DOI:
10.1039/C4MD00067F
(Concise Article)
Med. Chem. Commun., 2014,
5, 1130-1137
Sulfonium ions as inhibitors of the mycobacterial galactofuranosyltransferase GlfT2†
Received
19th February 2014
, Accepted 11th March 2014
First published on 12th March 2014
Abstract
The mycobacterial cell wall possesses a core galactan moiety composed of approximately 30 galactofuranosyl residues attached via alternating β-(1→5) and β-(1→6) linkages. A bifunctional galactofuranosyltransferase, GlfT2, is one of two essential enzymes for mycobacterial cell wall biosynthesis. The enzymatic reactions catalyzed by GlfT2 undoubtedly proceed by way of a transition state that has significant oxocarbenium-ion character. In this paper, a series of sulfonium ion compounds were designed and synthesized as analogues of the donor substrate, uridine diphosphate-galactofuranose, as potential inhibitors of GlfT2. The compounds contain moieties that mimic both galactofuranose and uridine diphosphate domains, and carry a permanent positive charge to mimic the oxocarbenium ion-like transition state. These compounds were evaluated against Glf2 using a coupled spectrophotometric assay, and some were shown to be weak inhibitors of the enzyme.
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis, and other mycobacterial species possess a unique cell wall structure containing an array of carbohydrates and lipids.1 The major constituent of this complex architecture is the mycolyl–arabinogalactan (mAG) complex, which provides the organism with significant protection from its environment.2,3 A notable feature of the mAG complex is that all of the galactose and arabinose residues in the arabinogalactan (AG) domain are in the furanose ring form.4 Glycoconjugates containing furanose residues are absent in humans and thus the glycosyltransferases involved in AG biosynthesis are of interest as targets for new antibacterial agents.4–6 The pathway by which the mAG complex is assembled involves the sequential addition of sugar residues to a polyprenol bound intermediate by a number of glycosyltransferases.3 Among the enzymes in this pathway that have been biochemically-characterized,5,7,8 are two bifunctional galactofuranosyltransferases, GlfT1 and GlfT2. Both enzymes transfer galactofuranose (Galf) from uridine diphosphate-galactofuranose (UDP-Galf, 1), to an acceptor oligosaccharide via an oxacarbenium ion transition state (2) to form the elongated oligosaccharide and in the process liberate UDP (Scheme 1).
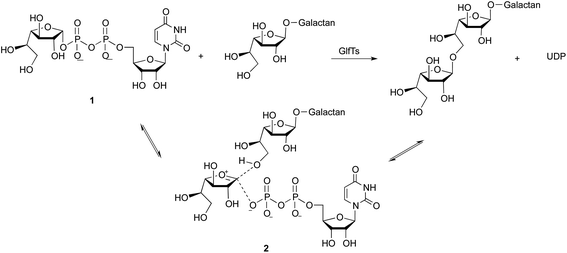 |
| | Scheme 1 Mycobacterial galactofuranosyltransferase-catalyzed galactan elongation. | |
Of these two galactofuranosyltransferases, GlfT2 has received the most investigation. The protein, an inverting glycosyltransferase, has been recombinantly expressed7,9 and shown to possess a single active site that is capable of carrying out two distinct glycosyl transfer reactions leading to either β-Galf-(1→5)-β-Galf or β-Galf-(1→6)-β-Galf linkages.10,11 The polymerase activity of GlfT2 proceeds via a processive mechanism12 and a recent crystal structure has revealed a novel tetrameric structure that led to a postulated model for chain length control.13 The design of effective inhibitors for GlfT2 has been limited to date, although this is an active area of investigation.14–20
A common strategy in developing inhibitors of enzymes that carry out glycosyl transfer reactions, in particular glycosidases, is to synthesize structures that mimic the positive character of the oxacarbenium ion transition state (2, Scheme 1).21,22 Iminosugars, which carry a positive charge at physiological pH, have been widely studied and used as inhibitors of glycosyltransferases and glycosidases.23 Another approach is to prepare molecules possessing a positively charged sulfur atom (sulfonium ions) to establish this electrostatic mimicry.24–28
Yuasa and coworkers developed the first carbohydrate-based sulfonium-ion derivative 324 (Fig. 1), as a β-glucosidase inhibitor. Since then, other carbohydrate-based sulfonium-ion derivatives were synthesized as inhibitors of various glycosidases. For example, sulfonium ion derivatives of the iminosugar glycosidase inhibitors swainsonine and castanospermine, 4 and 5, respectively, have been synthesized and evaluated for inhibitory activity against glycosidases.25,26 These studies were validated when two naturally-occurring sulfonium ion glycosidase inhibitors, salacinol (6)27 and kotalanol (7)28 were isolated in 1997. Both salacinol and kotalanol have significant inhibitory activity toward α-glucosidases.29,30 Due to their unique structural features and potential to become a lead drug candidate in the treatment of type II diabetes,31 an increasing amount of research on carbohydrate-based cyclic sulfonium compounds has since been carried out. Ponkoranol,32 salaprinol32 and de-O-sulfonated analogues33–35 were also obtained from Salacia genus plants. Their diasteriomers, nitrogen derivatives, selenium derivatives36–39 and maltose-extended analogues40 have been chemically synthesized and their biological activities evaluated.
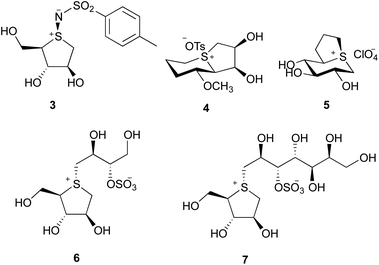 |
| | Fig. 1 Structures of some known sulfonium ion compounds. | |
Although sulfonium ions have been demonstrated to inhibit glycosidases, reports of their ability to inhibit glycosyltransferases are less common. Intrigued by the possibility of developing inhibitors of GlfT2 based upon a sulfonium ion scaffold, we designed a route to a suitable mimic of the postulated transition state in this glycosylation reaction. We describe here the synthesis of a panel of compounds with the general structure 8 (Scheme 2) and an evaluation of their ability to inhibit GlfT2.
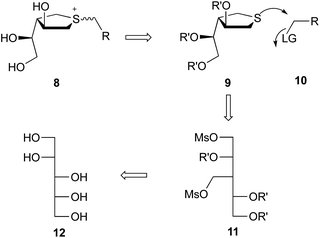 |
| | Scheme 2 Retrosynthetic analysis of sulfonium ions with general structure 8. | |
Results and discussion
Design considerations
Based on previous work on the synthesis of salacinol and derivatives29–39 we considered that the most direct route to the target sulfonium ions was through an intermolecular SN2 reaction between a cyclic sulfide (9, Scheme 2) and an appropriate electrophile (10). In selecting the cyclic sulfide scaffold to build the targets upon, it was necessary to choose a ring system in which both the ring oxygen and the hydroxyl group on C2 were removed to provide a stable species. Previous studies have demonstrated that GlfT2 will tolerate modification of the OH at C2 in UDP-Galf.14,40 The two substituents on the sulfide ring were designed to have the same stereochemistry as those of C3 and C4 on the galactofuranose ring. Therefore, we envisioned that 9 could be prepared by cyclization of the branched alditol derivative 11, which in turn could come from commercially available D-arabinitol (12). With regard to the electrophile, we chose to explore a number of simple alkyl halides, as well as more complex structures that could better mimic the uridine diphosphate portion of the postulated transition state structure.
Synthesis the cyclic sulfide
The synthesis of the cyclic sulfide was achieved in a nine-step sequence, starting from 12 (Scheme 3). First, reaction of 12 with 3,3-dimethoxypentane in the presence of camphorsulfonic acid afforded a diacetal intermediate, 13, which has the hydroxyl on C3 unprotected, in 72% yield. Oxidation of this hydroxyl group with SO3·pyridine complex in DMSO proceeded in 84% yield to give the C2-symmetric ketone 14, which was subsequently reacted with methyl triphenylphosphonium iodide and NaHMDS affording a 93% yield of olefin 15.41 Cleavage of the acetal protecting groups from 15 was achieved upon reaction with camphorsulfonic acid, providing compound 16 in 97% yield. This tetraol was then reacted with one equivalent of trityl chloride to produce the monoprotected compound 17 in 63% yield. The remaining hydroxyl groups were then protected as benzyl ethers by reaction of 17 with benzyl bromide and NaH to generate product 18 in 87% yield.
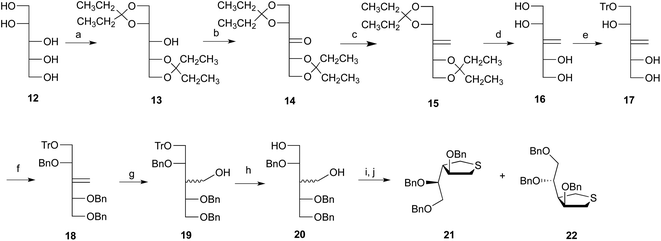 |
| | Scheme 3
Reagents and conditions: (a) 3,3-dimethoxypentane, CSA, THF, 72%; (b) SO3·Py, DMSO, 84%; (c) Ph3PCH3I, NaHMDS, 93%; (d) CSA, CH3OH 97%; (e) TrCl, DMAP, Et3N, DMF, 63%; (f) BnBr, NaH, DMF, 87%; (g) BH3·(CH3)2S, THF, H2O2, NaOH, 80%; (h) p-TSA, CH3OH, CH2Cl2, 96%; (i) MsCl, Et3N, CH2Cl2; (j) Na2S·9H2O, DMF, 2 steps 84%. | |
A hydroboration–oxidation sequence was explored to convert the alkene into a hydroxymethyl group. We initially used borane–dimethylsulfide (BH3·S(CH3)2) complex, which gave an organoborane that was subsequently oxidized to the stereoisomers 19 by treatment with H2O2 under basic conditions. The isomers, which could not be separated, were obtained in total yield of 80% in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4. To improve the stereoselectivity, other borane reagents (BH3·THF, BH3·pyridine, or BH3·NEt3) were examined, but none gave better results. More sterically-demanding hydroboration reagents, 9-BBN, disiamylborane and thexylborane, were also investigated. Unfortunately, the conversion of 18 into the corresponding organoborane was not successful when using these reagents.
:1.4. To improve the stereoselectivity, other borane reagents (BH3·THF, BH3·pyridine, or BH3·NEt3) were examined, but none gave better results. More sterically-demanding hydroboration reagents, 9-BBN, disiamylborane and thexylborane, were also investigated. Unfortunately, the conversion of 18 into the corresponding organoborane was not successful when using these reagents.
The stereoisomeric mixture of 19 was treated with p-toluenesulfonic acid, giving 20, also as an inseparable mixture of stereoisomers, in a combined yield of 96%. Treatment of the mixture of diols with methanesulfonyl chloride and triethylamine in CH2Cl2 produced the expected mesylated product, which was directly treated with sodium sulfide nonahydrate (Na2S·9H2O) in DMF at 100 °C to form, in 84% yield, a separable mixture of the cyclic sulfides 21 and 22, in a ratio of 1.4:1. In the ring forming reaction, it was necessary to heat the solution slowly to 100 °C, to allow the Na2S·9H2O to dissolve completely before the initiation of displacement. If heated quickly, dehydration of Na2S·9H2O occurs before Na2S dissolves in the solvent. It was found that substituted tetrahydrofuran side products 23 and 24 (Fig. 2) were generated from the reaction of 20 and when the reaction mixture was heated too rapidly.
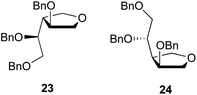 |
| | Fig. 2 Tetrahydrofuran byproducts 23 and 24. | |
It was not possible to determine the relative stereochemistry of two substituents on the cyclic thioethers 21 and 22 using NMR spectroscopy. In an effort to prove unequivocally the structures, we investigated the conversion of 21 and 22 into compounds that could be crystallized. Thus, 21 and 22 were oxidized by m-CPBA to give 25 and 26, which were subsequently hydrogenolyzed over Pd–C to form 27 and 28 in 92% yield over two steps (Scheme 4). Neither 27 nor 28 were crystalline. However, oxidation of 28 with NaIO4 produced an aldehyde that was condensed with 2,4-dinitrophenylhydrazine to give a yellow crystalline solid, 29. From the crystal structure of 29 (Fig. S1,† CCDC 986599), the two substituents on the ring were determined to be syn to each other. It can therefore be inferred that two substituents in 21 have an anti relationship. Thus, compound 21 has the “galactofuranose” configuration, and was used in the coupling reactions with alkyl iodides to prepare the sulfonium ion targets.
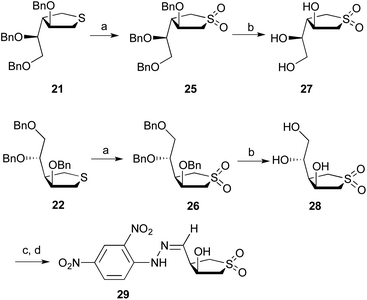 |
| | Scheme 4
Reagents and conditions: (a) m-CPBA, CH2Cl2, 94%; (b) H2, Pd–C, HOAc, CH3OH, 98%; (c) NaIO4, NaHCO3, THF, H2O; (d) 2,4-dibitrophenylhydrazine, CH3OH, 2 steps 30%. | |
Preparation of alkyl iodides
It has been reported previously that alkyl iodides37,42 and triflates43 are good electrophiles in the synthesis of sulfonium ions because the leaving groups are weakly nucleophilic anions, which reduce the possibility of decomposition of the sulfonium ion via nucleophilic substitution reactions.44 When these reactions are carried out with alkyl iodides in the presence of a silver salt (e.g., AgBF4), the iodine ion liberated in the reaction can be precipitated as AgI and thus is unable to act as a nucleophile. Based upon these reports, we chose alkyl iodides and bromides as the alkylating agents for the synthesis of the target molecules.
A range of alkyl iodides and bromides with diverse branching patterns were selected to prepare sulfonium ion analogues (Fig. 3). Compounds 30–33 contain linear or branched alkyl groups. 34 and 35 have an oxygen atom in the chain, which may form hydrogen-bonding interaction with the enzyme. Iodides 36–38 will provide the sulfonium ion analogs with a hydroxyl group after hydrogenolysis, which could also hydrogen bond with the enzyme. We also chose to synthesize sulfonium ion analogues containing benzylic groups from compounds 39–41. In addition, to mimic UDP-Galf, iodides containing a uridine moiety were prepared. In previous molecular modeling studies by van Boom and coworkers, a five-atom linker between the uridine and the sugar moiety was proposed to provide the required distance to span a pyrophosphate moiety.45 Thus, compounds 42–44, which contain a uridine moiety and five or six atoms separated from the iodide were selected. The synthesis of these alkyl halides can be found in the ESI (Scheme S1†).
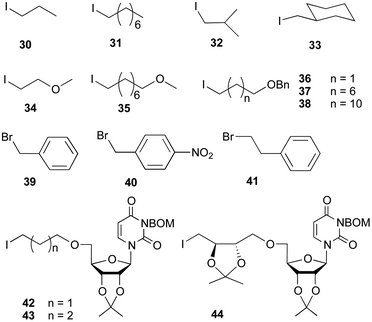 |
| | Fig. 3 Structures of halides 30–44. | |
Coupling and deprotection reactions
Reactions with simple alkyl halides.
With the cyclic sulfide and the various halides in hand, we explored their coupling to produce the targets (Schemes 5–7). These reactions were carried out as reported by Mohan et al.,37 by reacting the two substrates in CH3CN in the presence of AgBF4 at 65 °C to give the sulfonium ion tetrafluoroborate salt. It was necessary to carry out the coupling reaction under an argon atmosphere to minimize the formation of the sulfoxide byproduct (Fig. S2†), presumably resulting from air oxidation of the sulfide.
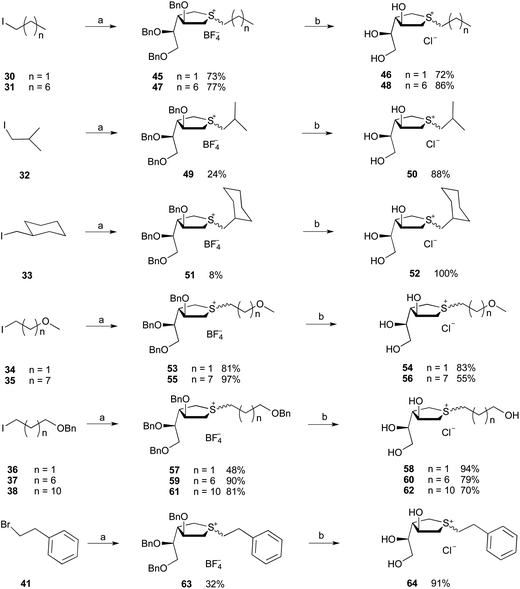 |
| | Scheme 5
Reagents and conditions: (a) 21, AgBF4, CH3CN, 65 °C; (b) BCl3, 1 M in CH2Cl2, then Amberlyst resin (Cl−). | |
The coupling reactions of 21 with the unhindered primary alkyl iodides (30, 31, 34, 35 and 36–38) gave the expected products in yields ranging from 48% to 97%. The more hindered alkyl iodides, 32 and 33, gave much poorer yield of 24% and 8%, respectively. Similarly, 1-bromo-2-phenylethane (41) was converted into compound 63 in modest (32%) yield. As shown, the coupling reactions gave mixtures of R/S stereoisomers, which was expected46–48 given the lack of substituents at the carbons adjacent to the sulfur. Such groups would be anticipated to influence the stereochemistry of the reaction. The ratios of stereoisomers ranged from 60:40 to 95:5, which were inseparable. Despite extensive efforts, determining the structure of the major isomer by NMR spectroscopy proved impossible, and none of the compounds were crystalline solids. Therefore, the products were characterized as mixtures. Subsequent, removal of the benzyl groups from the protected sulfonium ions was carried out upon treatment with boron trichloride (1 M in CH2Cl2) at −78 °C. The products were then treated with Amberlyst resin (Cl− form) to convert the tetrafluoroborate salt into the corresponding chloride salt. The final chloride salts 46, 48, 50, 52, 54, 56, 58, 60, 62 and 64 were obtained in 55–100% yield over this two-step transformation.
Reactions with benzylic halides.
An alternate approach was developed for the preparation of the sulfonium ion compounds from benzylic bromides 39 and 40, as the products would not be expected to be stable to BCl3-promoted debenzylation. Thus, as illustrated in Scheme 6, compound 21 was converted by Birch reduction into 65 in 90% yield. Subsequent reaction of 65 with benzyl bromide 39 in 1,1,1,3,3,3-hexafluoro-isopropanol37 at 50 °C afforded 66 in 57% yield. Using the same conditions, reaction of 65 with p-nitrobenzyl bromide 40 produced 67 in 58% yield.
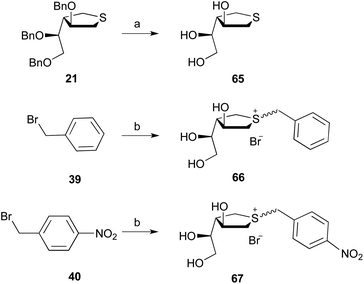 |
| | Scheme 6
Reagents and conditions: (a) Na, NH3, 90%; (b) 65, CF3CHOHCF3, 66:57%; 67:58%. | |
Reactions with uridine-derived iodides.
The preparation of analogues containing the uridine moiety is shown in Scheme 7. Reaction of 21 with iodides 42 and 43 under the same conditions as described above afforded 68 and 70, in 30% and 39% yield, respectively. Although the iodine was attached to linear alkyl group, the uridine-derived iodides showed substantially lower reactivity that the simple alkyl iodides 35 and 36. Treatment of 68 and 70 with BCl3 led to cleavage of all of the benzyl and BOM groups, as well as the isopropylidene acetal, to afford the expected products 69 and 71 in moderate yields of 55% and 54%, respectively. Iodide 44, which possess an isopropylidene acetal in the linker, proved extremely unreactive. Even after extended reactions times none of the desired substation product 72 was observed.
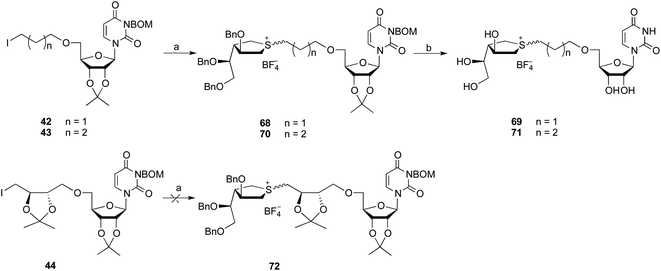 |
| | Scheme 7
Reagents and conditions: (a) 21, AgBF4, CH3CN, 65 °C, 68:30%; 70:39%; (b) BCl3, 1 M in CH2Cl2, then Amberlyst resin (Cl−), 69:55%; 71:54%. | |
Evaluation of sulfonium ions targets as inhibitors of GlfT2
The sulfonium ion analogues of UDP-Galf were investigated as potential inhibitors of GlfT2, using a reported coupled spectrophotometric assay.49,50 In these assays, an acceptor trisaccharide β-D-Galf-(1→5)-β-D-Galf-(1→6)-β-D-Galf-octyl and the donor substrate UDP-Galf (1) are incubated with a potential inhibitor. To obtain an indication of the potency of the various compounds, the analogues were initially screened at a concentration of 4 mM against the enzyme. The percentage activities compared to the no-inhibitor control are shown in Fig. 4. Under these conditions, most of the compounds show weak levels of inhibition against the enzyme, with the most potent compound, 62, leading to about a 60% inhibition of activity. Neither of the compounds possessing the uridine moiety, 69 or 71, demonstrated strong inhibition. Given the low levels of inhibition activity, additional studies to determine exact Ki values for these compounds were not carried out.
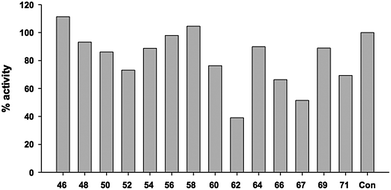 |
| | Fig. 4 Inhibition activity of the sulfonium ion analogues against GlfT2 at concentration of 4 mM. | |
Conclusions
In summary, we have synthesized a panel of sulfonium ions that were designed as potential inhibitors of the mycobacterial galactofuranosyltransferase GlfT2. The synthesis of the targets involved the preparation of a sulfide, 21, from D-arabinintol and its subsequent coupling with a range of alkyl halides followed by cleavage of the benzyl protecting groups under Lewis acidic conditions. The ability of these compounds to prevent GlfT2-mediated transfer of a galactofuranose residue to an acceptor reveled low levels of activity suggesting the limited potential of this class of compounds as inhibitors of mycobacterial AG biosynthesis.
Experimental section
Synthetic chemistry
General procedure for the preparation of sulfonium ions.
To a solution of the cyclic sulfide 21 (1.0 equiv.) in dry CH3CN (3 mL) was added the alkyl halide (1.0 equiv.) and AgBF4 (1.0 equiv.). The reaction mixture was covered with aluminum foil and stirred at 65 °C for 24 h. The mixture was then cooled to rt, concentrated and the resulting residue was purified by chromatography (toluene–CH3OH, 30:1) to give the sulfonium ion as mixture of isomers, which were inseparable.
General procedure for the deprotection of benzyl groups.
To a solution of benzylated sulfonium ion (1.0 equiv.) in CH2Cl2 (2 mL) was added BCl3 (1 M solution in CH2Cl2, 4 mL) at −78 °C under an Ar atmosphere. The mixture was stirred at −78 °C for 2 h and then warmed slowly to 0 °C and stirred for another 30 min. The solution was bubbled with air to remove the excess BCl3 and then was added CH3OH (3 mL). The solution was concentrated and the residue was coevaporated with CH3OH (2 × 5 mL). The deprotected product was then dissolved in CH3OH (5 mL) and stirred with ion exchange resin Amberlyst (Cl− form) for 2 h and filtered. The filtrate was concentrated and the resulting oil was passed through a short Iatrobead column (CH2Cl2–CH3OH 5:1 → 1:1) to give the target products.
(2S,4S)-3-Methylenepentane-1,2,4,5-tetraol (16).
A solution of 15 (1.88 g, 6.62 mmol) and CSA (0.14 g, 0.6 mmol) in CH3OH (12 mL) and CH2Cl2 (6 mL) was heated at reflux for 16 h, cooled to rt, neutralized with Et3N and then concentrated. The resulting residue was purified by chromatography (CH2Cl2–CH3OH 19:1 → 7:1) to give 16 as a colorless oil (0.95 g, 97%): Rf = 0.33 (CH2Cl2–CH3OH, 5:1); [α]20D +26.7 (c 1.0, CH3OH); 1H NMR (400 MHz, CD3OD): δ 5.28 (s, 2H), 4.14 (dd, J = 4.1, 7.3 Hz, 2H), 3.60 (dd, J = 4.1, 11.4 Hz, 2H), 3.48 (dd, J = 7.3, 11.4 Hz, 2H); 13C NMR (125 MHz, CD3OD): δ 151.1, 113.0, 74.4, 67.1; HRMS-ESI m/z [M + Na]+ calcd for C6H12O4Na: 171.0628, found: 171.0628.
(2S,4S)-3-Methylene-5-(trityloxy)pentane-1,2,4-triol (17).
Compound 16 (0.96 g, 6.49 mmol) and DMAP (0.08 g, 0.65 mmol) were dissolved in DMF (30 mL) and Et3N (1 mL). To this mixture was added a solution of trityl chloride (1.80 g, 6.49 mmol) in DMF (20 mL) dropwise. The reaction mixture was stirred overnight to give a cloudy solution, to which CH3OH (5 mL) was added. The solution was concentrated and the resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 30:1 → 5:1) to give 17 as an oil (1.60 g, 63%): Rf = 0.63 (CH2Cl2–CH3OH, 9:1); [α]20D +9.4 (c 1.9, CH2Cl2); 1H NMR (400 MHz, CD3OD): δ 7.47–7.44 (m, 6H), 7.31–7.26 (m, 6H), 7.23–7.19 (m, 3H), 5.24 (s, 1H), 5.20 (s, 1H), 4.24 (dd, J = 5.4, 5.7 Hz, 1H), 4.03 (dd, J = 3.5, 7.2 Hz, 1H), 3.52 (dd, J = 3.5, 11.4 Hz, 1H), 3.35 (dd, J = 7.2, 11.4 Hz, 1H), 3.21–3.14 (m, 2H), 2.95 (s, 1H), 2.84 (s, 2H); 13C NMR (100 MHz, CD3OD): δ 151.1, 145.5, 130.0, 128.8, 128.1, 112.7, 88.2, 74.3, 72.8, 69.2, 67.2; HRMS-ESI m/z [M + Na]+ calcd for C25H26O4Na: 413.1723, found: 413.1724.
(2S,4S)-3-Methylene-5-(trityloxy)-1,2,4-tribenzyloxypentane (18).
Compound 17 (94 mg, 0.24 mmol) and benzyl bromide (0.15 g, 0.88 mmol) were dissolved in DMF (3 mL) and cooled to 0 °C. To this mixture was added NaH (36 mg, 0.88 mmol, 60% in mineral oil) slowly and the solution was stirred for 1 h at 0 °C. The excess reagents were then quenched by the addition of H2O and the solution was extracted with EtOAc. The organic phase was washed with brine, dried (Na2SO4) and concentrated, and the resulting residue was purified by chromatography (EtOAc–hexane 1:15) to give 18 as an oil (0.14 g, 87%): Rf = 0.56 (EtOAc–hexane 1:4); [α]20D +27.5 (c 0.7, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.47–7.21 (m, 30H), 5.44 (s, 1H), 5.42 (s, 1H), 4.66 (d, J = 12.1 Hz, 1H), 4.51–4.35 (m, 4H), 4.21 (d, J = 12.1 Hz, 1H), 3.92 (dd, J = 3.1, 7.3 Hz, 1H), 3.84 (dd, J = 3.5, 7.3 Hz, 1H), 3.45 (dd, J = 7.3, 10.6 Hz, 1H), 3.38 (dd, J = 7.2, 10.4 Hz, 1H), 3.34 (dd, J = 3.5, 10.6 Hz, 1H), 3.10 (dd, J = 3.1, 10.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 144.2, 144.1, 138.7, 138.4, 138.3, 128.8, 128.4, 128.3, 128.2, 128.1, 127.7, 127.6, 127.5, 127.4, 127.3, 126.9, 115.3, 86.8, 79.1, 78.6, 73.3, 73.1, 70.9, 70.6, 66.9; HRMS-ESI m/z [M + Na]+ calcd for C46H44O4Na: 683.3131, found: 683.3131.
(2S,3S)-3,4-Bis(benzyloxy)-2-((S)-1-(benzyloxy)-2-(trityloxy)ethyl)butan-1-ol and (2R,3S)-3,4-bis(benzyloxy)-2-((S)-1-(benzyloxy)-2-(trityloxy)ethyl)butan-1-ol (19).
To a solution of 18 (4.33 g, 6.56 mmol) in THF (30 mL) at 0 °C was added BH3·S(CH3)2 (10.0 mL, 19.8 mmol, 2 M in THF), and then the solution was warmed to rt and stirred for 16 h under argon. The solution was then added dropwise to a mixture of H2O–THF–NaOH (2M)–H2O2 (30%) (1:1:3:1.5, 20 mL) at 0 °C and stirred for 1 h. The mixture was extracted with EtOAc, and the organic layer was washed with brine, dried (Na2SO4) and concentrated. The residue was purified by chromatography (EtOAc–hexane 1:6) to give a inseparable mixture of stereoisomers 19 with ratio 60:40, as as a colorless oil (3.60 g, 80%): Rf = 0.34 (EtOAc–hexane 1:4); HRMS-ESI m/z [M + Na]+ calcd for C46H46O5Na: 701.3238, found: 701.3240. The compound was used in the next reaction without further characterization.
(2S,3R)-2-(Benzyloxy)-3-((S)-1,2-bis(benzyloxy)ethyl)butane-1,4-diol and (2S,3S)-2-(benzyloxy)-3-((S)-1,2-bis(benzyloxy)ethyl)butane-1,4-diol (20).
To a solution of 19 (3.51 g, 5.18 mmol) in CH2Cl2 (10 mL) and CH3OH (10 mL) was added p-TsOH (0.35 g, 1.84 mmol). The mixture was stirred for 3 h, neutralized with Et3N and concentrated. The resulting residue was purified by chromatography (EtOAc–hexane 1:2) to give inseparable stereoisomers 20 as a colorless oil (2.16 g, 96%): Rf = 0.29 (EtOAc–hexane 1:1); 1H NMR (500 MHz, CDCl3): δ 7.39–7.22 (m, 15H), 4.71 (d, J = 11.5 Hz, 0.6H), 4.68 (d, J = 11.5 Hz, 0.4H), 4.63–4.52 (m, 3H), 4.42 (d, J = 11.5 Hz, 0.8H), 4.37 (d, J = 11.6 Hz, 0.6H), 4.34 (d, J = 11.6 Hz, 0.6H), 4.12 (ddd, J = 2.8, 5.3, 5.3 Hz, 0.6H), 3.98 (dd, J = 4.8, 8.6 Hz, 0.4H), 3.95–3.64 (m, 7H), 2.62 (br, 2H), 2.12 (dddd, J = 3.6, 3.6, 5.2, 7.2 Hz, 0.4H), 2.04 (dddd, J = 2.9, 2.9, 4.2, 5.9 Hz, 0.6H); 13C NMR (125 MHz, CDCl3): δ 138.3, 138.1, 138.0, 137.9, 128.6, 128.5, 128.4, 128.0, 127.9, 127.7, 127.8, 127.6, 79.2, 78.0, 77.8, 76.9, 73.6, 73.4, 73.0, 72.9, 72.0, 71.7, 71.6, 69.7, 63.4, 60.8, 60.5, 60.1, 44.5, 43.8; HRMS-ESI m/z [M + Na]+ calcd for C27H32O5Na: 459.2142, found: 459.2140.
(3R,4S)-3-(Benzyloxy)-4-((S)-1,2-bis(benzyloxy)ethyl)tetrahydrothiophene (21) and (3R,4R)-3-(benzyloxy)-4-((S)-1,2-bis(benzyloxy)ethyl)tetrahydrothiophene (22).
To a solution of isomers 20 (1.85 g, 4.24 mmol) and Et3N (2 mL) in dry CH2Cl2 (40 mL) at −30 °C was added methanesulfonyl chloride (0.90 mL, 10.32 mmol). The reaction mixture was warmed to 0 °C steadily and stirred for 1 h and the excess reagent was quenched by the addition of ice. The organic layer was washed with a saturated aqueous NaHCO3 solution (3 × 10 mL), followed by brine, and then dried (Na2SO4) and concentrated. The residue was coevaporated twice with toluene and dissolved in dry DMF (30 mL). To this solution was added Na2S·9H2O (1.25 g, 5.21 mmol) and the mixture was heated at 100 °C for 2 h. After being cooled to rt, the mixture was diluted with Et2O and the organic layer was washed with H2O (3 × 10 mL), followed by brine and then dried (Na2SO4) and concentrated. The resulting residue was purified by chromatography (EtOAc–hexane 1:10) to give 21 (0.90 g) and 22 (0.64 g) (1.4:1, 84% in total) both as yellowish oils: (21) Rf = 0.40 (EtOAc–hexane 1:6); [α]20D −95.2 (c 1.6, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ = 7.37–7.26 (m, 15H), 4.74 (d, J = 11.7 Hz, 1H), 4.55–4.49 (m, 3H), 4.45 (d, J = 11.7 Hz, 1H), 4.38 (d, J = 11.7 Hz, 1H), 4.07 (ddd, J = 6.3, 7.9, 7.9 Hz, 1H), 3.86 (ddd, J = 5.0, 5.0, 5.0 Hz, 1H), 3.60 (dd, J = 5.0, 10.1 Hz, 1H), 3.56 (dd, J = 5.0, 10.1 Hz, 1H), 3.04–3.96 (m, 2H), 2.84–2.74 (m, 2H), 2.42 (dddd, J = 3.7, 7.9, 8.3, 9.2 Hz, 1H); 13 C NMR (125 MHz, CDCl3): δ 26.9, 33.8, 50.2, 72.2, 72.3, 73.2, 73.7, 76.0, 82.0, 127.5, 127.6, 127.7, 127.8, 128.3, 128.4, 138.1, 138.7; HRMS-ESI m/z [M + Na]+ calcd for C27H30O3SNa: 457.1808, found: 457.1805. (22) Rf = 0.33 (EtOAc–hexane 1:6); [α]20D −76.2 (c 0.37, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.37–7.26 (m, 15H), 4.71 (d, J = 11.3 Hz, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.59 (d, J = 11.8 Hz, 1H), 4.55–4.53 (m, 2H), 4.45 (d, J = 11.3 Hz, 1H), 4.38 (d, J= 1.6 Hz, 1H), 3.92 (ddd, J = 2.9, 4.2, 10.0 Hz, 1H), 3.73 (dd, J = 2.9, 10.6 Hz, 1H), 3.48 (dd, J = 4.2, 10.6 Hz, 1H), 3.14–3.08 (m, 1H), 2.94–2.90 (m, 1H), 2.81–2.74 (m, 2H), 2.45 (dddd, J = 3.0, 7.3, 10.0, 12.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 138.7, 138.4, 138.1, 128.4, 128.3, 127.7, 127.6, 127.5, 81.1, 77.4, 73.5, 72.3, 70.9, 70.7, 51.9, 34.8, 30.4; HRMS-ESI m/z [M + Na]+ calcd for C27H30O3SNa: 457.1808, found: 457.1810.
Evaluation of inhibitory activity
Inhibition activity test.
Solutions of KCl (2 M), MgCl2 (1 M), and MOPS (1 M, pH 7.6) were prepared in de-ionized distilled (MilliQ, MQ) water, filtered and stored at 4 °C. Recombinant GlfT2, prepared and stored as previously reported were used in the assay.7 On the day of experiment, donor analogues were reconstituted in filtered MQ water to give a 32 mM stock. Solutions of 15 mM NADH, 5 U mg−1 PK, 16.8 U mg−1 LDH, and 40 mM UDP-Galf were prepared in 50 mM MOPS (pH 7.6); 100 mM PEP was prepared in 250 mM MOPS (pH 7.6); 40 mM trisaccharide was prepared in filtered MQ water. All solutions were stored on ice during operation. Reactions to screen the ability of analogues to inhibit GlfT2 were initiated with the addition of GlfT2 (0.5 μg) to assays to give a final volume of 40 μL containing 50 mM MOPS (pH 7.6), 50 mM KCl, 20 mM MgCl2, 1.1 mM NADH, 3.5 mM PEP, 7.5 U PK, 16.8 U LDH, 2 mM trisaccharide acceptor, 4 mM analogues and 0.75 mM donor UDP-Galf. Reactions were incubated at 37 °C and monitored at 340 nm at 10–15 s intervals for 20 min using a Spectra Max 340 PC microplate reader. The inhibition screening assays were repeated at two-times linking enzyme levels (15 U PK and 33.6 U LDH), to rule out inhibition of the linking enzymes by the analogues.
Acknowledgements
This work was supported by the Alberta Glycomics Centre and the Natural Sciences and Engineering Research Council of Canada. JL was supported by a fellowship from Alberta Innovates Health Solutions.
References
- P. J. Brennan and H. Nikaido, Annu. Rev. Biochem., 1995, 64, 29–63 CrossRef CAS PubMed.
- L. G. Dover, A. M. Cerdeno-Tarraga, M. J. Pallen, J. Parkhill and G. S. Besra, FEMS Microbiol. Rev., 2004, 28, 225–250 CrossRef CAS PubMed.
- S. Berg, D. Kaur, M. Jackson and P. J. Brennan, Glycobiology, 2007, 17, 35R–56R CrossRef CAS PubMed.
- T. L. Lowary, Mini-Rev. Med. Chem., 2003, 3, 689–702 CrossRef CAS PubMed.
- M. R. Richards and T. L. Lowary, ChemBioChem, 2009, 19, 1920–1938 CrossRef PubMed.
- J. S. Blanchard, Annu. Rev. Biochem., 1996, 65, 215–239 CrossRef CAS PubMed.
- N. L. Rose, G. C. Completo, S. Lin, M. R. McNeil, M. M. Palcic and T. L. Lowary, J. Am. Chem. Soc., 2006, 128, 6721–6729 CrossRef CAS PubMed.
- C. Breton, L. Snajdrova, C. Jeanneau, J. Koca and A. Imberty, Glycobiology, 2006, 16, 29R–37R CrossRef CAS PubMed.
- L. Alderwick, L. Dover, N. Veerapen, S. Gurcha, L. Kremer, D. Roper, A. Pathak, R. Reynolds and G. Besra, Protein Expression Purif., 2008, 58, 332–341 CrossRef CAS PubMed.
- M. G. Szczepina, R. B. Zheng, G. C. Completo, T. L. Lowary and B. M. Pinto, ChemBioChem, 2009, 10, 2052–2059 CrossRef CAS PubMed.
- J. F. May, M. R. Levengood, R. A. Splain, C. D. Brown and L. L. Kiessling, Biochemistry, 2012, 51, 1148–1159 CrossRef CAS PubMed.
- M. R. Levengood, R. A. Splain and L. L. Kiessling, J. Am. Chem. Soc., 2011, 133, 12758–12766 CrossRef CAS PubMed.
- R. W. Wheatley, R. B. Zheng, M. R. Richards, T. L. Lowary and K. K. S. Ng, J. Biol. Chem., 2012, 287, 28132–28143 CrossRef CAS PubMed.
- M. B. Poulin, R. Zhou and T. L. Lowary, Org. Biomol. Chem., 2012, 10, 4074–4087 CAS.
- A. K. Pathak, V. Pathak, L. Seitz, J. A. Maddry, S. S. Gurcha, G. S. Besra, W. J. Suling and R. C. Reynolds, Bioorg. Med. Chem., 2001, 9, 3129–3143 CrossRef CAS PubMed.
- J. Frigell, J. A. Pearcey, T. L. Lowary and I. Cumpstey, Eur. J. Org. Chem., 2011, 1367–1375 CrossRef CAS.
- S. Cren, S. S. Gurcha, A. J. Blake, G. S. Besra and N. R. Thomas, Org. Biomol. Chem., 2004, 2, 2418–2420 CAS.
- S. Cren, C. Wilson and N. R. Thomas, Org. Lett., 2005, 7, 3521–3523 CrossRef CAS PubMed.
- A. E. Trunkfield, S. S. Gurcha, G. S. Besra and T. D. H. Bugg, Bioorg. Med. Chem., 2010, 18, 2651–2663 CrossRef CAS PubMed.
- K. Vembaiyan, J. A. Pearcey, M. Bhasin, T. L. Lowary and W. Zou, Bioorg. Med. Chem., 2011, 19, 58–66 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed.
- C. Breton, S. Fournel-Gigleux and M. M. Palcic, Curr. Opin. Struct. Biol., 2012, 2, 540–549 CrossRef PubMed.
- R. J. Nash, A. Kato, C. Y. Yu and G. W. Fleet, Future Med. Chem., 2011, 3, 1513–1521 CrossRef CAS PubMed.
- H. Yuasa, T. Kajimoto and C. H. Wong, Tetrahedron Lett., 1994, 35, 8243–8246 CrossRef CAS.
- I. Izquierdo, M. T. Plaza and F. Aragon, Tetrahedron: Asymmetry, 1996, 7, 2567–2575 CrossRef CAS.
- L. Svansson, B. D. Johnston, J. H. Gu, B. Patrick and B. M. Pinto, J. Am. Chem. Soc., 2000, 122, 10769–10775 CrossRef CAS.
- M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G. Tanabe and O. Muraoka, Tetrahedron Lett., 1997, 38, 8367–8370 CrossRef CAS.
- M. Yoshikawa, T. Murakami, K. Yashiro and H. Matsuda, Chem. Pharm. Bull., 1998, 46, 1339–1340 CrossRef CAS PubMed.
- H. Matruda, T. Morikawa and M. Yoshikawa, Pure Appl. Chem., 2002, 74, 1301–1308 Search PubMed.
- S. Mohan and B. M. Pinto, Carbohydr. Res., 2007, 342, 1551–1580 CrossRef CAS PubMed.
- E. J. Rossi, L. Sim, D. A. Kuntz, D. Hahn, B. D. Johnston, A. Ghavami, M. G. Szczepina, N. S. Kumar, E. E. Sterchi, B. L. Nichols, B. M. Pinto and D. R. Rose, FEBS J., 2006, 273, 2673–2683 CrossRef CAS PubMed.
- M. Yoshikawa, F. M. Xu, S. Nakamura, T. Wang, H. Matsuda, G. Tanabe and O. Muraoka, Heterocycles, 2008, 75, 1397–1405 CrossRef CAS.
- G. Tanabe, W. J. Xie, A. Ogawa, C. N. Cao, T. Minematsu, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem. Lett., 2009, 19, 2195–2198 CrossRef CAS PubMed.
- K. Jayakanthan, S. Mohan and B. M. Pinto, J. Am. Chem. Soc., 2009, 131, 5621–5626 CrossRef CAS PubMed.
- W. J. Xie, G. Tanabe, J. Akaki, T. Morikawa, K. Ninomiya, T. Minematsu, M. Yoshikawa, X. M. Wu and O. Muraoka, Bioorg. Med. Chem., 2011, 19, 2015–2022 CrossRef CAS PubMed.
- B. D. Johnston, A. Ghavami, M. T. Jensen, B. Svensson and B. M. Pinto, J. Am. Chem. Soc., 2002, 124, 8245–8250 CrossRef CAS PubMed.
- S. Mohan, L. Sim, D. R. Rose and B. M. Pinto, Carbohydr. Res., 2007, 342, 901–912 CrossRef CAS PubMed.
-
(a) S. Mohan and B. M. Pinto, Nat. Prod. Rep., 2010, 27, 481–488 RSC;
(b) S. Mohan, R. Eskandari and B. M. Pinto, Acc. Chem. Res., 2014, 47, 211–225 CrossRef CAS PubMed.
- R. Eskandari, K. Jones, K. R. Reddy, K. Jayakanthan, M. Chaudet, D. R. Rose and B. M. Pinto, Chem. – Eur. J., 2011, 17, 14817–14825 CrossRef CAS PubMed.
- R. B. Snitynsky and T. L. Lowary, Org. Lett., 2014, 16, 212–215 CrossRef CAS PubMed.
- R. E. Maleczka Jr, L. R. Terrell, F. Geng and J. S. Ward III, Org. Lett., 2002, 4, 2841–2844 CrossRef.
- M. Hori, T. Kataoka, H. Shimizu, O. Komatsu and K. Hamada, J. Org. Chem., 1987, 52, 3668–3673 CrossRef CAS.
- M. Oki, Y. Yamada and S. Murata, Bull. Chem. Soc. Jpn., 1988, 61, 707–714 CrossRef CAS.
- L. Svansson, B. D. Johnston, J. H. Gu, B. Patrick and B. M. Pinto, J. Am. Chem. Soc., 2000, 122, 10769–10775 CrossRef CAS.
- H. J. G. Broxterman, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 1988, 29, 4893–4896 CrossRef CAS.
- V. Ulgar, J. G. Fernández-Bolanos and M. Bols, J. Chem. Soc., Perkin Trans. 1, 2002, 1242–1246 RSC.
- E. Gallienne, M. Benazza, G. Demailly, J. Bolte and M. Lemaire, Tetrahedron, 2005, 61, 4557–4568 CrossRef CAS.
- H. Yuasa, J. Takada and H. Hashimoto, Bioorg. Med. Chem. Lett., 2001, 11, 1137–1139 CrossRef CAS PubMed.
- N. L. Rose, R. B. Zheng, J. Pearcey, R. Zhou, G. C. Completo and T. L. Lowary, Carbohydr. Res., 2008, 343, 2130–2139 CrossRef CAS PubMed.
- G. C. Completo and T. L. Lowary, J. Org. Chem., 2008, 73, 4513–4525 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4md00067f |
|
| This journal is © The Royal Society of Chemistry 2014 |
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence









