. Chem., 1975, 250, 1926–1932 CAS.
- G. Arakere and C. E. Frasch, Infect. Immun., 1991, 59, 4349–4356 CAS.
- F. Michon, C. H. Huang, E. K. Farley, L. Hronowski, J. Di and P. C. Fusco, Dev. Biol., 2000, 103, 151–160 CAS.
- P. A. García-Ojeda, S. Hardy, S. Kozlowski, K. E. Stein and I. M. Feavers, Infect. Immun., 2004, 72, 3451–3460 CrossRef PubMed.
- J. Häyrinen, S. Haseley, P. Talaga, M. Mühlenhoff, J. Finne and J. F. G. Vliegenthart, Mol. Immunol., 2002, 39, 399–411 CrossRef.
- F. Broecker, J. Aretz, Y. Yang, J. Hanske, X. Guo, A. Reinhardt, A. Wahlbrink, C. Rademacher, C. Anish and P. H. Seeberger, ACS Chem. Biol., 2014, 9, 867–873 CrossRef CAS PubMed.
- K.-F. Mo, X. Li, H. Li, L. Y. Low, C. P. Quinn and G.-J. Boons, J. Am. Chem. Soc., 2012, 134, 15556–15562 CrossRef CAS PubMed.
- J. Ganguly, L. Y. Low, N. Kamal, E. Saile, L. S. Forsberg, G. Gutierrez-Sanchez, A. R. Hoffmaster, R. Liddington, C. P. Quinn, R. W. Carlson and E. L. Kannenberg, Glycobiology, 2013, 23, 820–832 CrossRef CAS PubMed.
- P. M. Fratamico, T. P. Strobaugh, M. B. Medina and A. G. Gehring, Biotechnol. Tech., 1998, 12, 571–576 CrossRef CAS.
- Y. Wang, Z. Ye, C. Si and Y. Ying, Food Chem., 2013, 136, 1303–1308 CrossRef CAS PubMed.
- E. Duverger, N. Lamerant-Fayel, N. Frison and M. Monsigny, in Surface plasmon resonance: methods and protocols, (methods in molecular biology), ed. N. J. De Mol and M. J. E. Fischer, Springer, Utrecht, 2010, vol. 627, pp. 157–178 Search PubMed.
- K. Drickamer, Nat. Struct. Biol., 1995, 2, 437–439 CrossRef CAS PubMed.
- WHO, technical report N°897, Annex 1, 2000, pp.27–60.
- J. W. Conlan, H. Shen, A. Webb and M. B. Perry, Vaccine, 2002, 20, 3465–3471 CrossRef CAS.
- J. S. Gunn and R. K. Ernst, Ann. N. Y. Acad. Sci., 2007, 1105, 202–218 CrossRef CAS PubMed.
- M. B. Perry, L. MacLean and D. W. Griffith, Biochem. Cell Biol., 1986, 64, 21–28 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |